
Elucidation of Structures, Electronic Properties, and Chemical Bonding of Monophosphorus-Substituted Boron Clusters in Neutral, Negative, and Positively Charged PBn/


Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Growth Behavior of Different Sized Phosphorus-Doped Boron Clusters
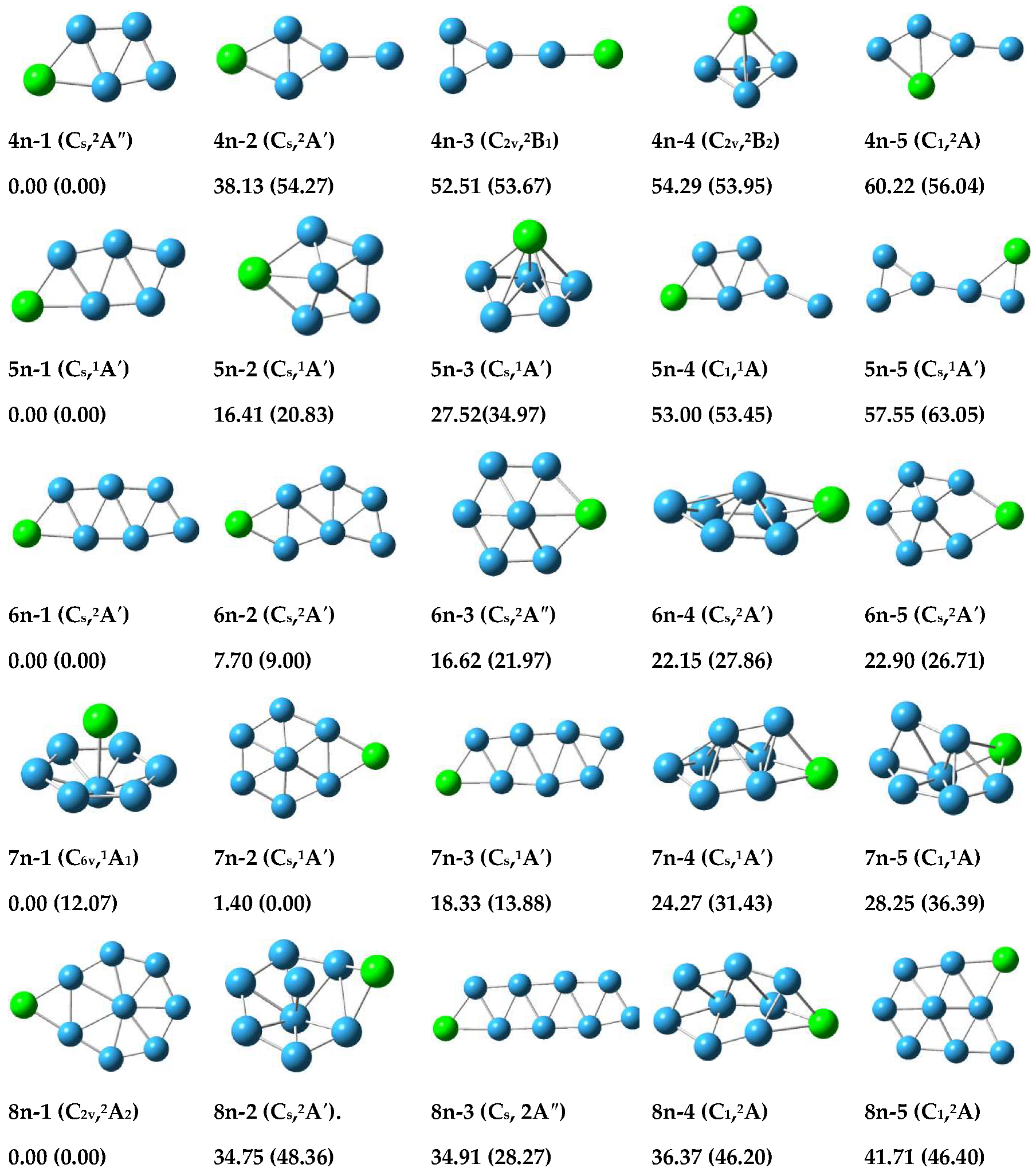
