
Introducing an Efficient In Vitro Cornea Mimetic Model for Testing Drug Permeability

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Characterization of Hydrogels
2.2. Cell Culture of Human Corneal Epithelial Cells
2.3. Permeability Testing
2.4. Quantification of Brinzolamide, Dexamethasone, Chloramphenicol, Timolol, Pilocarpine, and Betaxolol
2.5. Histology
2.6. Immunofluorescent Staining of Tight Junction Proteins
2.7. Data Analysis
2.8. Statistical Analysis
3. Results and Discussions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cholkar, K.; Patel, S.P.; Vadlapudi, A.D.; Mitra, A.K. Novel Strategies for Anterior Segment Ocular Drug Delivery. J. Ocul. Pharmacol. Ther. 2013, 29, 106–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantelli, F.; Mauris, J.; Argüeso, P. The ocular surface epithelial barrier and other mechanisms of mucosal protection: From allergy to infectious diseases. Curr. Opin. Allergy Clin. Immunol. 2013, 13, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Directive of the European Parliament and of the Council on the Protection of Animals Used for Scientific Purposes. Directive 2010/63/EU. 2010. Available online: https://eur-lex.europa.eu/eli/dir/2010/63/oj (accessed on 10 January 2021).
- Kouchak, M.; Bahmandar, R.; Bavarsad, N.; Farrahi, F. Ocular Dorzolamide Nanoliposomes for Prolonged IOP Reduction: In-vitro and in-vivo Evaluation in Rabbits. Iran. J. Pharm. Res. 2016, 15, 205–212. [Google Scholar] [PubMed]
- Palma, S.D.; Tártara, L.I.; Quinteros, D.; Allemandi, D.A.; Longhi, M.R.; Granero, G.E. An efficient ternary complex of acetazolamide with HP-ß-CD and TEA for topical ocular administration. J. Control. Release 2009, 138, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Makhmalzadeh, B.S.; Salimi, A.; Niroomand, A. Loratadine-Loaded Thermoresponsive Hydrogel: Characterization and Ex-vivo Rabbit Cornea Permeability Studies. Iran. J. Pharm. Res. 2018, 17, 460–469. [Google Scholar]
