
A PCR-RFLP Technique to Assess the Geographic Origin of Plasmodium falciparum Strains in Central America





Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico Analysis of the pfs47 Gene and Search for Candidate Restriction Enzymes
2.2. Construction of Phylogenetic Trees Based on Nucleotide and Amino Acid Sequences of pfs47
2.3. Amplification and Sequencing of the pfs47, pfs48/45, and pvs47 Genes
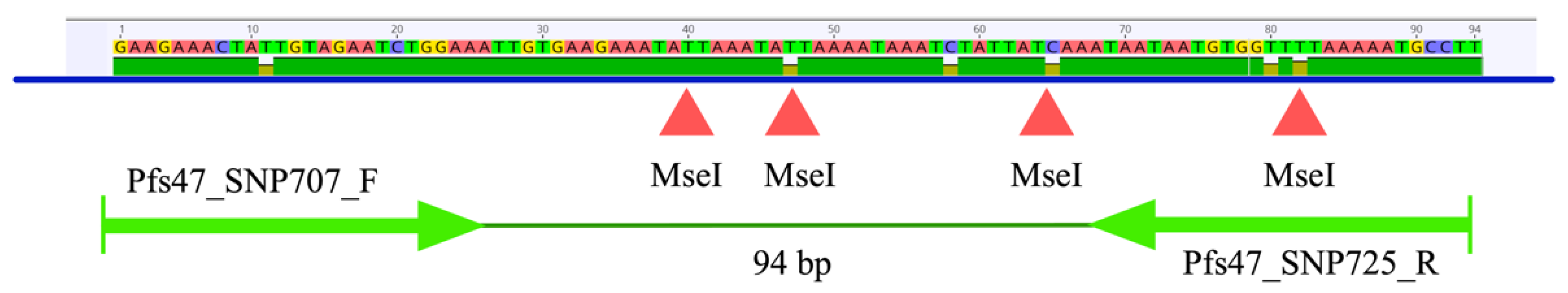
2.4. PCR-RFLP of pfs47
3. Results
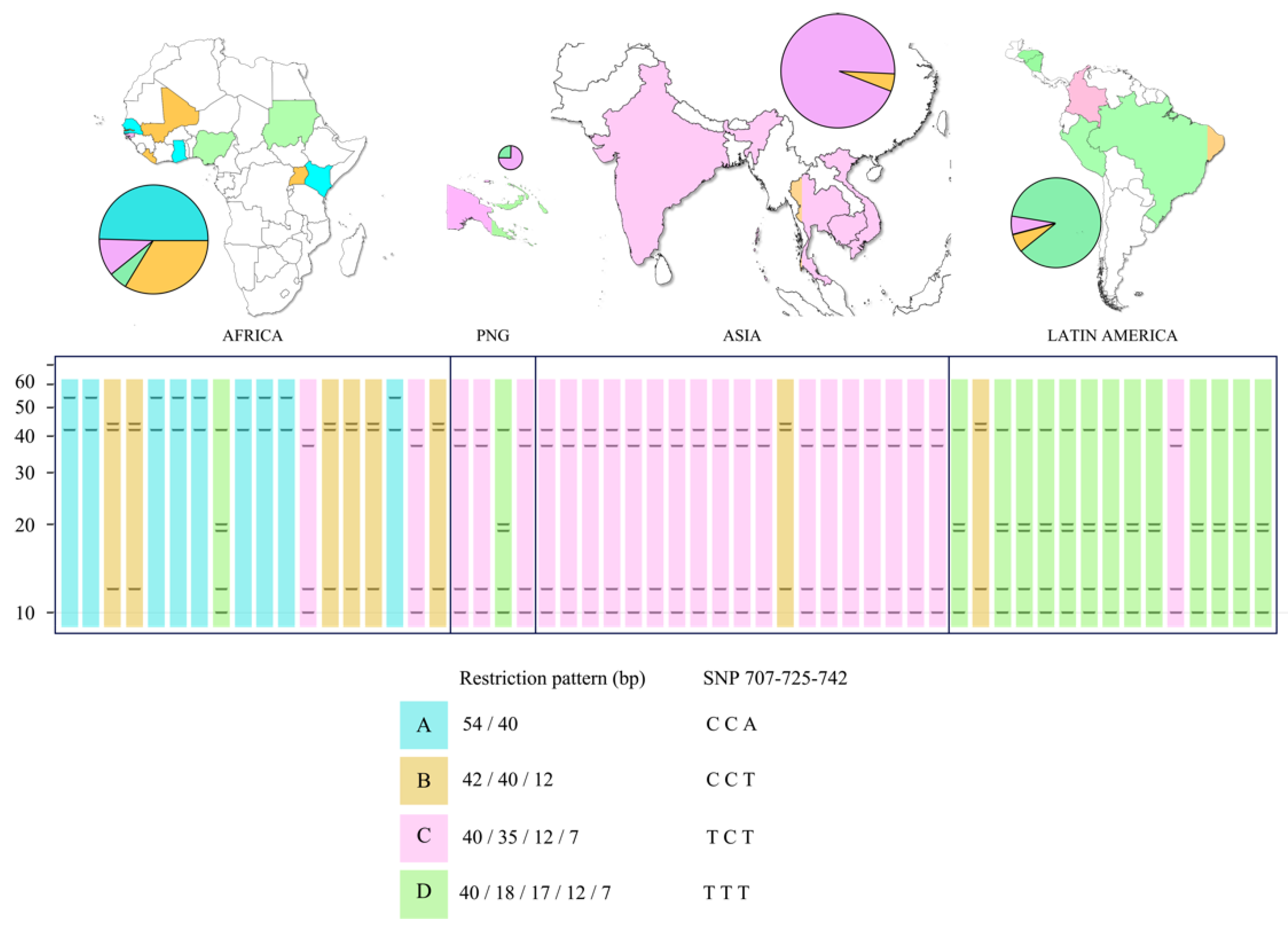
3.1. In Silico Analysis of the pfs47 Gene and PCR-RFLP Using Tru1I (MseI) to Reveal Informative Polymorphic Sites
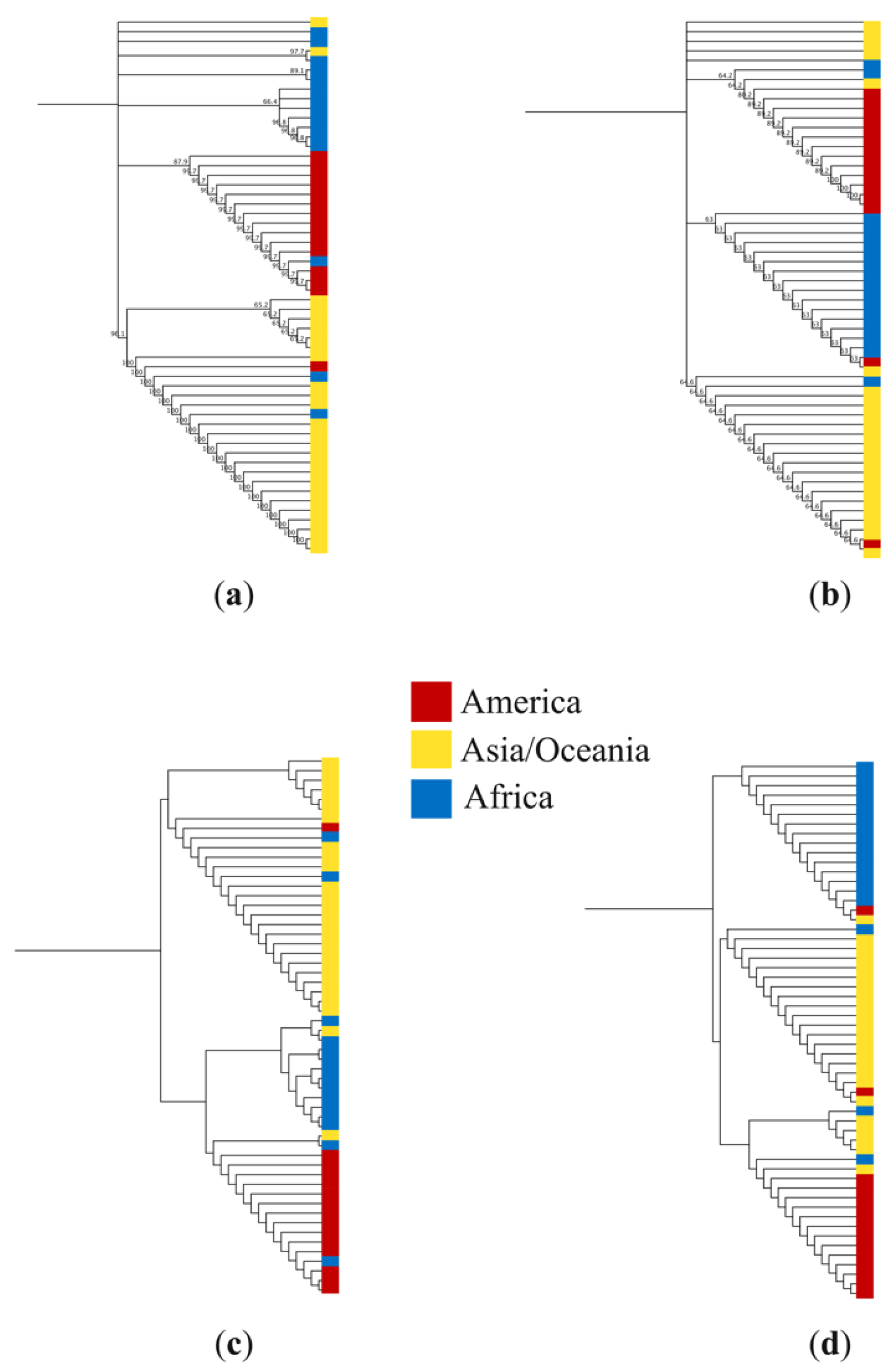
3.2. Phylogenetic Trees Based on Nucleotide and Amino Acid Sequences of pfs47
3.3. Sequencing of the pfs47, pfs48/45, and pvs47 Genes
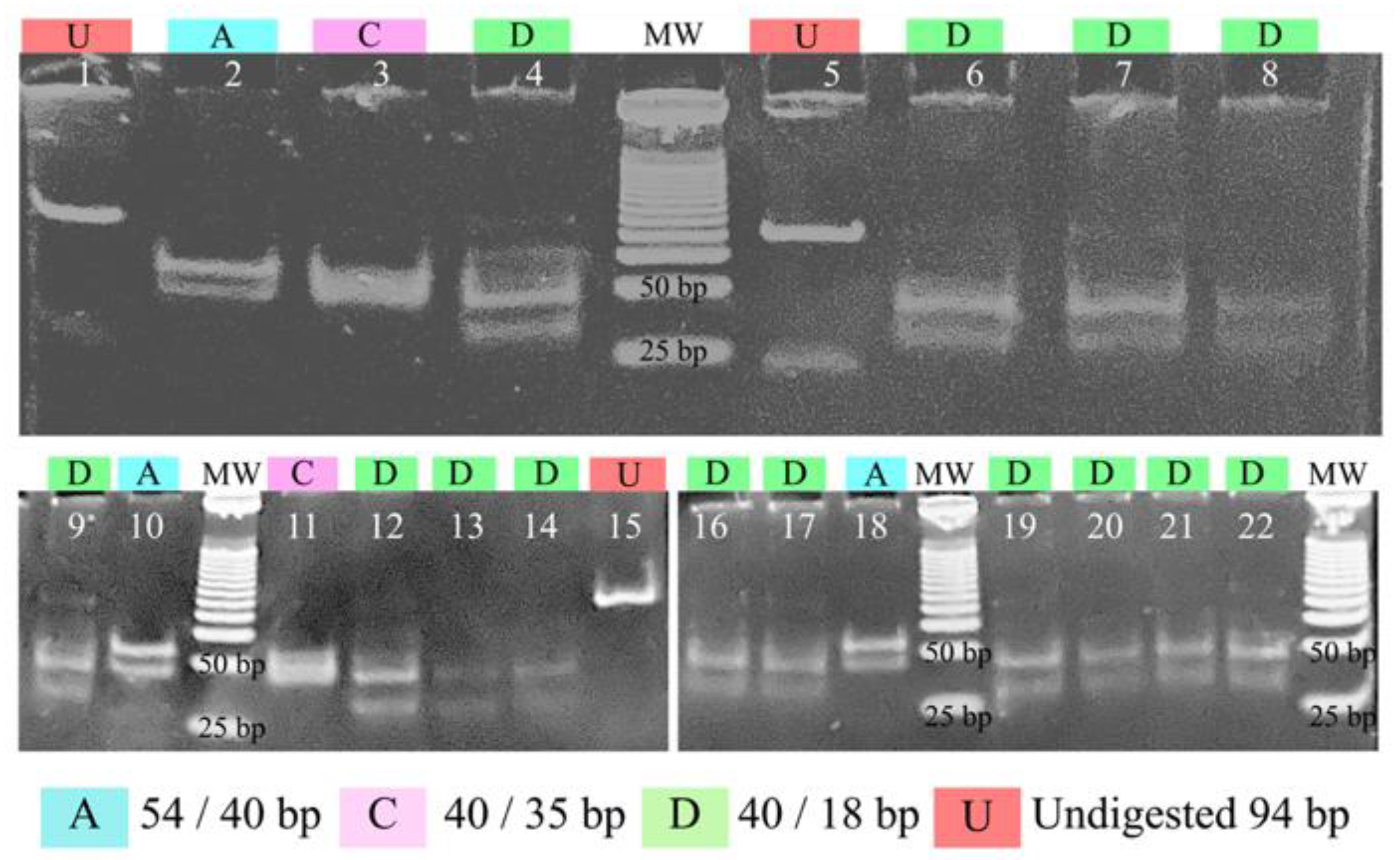
3.4. PCR-RFLP of pfs47
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2021; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- World Health Organization. World Malaria Report 2014; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Nieto-Zelaya, V.; Vanessa, G.; Alvarado-Claros, J.; García, J.; Alger, S.; Tovar-Calderón, J. Navarro, School child with Plasmodium falciparum malaria from Africa: Public health risk in Honduras. Rev. Med. Hondur. 2019, 87, 20–26. [Google Scholar]
- Valdivia, H.O.; Villena, F.E.; Lizewski, S.E.; Garcia, J.; Alger, J.; Bishop, D.K. Genomic surveillance of Plasmodium falciparum and Plasmodium vivax cases at the University Hospital in Tegucigalpa, Honduras. Sci. Rep. 2020, 10, 20975. [Google Scholar] [CrossRef]
- Higuita, N.A.; Suarez, J.A.; Millender, E.; Creighton, E.G.; Corbisiero, M.F.; Freites, C.O.; Cordero, J.H.; Kousari, A.; Unterborn, R.; Marcos, L.A.; et al. bound journey of migrant peoples InTransit across Dante’s Inferno and Purgatory in the Americas. Travel Med. Infect. Dis. 2022, 47, 102317. [Google Scholar] [CrossRef]
- Fontecha, G.; Pinto, A.; Archaga, O.; Betancourth, S.; Escober, L.; Henriquez, J.; Valdivia, H.O.; Montoya, A.; Mejía, R.E. Assessment of Plasmodium falciparum anti-malarial drug resistance markers in pfcrt and pfmdr1 genes in isolates from Honduras and Nicaragua, 2018–2021. Malar. J. 2021, 20, 465. [Google Scholar] [CrossRef] [PubMed]
- Preston, M.D.; Campino, S.; Assefa, S.A.; Echeverry, D.F.; Ocholla, H.; Amambua-Ngwa, A.; Stewart, L.B.; Conway, D.J.; Borrmann, S.; Michon, P.; et al. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat. Commun. 2014, 5, 4052. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Zou, Q.; Li, Y.; Zhu, G.; Zhou, H.; Zhang, M.; Tian, F.; Liu, Y.; Lu, F. A PCR-Based Technique to Track the Geographic Origin of Plasmodium falciparum with 23-SNP Barcode Analysis. Front. Public Health 2021, 9, 649170. [Google Scholar] [CrossRef] [PubMed]
- Diez Benavente, E.; Campos, M.; Phelan, J.; Nolder, D.; Dombrowski, J.G.; Marinho, C.R.F.; Sriprawat, K.; Taylor, A.R.; Watson, J.; Roper, C.; et al. A molecular barcode to inform the geographical origin and transmission dynamics of Plasmodium vivax malaria. PLoS Genet. 2020, 16, e1008576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, P.T.; Alves, J.M.; Santamaria, A.M.; Calzada, J.E.; Xayavong, M.; Parise, M.; da Silva, A.J.; Ferreira, M.U. Using mitochondrial genome sequences to track the origin of imported Plasmodium vivax infections diagnosed in the United States. Am. J. Trop. Med. Hyg. 2014, 90, 1102–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Tessema, S.K.; Murphy, M.; Xu, S.; Schwartz, A.; Wang, W.; Cao, Y.; Lu, F.; Tang, J.; Gu, Y.; et al. Confirmation of the absence of local transmission and geographic assignment of imported falciparum malaria cases to China using microsatellite panel. Malar. J. 2020, 19, 244. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cruz, A.; Raytselis, N.; Withers, R.; Dwivedi, A.; Crompton, P.D.; Traore, B.; Carpi, G.; Silva, J.C.; Barillas-Mury, C. A genotyping assay to determine geographic origin and transmission potential of Plasmodium falciparum malaria cases. Commun. Biol. 2021, 4, 1145. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, S.A.; Kappe, S.H.I. The s48/45 six-cysteine proteins: Mediators of interaction throughout the Plasmodium life cycle. Int. J. Parasitol. 2017, 47, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Nikolaeva, D.; Draper, S.J.; Biswas, S. Toward the development of effective transmission-blocking vaccines for malaria. Expert Rev. Vaccines 2015, 14, 653–680. [Google Scholar] [CrossRef]
- Sauerwein, R.W.; Bousema, T. Transmission blocking malaria vaccines: Assays and candidates in clinical development. Vaccine 2015, 33, 7476–7482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dijk, M.R.; Janse, C.J.; Thompson, J.; Waters, A.P.; Braks, J.A.; Dodemont, H.J.; Stunnenberg, H.G.; van Gemert, G.J.; Sauerwein, R.W.; Eling, W. A central role for P48/45 in malaria parasite male gamete fertility. Cell 2001, 104, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Ramphul, U.N.; Garver, L.S.; Molina-Cruz, A.; Canepa, G.E.; Barillas-Mury, C. Plasmodium falciparum evades mosquito immunity by disrupting JNK-mediated apoptosis of invaded midgut cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Cruz, A.; Garver, L.S.; Alabaster, A.; Bangiolo, L.; Haile, A.; Winikor, J.; Ortega, C.; van Schaijk, B.C.; Sauerwein, R.W.; Taylor-Salmon, E.; et al. The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Science 2013, 340, 984–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canepa, G.E.; Molina-Cruz, A.; Barillas-Mury, C. Molecular Analysis of Pfs47-Mediated Plasmodium Evasion of Mosquito Immunity. PLoS ONE 2016, 11, e0168279. [Google Scholar] [CrossRef]
- Molina-Cruz, A.; Barillas-Mury, C. The remarkable journey of adaptation of the Plasmodium falciparum malaria parasite to New World anopheline mosquitoes. Mem. Inst. Oswaldo Cruz 2014, 109, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cruz, A.; Canepa, G.E.; Kamath, N.; Pavlovic, N.V.; Mu, J.; Ramphul, U.N.; Ramirez, J.L.; Barillas-Mury, C. Plasmodium evasion of mosquito immunity and global malaria transmission: The lock-and-key theory. Proc. Natl. Acad. Sci. USA 2015, 112, 15178–15183. [Google Scholar] [CrossRef] [Green Version]