- Kaluzhny, Y.; Kinuthia, M.W.; Truong, T.; Lapointe, A.M.; Hayden, P.; Klausner, M. New Human Organotypic Corneal Tissue Model for Ophthalmic Drug Delivery Studies. Investig. Opthalmol. Vis. Sci. 2018, 59, 2880–2898. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ghezzi, C.E.; Gomes, R.; Pollard, R.E.; Funderburgh, J.L.; Kaplan, D.L. In vitro 3D corneal tissue model with epithelium, stroma, and innervation. Biomaterials 2017, 112, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hahne, M.; Zorn-Kruppa, M.; Guzman, G.; Brandner, J.M.; Haltner-Ukomado, E.; Wätzig, H.; Reichl, S. Prevalidation of a Human Cornea Construct as an Alternative to Animal Corneas for In Vitro Drug Absorption Studies. J. Pharm. Sci. 2012, 101, 2976–2988. [Google Scholar] [CrossRef]
- Hahne, M.; Reichl, S. Development of a serum-free human cornea construct for in vitro drug absorption studies: The influence of varying cultivation parameters on barrier characteristics. Int. J. Pharm. 2011, 416, 268–279. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Takezawa, T. Fabrication of a Corneal Model Composed of Corneal Epithelial and Endothelial Cells via a Collagen Vitrigel Membrane Functioned as an Acellular Stroma and Its Application to the Corneal Permeability Test of Chemicals. Drug Metab. Dispos. 2018, 46, 1684–1691. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.-E.; Basu, S.K.; Lee, V.H.L. Air-interface condition promotes the formation of tight corneal epithelial cell layers for drug transport studies. Pharm. Res. 2000, 17, 670–676. [Google Scholar] [CrossRef]
- Scholz, M.; Lin, J.-E.C.; Lee, V.H.L.; Keipert, S. Pilocarpine Permeability across Ocular Tissues and Cell Cultures: Influence of Formulation Parameters. J. Ocul. Pharmacol. Ther. 2002, 18, 455–468. [Google Scholar] [CrossRef]
- Reichl, S.; Kölln, C.; Hahne, M.; Verstraelen, J. In vitro cell culture models to study the corneal drug absorption. Expert Opin. Drug Metab. Toxicol. 2011, 7, 559–578. [Google Scholar] [CrossRef]