- Padley, D.J.; Heath, A.B.; Sutherland, C.; Chiodini, P.L.; Baylis, S.A.; Collaborative Study, G. Establishment of the 1st World Health Organization International Standard for Plasmodium falciparum DNA for nucleic acid amplification technique (NAT)-based assays. Malar. J. 2008, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Brito-Sousa, J.D.; Santos, T.C.; Avalos, S.; Fontecha, G.; Melo, G.C.; Val, F.; Siqueira, A.M.; Alecrim, G.C.; Bassat, Q.; Lacerda, M.V.; et al. Clinical Spectrum of Primaquine-induced Hemolysis in Glucose-6-Phosphate Dehydrogenase Deficiency: A 9-Year Hospitalization-based Study from the Brazilian Amazon. Clin Infect Dis 2019, 69, 1440–1442. [Google Scholar] [CrossRef] [PubMed]
- Anthony, T.G.; Polley, S.D.; Vogler, A.P.; Conway, D.J. Evidence of non-neutral polymorphism in Plasmodium falciparum gamete surface protein genes Pfs47 and Pfs48/45. Mol. Biochem. Parasitol. 2007, 156, 117–123. [Google Scholar] [CrossRef]
- Woo, M.K.; Kim, K.A.; Kim, J.; Oh, J.S.; Han, E.T.; An, S.S.; Lim, C.S. Sequence polymorphisms in Pvs48/45 and Pvs47 gametocyte and gamete surface proteins in Plasmodium vivax isolated in Korea. Mem. Inst. Oswaldo Cruz 2013, 108, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangdi, K.; Gatton, M.L.; Kelly, G.C.; Clements, A.C. Cross-border malaria: A major obstacle for malaria elimination. Adv. Parasitol. 2015, 89, 79–107. [Google Scholar] [PubMed] [Green Version]
- Iqbal, J.; Al-Awadhi, M.; Ahmad, S. Decreasing trend of imported malaria cases but increasing influx of mixed P. falciparum and P. vivax infections in malaria-free Kuwait. PLoS ONE 2020, 15, e0243617. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.A.; Mansur, F.A.F.; Abdul Rahman, N. Imported Falciparum Malaria: A case series in a tertiary hospital. Malays. J. Pathol. 2020, 42, 107–110. [Google Scholar]