- Almubrad, T.; Akhtar, S. Structure of corneal layers, collagen fibrils, and proteoglycans of tree shrew cornea. Mol. Vis. 2011, 17, 2283–2291. [Google Scholar]
- Ihanamäki, T.; Pelliniemi, L.J.; Vuorio, E. Collagens and collagen-related matrix components in the human and mouse eye. Prog. Retin. Eye Res. 2004, 23, 403–434. [Google Scholar] [CrossRef] [PubMed]
- Korjamo, T.; Heikkinen, A.; Waltari, P.; Mönkkönen, J. The Asymmetry of the Unstirred Water Layer in Permeability Experiments. Pharm. Res. 2008, 25, 1714–1722. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.A.; Donato, D.M.; Balcioglu, H.E.; Schmidt, T.; Danen, E.H.; Koenderink, G.H. A guide to mechanobiology: Where biology and physics meet. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1853, 3043–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerholm, P.; Lagali, N.S.; Ong, J.A.; Merrett, K.; Jackson, W.B.; Polarek, J.W.; Suuronen, E.J.; Liu, Y.; Brunette, I.; Griffith, M. Stable corneal regeneration four years after implantation of a cell-free recombinant human collagen scaffold. Biomaterials 2014, 35, 2420–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gan, L.; Carlsson, D.J.; Fagerholm, P.; Lagali, N.; Watsky, M.A.; Munger, R.; Hodge, W.G.; Priest, D.; Griffith, M. A Simple, Cross-linked Collagen Tissue Substitute for Corneal Implantation. Investig. Opthalmol. Vis. Sci. 2006, 47, 1869–1875. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Deng, C.; McLaughlin, C.R.; Fagerholm, P.; Lagali, N.; Heyne, B.; Scaiano, J.; Watsky, M.A.; Kato, Y.; Munger, R.; et al. Collagen–phosphorylcholine interpenetrating network hydrogels as corneal substitutes. Biomaterials 2009, 30, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Haagdorens, M.; Cėpla, V.; Melsbach, E.; Koivusalo, L.; Skottman, H.; Griffith, M.; Valiokas, R.; Zakaria, N.; Pintelon, I.; Tassignon, M.-J. In Vitro Cultivation of Limbal Epithelial Stem Cells on Surface-Modified Crosslinked Collagen Scaffolds. Stem Cells Int. 2019, 2019, 7867613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domke, J.; Radmacher, M. Measuring the Elastic Properties of Thin Polymer Films with the Atomic Force Microscope. Langmuir 1998, 14, 3320–3325. [Google Scholar] [CrossRef]
- Sokolov, I.; Dokukin, M.E.; Guz, N.V. Method for quantitative measurements of the elastic modulus of biological cells in AFM indentation experiments. Methods 2013, 60, 202–213. [Google Scholar] [CrossRef]
- Hutter, J.L.; Bechhoefer, J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 1993, 64, 1868–1873. [Google Scholar] [CrossRef] [Green Version]
- Araki-Sasaki, K.; Ohashi, Y.; Sasabe, T.; Hayashi, K.; Watanabe, H.; Tano, Y.; Handa, H. An SV40-immortalized human corneal epithelial cell line and its characterization. Investig. Ophthalmol. Vis. Sci. 1995, 36, 614–621. [Google Scholar]
- Hakkarainen, J.J.; Reinisalo, M.; Ragauskas, S.; Seppänen, A.; Kaja, S.; Kalesnykas, G. Acute cytotoxic effects of marketed ophthalmic formulations on human corneal epithelial cells. Int. J. Pharm. 2016, 511, 73–78. [Google Scholar] [CrossRef]
- Toropainen, E.; Ranta, V.P.; Talvitie, A.; Suhonen, P.; Urtti, A. Culture model of human corneal epithelium for prediction of ocular drug absorption. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2942–2948. [Google Scholar]
- D’Este, M.; Eglin, D.; Alini, M. A systematic analysis of DMTMM vs EDC/NHS for ligation of amines to Hyaluronan in water. Carbohydr. Polym. 2014, 108, 239–246. [Google Scholar] [CrossRef]
- Balion, Z.; Cėpla, V.; Svirskiene, N.; Svirskis, G.; Druceikaitė, K.; Inokaitis, H.; Rusteikaitė, J.; Masilionis, I.; Stankevičienė, G.; Jelinskas, T.; et al. Cerebellar Cells Self-Assemble into Functional Organoids on Synthetic, Chemically Crosslinked ECM-Mimicking Peptide Hydrogels. Biomolecules 2020, 10, 754. [Google Scholar] [CrossRef]