- Oncel, K.; Sahin, A.; Esmer, F. Two Imported Plasmodium falciparum Malaria Cases in Sanliurfa. Turk. Parazitol. Derg. 2021, 45, 153–156. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, K.; Cheng, W.; Jiang, T.; Yao, Y.; Xu, M.; Yang, Y.; Tan, H.; Li, J. Molecular epidemiological surveillance of Africa and Asia imported malaria in Wuhan, Central China: Comparison of diagnostic tools during 2011–2018. Malar. J. 2020, 19, 321. [Google Scholar] [CrossRef]
- Chen, F.; He, J.; Dong, R.; Yang, F.; Liu, H.; Gu, D.; Wang, W. Application of SPR protein chip in screening for imported malaria. Sheng Wu Gong Cheng Xue Bao Chin. J. Biotechnol. 2021, 37, 1360–1367. [Google Scholar]
- Saoud, M.Z.; Ezzariga, N.; Benaissa, E.; Moustachi, A.; Lyagoubi, M.; Aoufi, S. Imported malaria: 54 cases diagnosed at the Ibn Sina Hospital Center in Rabat, Morocco. Med. Sante Trop. 2019, 29, 159–163. [Google Scholar] [PubMed]
- Seck, M.C.; Thwing, J.; Fall, F.B.; Gomis, J.F.; Deme, A.; Ndiaye, Y.D.; Daniels, R.; Volkman, S.K.; Ndiop, M.; Ba, M.; et al. Malaria prevalence, prevention and treatment seeking practices among nomadic pastoralists in northern Senegal. Malar. J. 2017, 16, 413. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.F.; Schaffner, S.F.; Dieye, Y.; Dieng, G.; Hainsworth, M.; Fall, F.B.; Diouf, C.N.; Ndiop, M.; Cisse, M.; Gueye, A.B.; et al. Genetic evidence for imported malaria and local transmission in Richard Toll, Senegal. Malar. J. 2020, 19, 276. [Google Scholar] [CrossRef] [PubMed]
- Escobar, D.F.; Lucchi, N.W.; Abdallah, R.; Valenzuela, M.T.; Udhayakumar, V.; Jercic, M.I.; Chenet, S.M. Molecular and epidemiological characterization of imported malaria cases in Chile. Malar. J. 2020, 19, 289. [Google Scholar] [CrossRef] [PubMed]
- Louzada, J.; de Almeida, N.C.V.; de Araujo, J.L.P.; Silva, J.; Carvalho, T.M.; Escalante, A.A.; Oliveira-Ferreira, J. The impact of imported malaria by gold miners in Roraima: Characterizing the spatial dynamics of autochthonous and imported malaria in an urban region of Boa Vista. Mem. Inst. Oswaldo Cruz 2020, 115, e200043. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Morales, A.J.; Suarez, J.A.; Risquez, A.; Villamil-Gomez, W.E.; Paniz-Mondolfi, A. Consequences of Venezuela’s massive migration crisis on imported malaria in Colombia, 2016–2018. Travel Med. Infect. Dis. 2019, 28, 98–99. [Google Scholar] [CrossRef] [PubMed]