- Taylor, Z.D.; Garritano, J.; Sung, S.; Bajwa, N.; Bennett, D.B.; Nowroozi, B.; Tewari, P.; Sayre, J.W.; Hubschman, J.-P.; Deng, S.X.; et al. THz and mm-Wave Sensing of Corneal Tissue Water Content: In Vivo Sensing and Imaging Results. IEEE Trans. Terahertz Sci. Technol. 2015, 5, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Tian, L.; Zhang, H.; Chen, X.; Li, L. Evaluation of corneal elastic modulus based on Corneal Visualization Scheimpflug Technology. Biomed. Eng. Online 2019, 18, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, C.; Gay, S.; Rhodes, R.; Pfister, R.; Miller, E.J. Collagen heterogeneity in normal rabbit cornea. I. Isolation and biochemical characterization of the genetically-distinct collagens. Biochim. Biophys. Acta (BBA) Protein Struct. 1980, 625, 78–88. [Google Scholar] [CrossRef]
- Kimura, K.; Mori, T.; Hadachi, H.; Saito, T.; Nishida, T.; Kawano, S.; Inoue, J. Quantitative Analysis of the Effects of Extracellular Matrix Proteins on Membrane Dynamics Associated with Corneal Epithelial Cell Motility. Investig. Opthalmol. Vis. Sci. 2010, 51, 4492–4499. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Gong, H.; Richman, R.; Freddo, T.F. Localization of occludin, ZO-1, and pan-cadherin in rabbit ciliary epithelium and iris vascular endothelium. Histochem. Cell Biol. 2000, 114, 303–310. [Google Scholar] [CrossRef]
- Huang, A.J.; Tseng, S.C.; Kenyon, K.R. Paracellular permeability of corneal and conjunctival epithelia. Investig. Ophthalmol. Vis. Sci. 1989, 30, 684–689. [Google Scholar]
- Sasaki, H.; Yamamura, K.; Tei, C.; Nishida, K.; Nakamura, J. Ocular Permeability of FITC-Dextran with Absorption Promoter for Ocular Delivery of Peptide Drug. J. Drug Target. 1995, 3, 129–135. [Google Scholar] [CrossRef]
- Becker, U.; Ehrhardt, C.; Daum, N.; Baldes, C.; Schaefer, U.F.; Ruprecht, K.W.; Kim, K.-J.; Lehr, C.-M. Expression of ABC-Transporters in Human Corneal Tissue and the Transformed Cell Line, HCE-T. J. Ocul. Pharmacol. Ther. 2007, 23, 172–181. [Google Scholar] [CrossRef]
- Schoenwald, R.D. Ocular Drug Delivery. Clin. Pharmacokinet. 1990, 18, 255–269. [Google Scholar] [CrossRef]
- Prausnitz, M.R.; Noonan, J.S. Permeability of cornea, sclera, and conjunctiva: A literature analysis for drug delivery to the eye. J. Pharm. Sci. 1998, 87, 1479–1488. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary. 2021. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 13 February 2021).
- Tomita, M.; Menconi, M.J.; Delude, R.L.; Fink, M.P. Polarized transport of hydrophilic compounds across rat colonic mucosa from serosa to mucosa is temperature dependent. Gastroenterology 2000, 118, 535–543. [Google Scholar] [CrossRef]
- Duvvuri, M.; Gong, Y.; Chatterji, D.; Krise, J.P. Weak Base Permeability Characteristics Influence the Intracellular Sequestration Site in the Multidrug-resistant Human Leukemic Cell Line HL-60. J. Biol. Chem. 2004, 279, 32367–32372. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Hakkarainen, J.J.; Pajander, J.; Laitinen, R.; Suhonen, M.; Forsberg, M. Similar molecular descriptors determine the in vitro drug permeability in endothelial and epithelial cells. Int. J. Pharm. 2012, 436, 426–443. [Google Scholar] [CrossRef]
- Liu, X.; Testa, B.; Fahr, A. Lipophilicity and Its Relationship with Passive Drug Permeation. Pharm. Res. 2010, 28, 962–977. [Google Scholar] [CrossRef]
- Mitra, A.K.; Mikkelson, T.J. Mechanism of Transcorneal Permeation of Pilocarpine. J. Pharm. Sci. 1988, 77, 771–775. [Google Scholar] [CrossRef]
- Verstraelen, J.; Reichl, S. Multidrug Resistance-Associated Protein (MRP1, 2, 4 and 5) Expression in Human Corneal Cell Culture Models and Animal Corneal Tissue. Mol. Pharm. 2014, 11, 2160–2171. [Google Scholar] [CrossRef]
- Rubelowski, A.-K.; Latta, L.; Katiyar, P.; Stachon, T.; Käsmann-Kellner, B.; Seitz, B.; Szentmáry, N. HCE-T cell line lacks cornea-specific differentiation markers compared to primary limbal epithelial cells and differentiated corneal epithelium. Graefe’s Arch. Clin. Exp. Ophthalmol. 2020, 258, 565–575. [Google Scholar] [CrossRef]

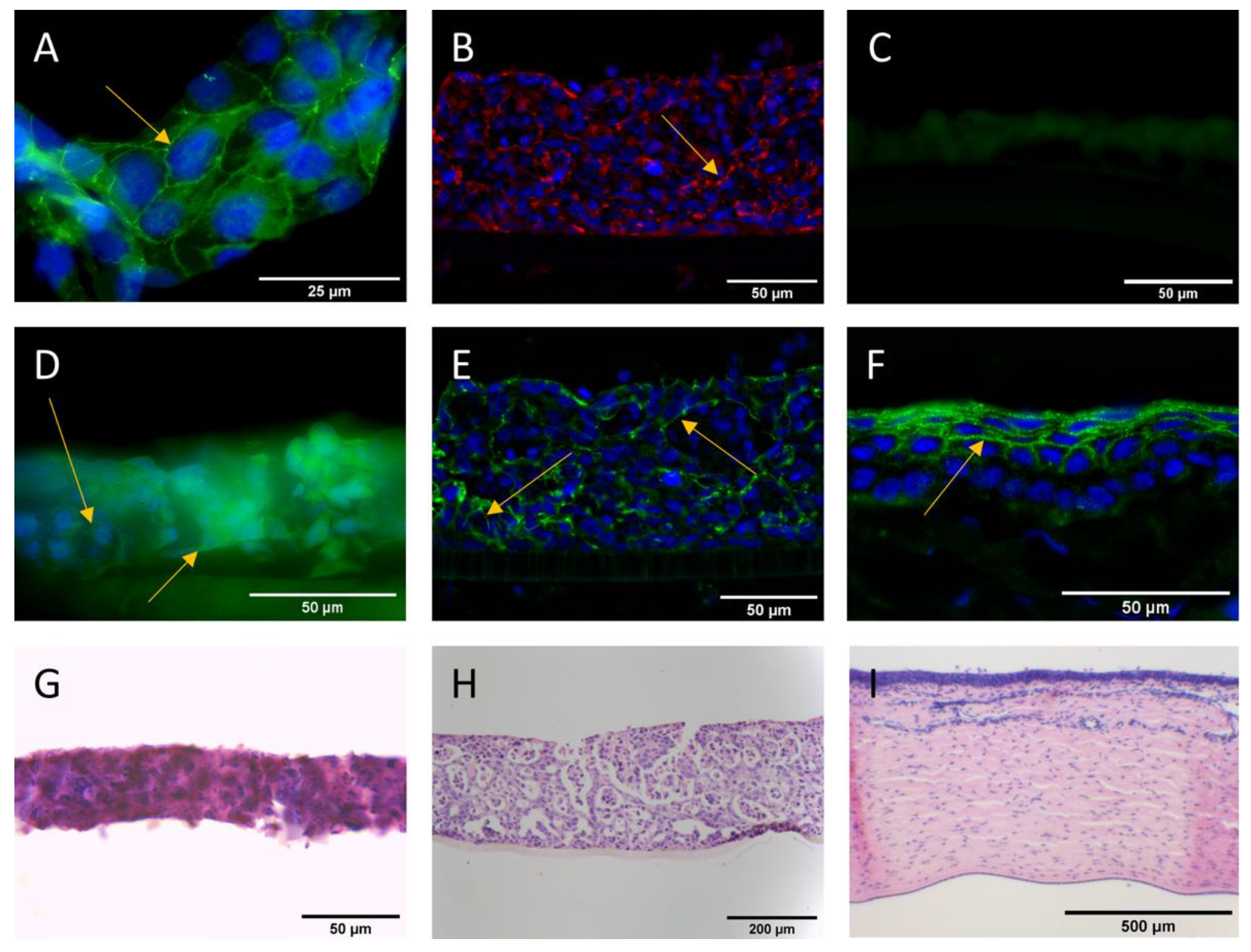

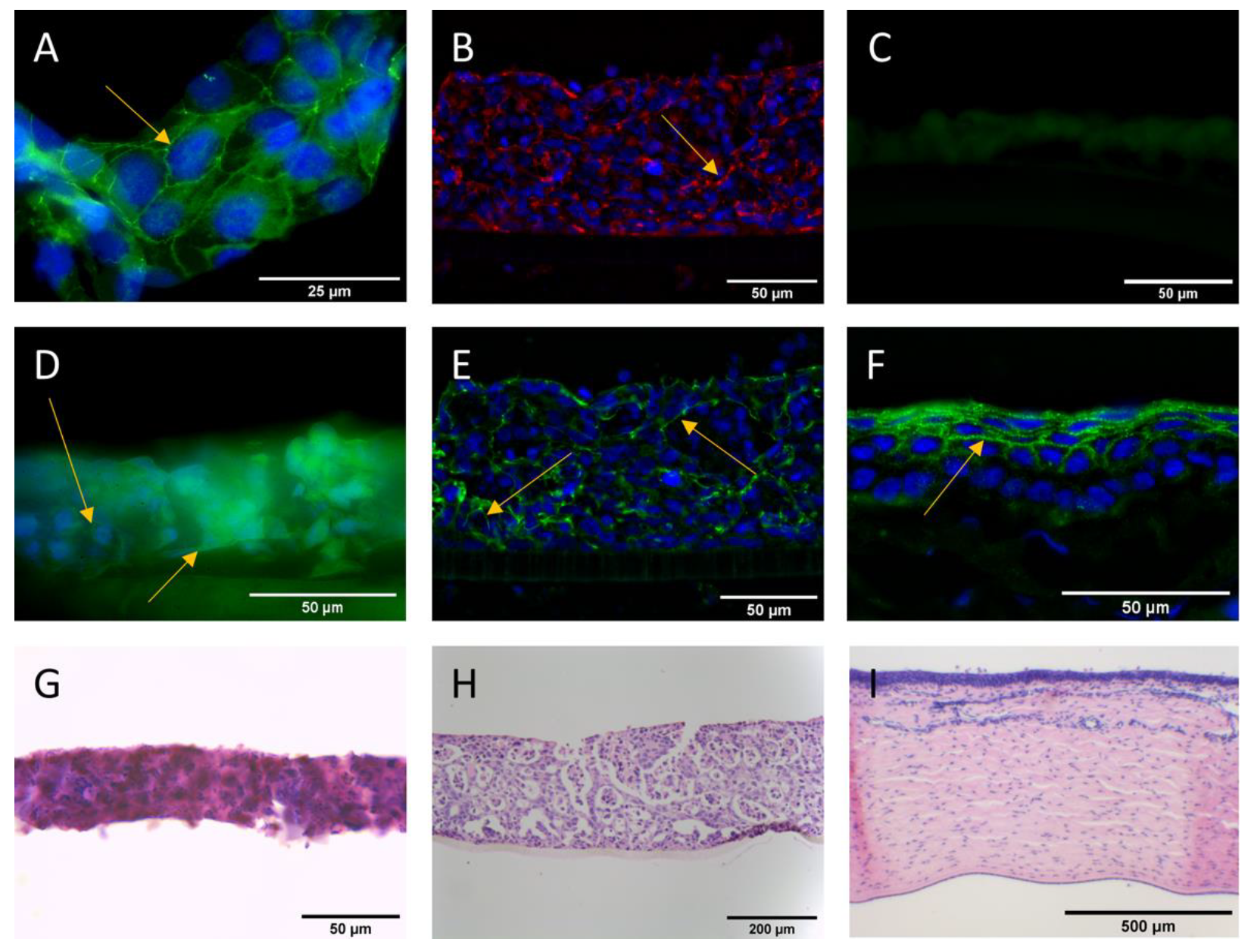
{kind=link}
{kind=link}
{kind=link}
| Ophthalmic Drug/Reference Molecule | Supplier | Concentration Tested |