- Buyon, L.E.; Santamaria, A.M.; Early, A.M.; Quijada, M.; Barahona, I.; Lasso, J.; Avila, M.; Volkman, S.K.; Marti, M.; Neafsey, D.E.; et al. Population genomics of Plasmodium vivax in Panama to assess the risk of case importation on malaria elimination. PLoS Negl. Trop. Dis. 2020, 14, e0008962. [Google Scholar] [CrossRef]
- Albuquerque, H.G.; Peiter, P.C.; Toledo, L.M.; Sabroza, P.C.; Pereira, R.D.S.; Caldas, J.P.; Angelo, J.R.; Dias, C.G.; Suárez-Mutis, M.C. Imported malaria in Rio de Janeiro state between 2007 and 2015: An epidemiologic approach. Mem. Inst. Oswaldo Cruz 2019, 114, e190064. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, N.C. Electrophoresis of DNA in agarose gels, polyacrylamide gels and in free solution. Electrophoresis 2009, 30 (Suppl. S1), S188–S195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.; Archaga, O.; Mejia, A.; Escober, L.; Henriquez, J.; Montoya, A.; Valdivia, H.O.; Fontecha, G. Evidence of a Recent Bottleneck in Plasmodium falciparum Populations on the Honduran-Nicaraguan Border. Pathogens 2021, 10, 1432. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Suwanabun, N.; Kaneko, O.; Iriko, H.; Otsuki, H.; Sattabongkot, J.; Kaneko, A.; Herrera, S.; Torii, M.; Tsuboi, T. Plasmodium vivax gametocyte proteins, Pvs48/45 and Pvs47, induce transmission-reducing antibodies by DNA immunization. Vaccine 2015, 33, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- de Jong, R.M.; Meerstein-Kessel, L.; Da, D.F.; Nsango, S.; Challenger, J.D.; van de Vegte-Bolmer, M.; van Gemert, G.J.; Duarte, E.; Teyssier, N.; Sauerwein, R.W.; et al. Monoclonal antibodies block transmission of genetically diverse Plasmodium falciparum strains to mosquitoes. NPJ Vaccines 2021, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Conway, D.J.; Machado, R.L.; Singh, B.; Dessert, P.; Mikes, Z.S.; Povoa, M.M.; Oduola, A.M.; Roper, C. Extreme geographical fixation of variation in the Plasmodium falciparum gamete surface protein gene Pfs48/45 compared with microsatellite loci. Mol. Biochem. Parasitol. 