|---|---|---|
| 6-carboxyfluorescein (6-CF) | Sigma-Aldrich | 50 µM |
| Rhodamine B (Rho B) | Sigma-Aldrich | 50 µM |
| FITC-dextran, 4 kDa (FD4) | Sigma-Aldrich | 50 µM |
| FITC-dextran, 70 kDa (FD70) | Sigma-Aldrich | 200 µg/mL |
| Rhodamine 123 (Rho 123) | Sigma-Aldrich | 10 µM |
| Betaxolol (Beta) | Cayman Chemicals | 10 µM |
| (+)-Pilocarpine HCl (Pilo) | Cayman Chemicals | 10 µM |
| Timolol maleate (Timo) | Cayman Chemicals | 10 µM |
| Chloramphenicol (Chlora) | BioChemica, AppliChem | 80 µM |
| Dexamethasone (Dexa) | Cayman Chemicals | 100 µM |
| Brinzolamide (Brinzo) | Cayman Chemicals | 89 µM |
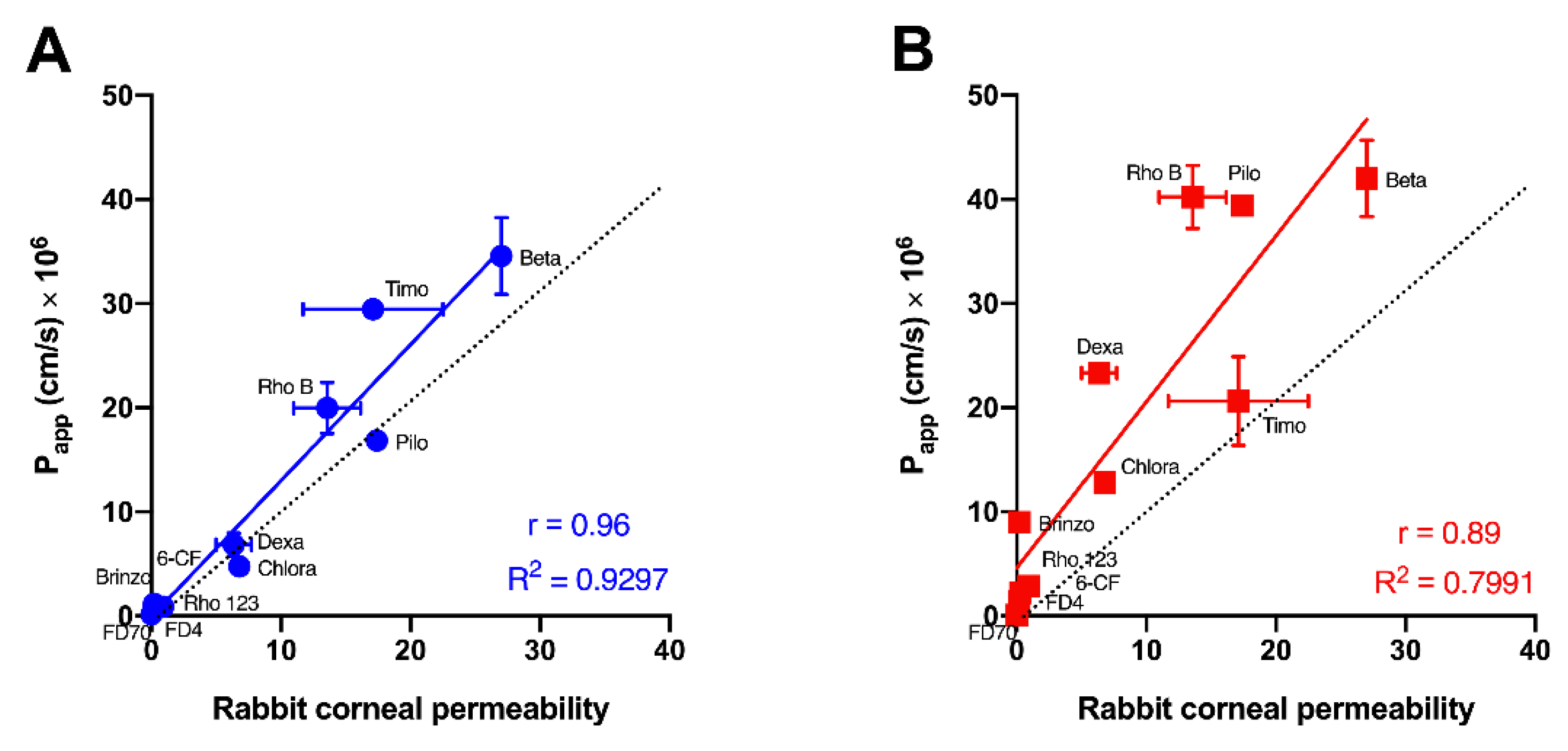
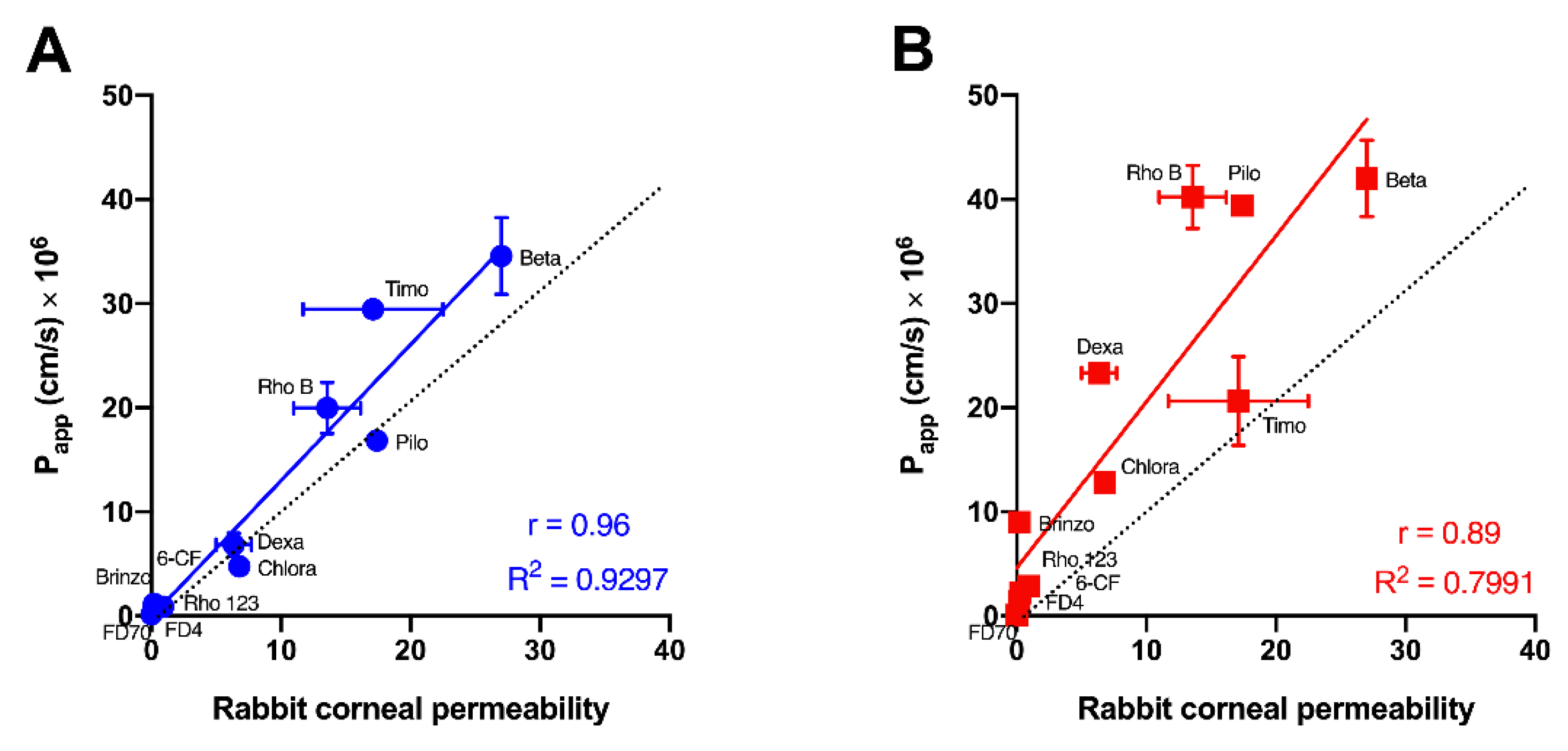
| Test Molecule | Molecular Weight * | LogP | Papp (cm/s) × 106 (Mean ± SEM) | ||||
|---|---|---|---|---|---|---|---|
| Hydrogel-Based Human Corneal Model | Cells Grown in Inserts | Rabbit Cornea | Rabbit Cornea (Values from Literature) | Ref. | |||
| FITC-dextran 70 kDa | 70,000 | −3.29 # | 0.16 ± 0.08 | 0.07 ± 0.01 | nd | Impermeable | [36] |
| FITC-dextran 4 kDa | 4000 | −3.41 # | 0.80 ± 0.52 | 1.43 ± 0.25 | 0.40 ± 0.09 | 0.056, 0.09 | [10,37] |
| Rhodamine 123 | 380.8 | 1.06 % | 0.96 ± 0.27 | 2.22 ± 0.05 | 0.42 ± 0.17 | 0.15 | [38] |
| 6-carboxy-fluorescein | 376.3 | −3.1 $ | 0.89 ± 0.16 | 2.87 ± 0.30 | 1.4 ± 0.2 | 0.46 | [28] |
| Brinzolamide | 383.5 | −1.8 * | 1.17 ± 0.36 | 8.99 ± 0.05 | nd | 0.2 | [5] |
| Dexamethasone | 392.5 | 1.83 * | 6.86 ± 1.13 | 23.32 ± 0.20 | nd | 5, 7.7 | [9,39] |
| Chloramphenicol | 323.1 | 1.14 * | 4.75 ± 0.39 | 12.79 ± 0.54 | nd | 6.8 | [39] |
| Timolol maleate | 316.4 | 1.1 * | 29.47 ± 1.05 | 20.62 ± 4.27 | nd | 11.7, 22.5 | [9,39] |
| Pilocarpine | 208.3 | 1.1 * | 16.86 ± 0.58 | 39.4 ± 0.97 | nd | 17.4 | [39] |
| Rhodamine B | 479.0 | 2.3 * | 19.98 ± 2.47 | 40.22 ± 3.02 | 9.1 ± 0.9 | 13.5, 18.1 | [10,28] |
| Betaxolol | 307.4 | 2.81 * | 34.56 ± 3.69 | 42.00 ± 3.66 | nd | 27 | [40] |
| Test Molecule | Hydrogel-Based Human Corneal Model vs. Rabbit Corneal Permeability | Cells Grown in Inserts vs. Rabbit Corneal Permeability | Hydrogel-Based Human Corneal Model vs. Cells Grown in Inserts |
|---|---|---|---|
| FITC-dextran 70 kDa | 0.0219 * | 0.3789 ns | 0.0077 ** |
| FITC-dextran 4 kDa | 0.0081 ** | <0.0001 **** | 0.0512 ns |
| Rhodamine 123 | 0.0044 ** | <0.0001 **** | <0.0001 **** |
| 6-carboxy-fluorescein | 0.9956 ns | <0.0001 **** | <0.0001 **** |
| Brinzolamide | 0.0650 ns | <0.0001 **** | <0.0001 **** |
| Dexamethasone | 0.7991 ns | <0.0001 **** | <0.0001 **** |
| Chloramphenicol | 0.0229 * | 0.0002 *** | <0.0001 **** |
| Timolol maleate | 0.0084 ** | 0.4509 ns | 0.0488 * |
| Pilocarpine | 0.8231 ns | <0.0001 **** | <0.0001 **** |
| Rhodamine B | 0.0089 ** | <0.0001 **** | <0.0001 **** |
| Betaxolol | 0.2858 ns | 0.0513 ns | 0.1389 ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Žiniauskaitė, A.; Cėpla, V.; Jelinskas, T.; Eimont, R.; Ulčinas, A.; Aldonytė, R.; Valiokas, R.; Kalesnykas, G.; Hakkarainen, J.J. Introducing an Efficient In Vitro Cornea Mimetic Model for Testing Drug Permeability. Sci 2021, 3, 30. https://doi.org/10.3390/sci3030030
Žiniauskaitė A, Cėpla V, Jelinskas T, Eimont R, Ulčinas A, Aldonytė R, Valiokas R, Kalesnykas G, Hakkarainen JJ. Introducing an Efficient In Vitro Cornea Mimetic Model for Testing Drug Permeability. Sci. 2021; 3(3):30. https://doi.org/10.3390/sci3030030
Chicago/Turabian StyleŽiniauskaitė, Agnė, Vytautas Cėpla, Tadas Jelinskas, Romuald Eimont, Artūras Ulčinas, Rūta Aldonytė, Ramūnas Valiokas, Giedrius Kalesnykas, and Jenni J. Hakkarainen. 2021. "Introducing an Efficient In Vitro Cornea Mimetic Model for Testing Drug Permeability" Sci 3, no. 3: 30. https://doi.org/10.3390/sci3030030
APA StyleŽiniauskaitė, A., Cėpla, V., Jelinskas, T., Eimont, R., Ulčinas, A., Aldonytė, R., Valiokas, R., Kalesnykas, G., & Hakkarainen, J. J. (2021). Introducing an Efficient In Vitro Cornea Mimetic Model for Testing Drug Permeability. Sci, 3(3), 30. https://doi.org/10.3390/sci3030030