2001, 115, 145–156. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | 5′-3′Sequence | Gene | Product (bp) | References |
|---|---|---|---|---|
| Pfs47_SNP707_F | GAAGAAACTATTGTAGAATCTGGAAA | pfs47 | 94 | Molina Cruz et al. 2021 [12] |
| Pfs47SNP725_R | AAGGCATTTTTATAACCACATTATTA | |||
| Pfs48/45_F1 | GATCTTTTTACATATTTGCCG | pfs48/45 1st round | 577 | Anthony et al. 2007 [24] |
| Pfs48/45_R | CTTCATAATATTCAATATCTCC | |||
| Pfs48/45_F2 | GATCTTTTTACATATTTGCCG | pfs48/45 2nd round | 532 | Anthony et al. 2007 [24] |
| Pfs48/45_R | CTTCATAATATTCAATATCTCC | |||
| Pvs47_F | CACACCACCGCAAACAGG | pvs47 | 1525 | Woo et al. 2013 [25] |
| Pvs47_R | GTGCACATTCCGCGGTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontecha, G.; Escobar, D.; Ortiz, B.; Pinto, A. A PCR-RFLP Technique to Assess the Geographic Origin of Plasmodium falciparum Strains in Central America. Trop. Med. Infect. Dis. 2022, 7, 149. https://doi.org/10.3390/tropicalmed7080149
Fontecha G, Escobar D, Ortiz B, Pinto A. A PCR-RFLP Technique to Assess the Geographic Origin of Plasmodium falciparum Strains in Central America. Tropical Medicine and Infectious Disease. 2022; 7(8):149. https://doi.org/10.3390/tropicalmed7080149
Chicago/Turabian StyleFontecha, Gustavo, Denis Escobar, Bryan Ortiz, and Alejandra Pinto. 2022. "A PCR-RFLP Technique to Assess the Geographic Origin of Plasmodium falciparum Strains in Central America" Tropical Medicine and Infectious Disease 7, no. 8: 149. https://doi.org/10.3390/tropicalmed7080149
APA StyleFontecha, G., Escobar, D., Ortiz, B., & Pinto, A. (2022). A PCR-RFLP Technique to Assess the Geographic Origin of Plasmodium falciparum Strains in Central America. Tropical Medicine and Infectious Disease, 7(8), 149. https://doi.org/10.3390/tropicalmed7080149