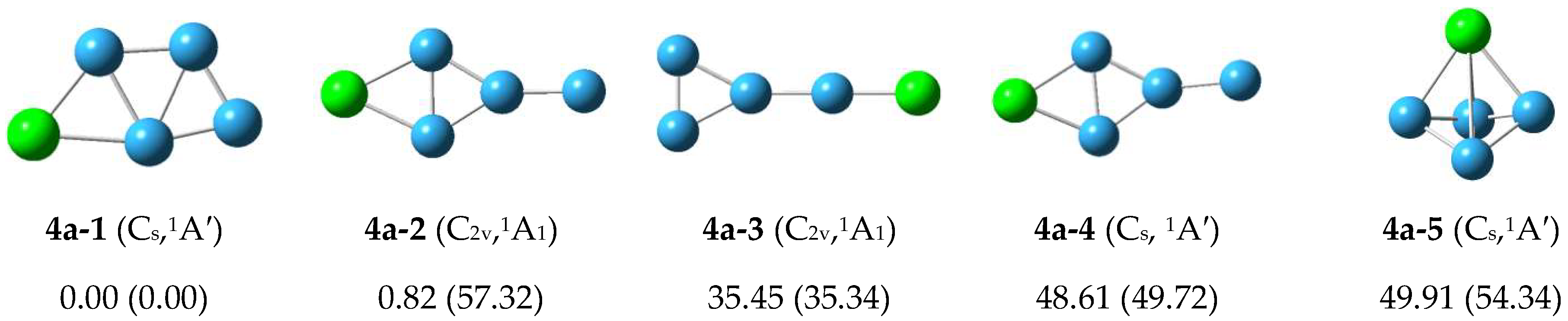
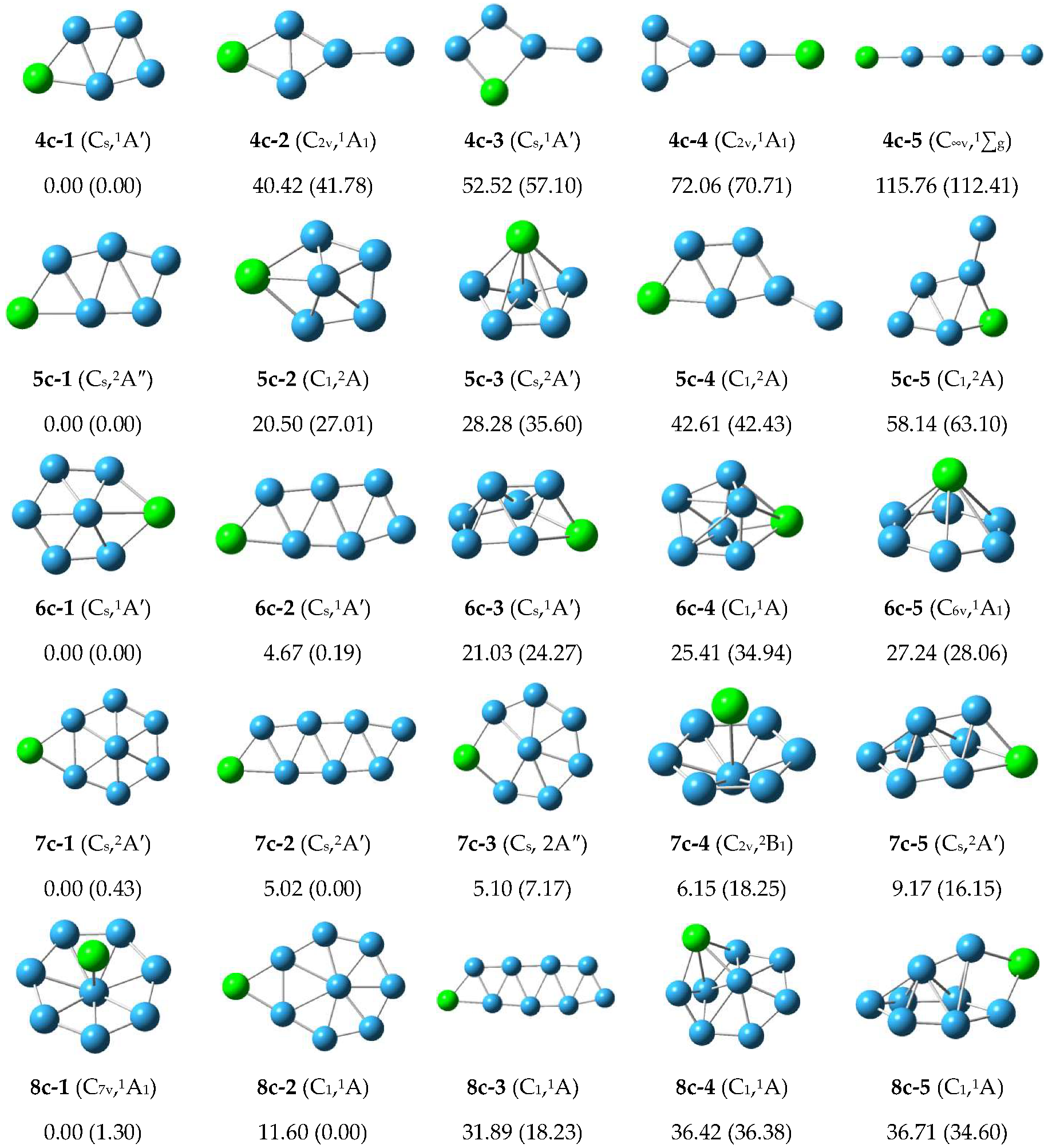
3.1.1. Structure of Neutral PBn (n = 4–8) Clusters
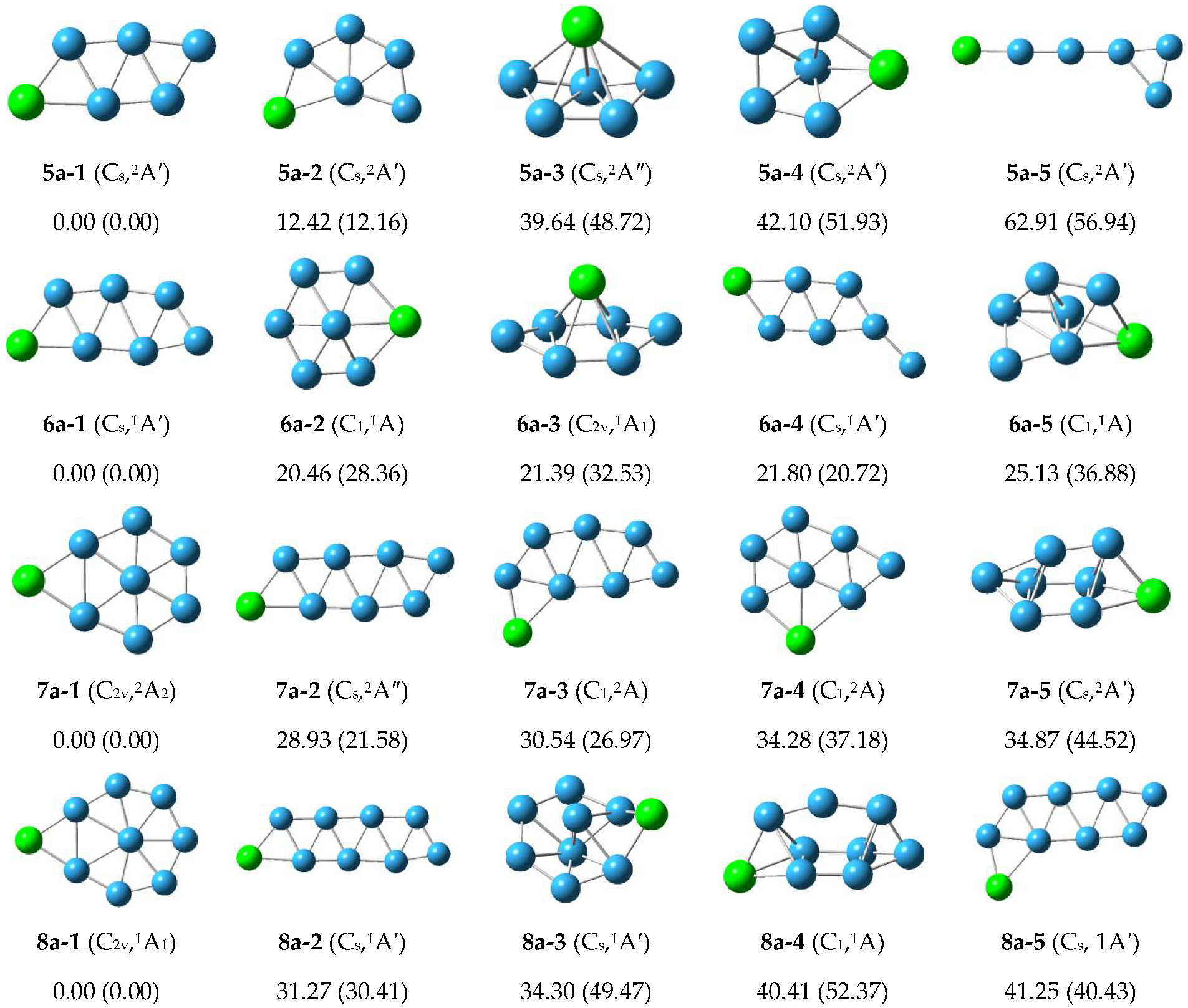
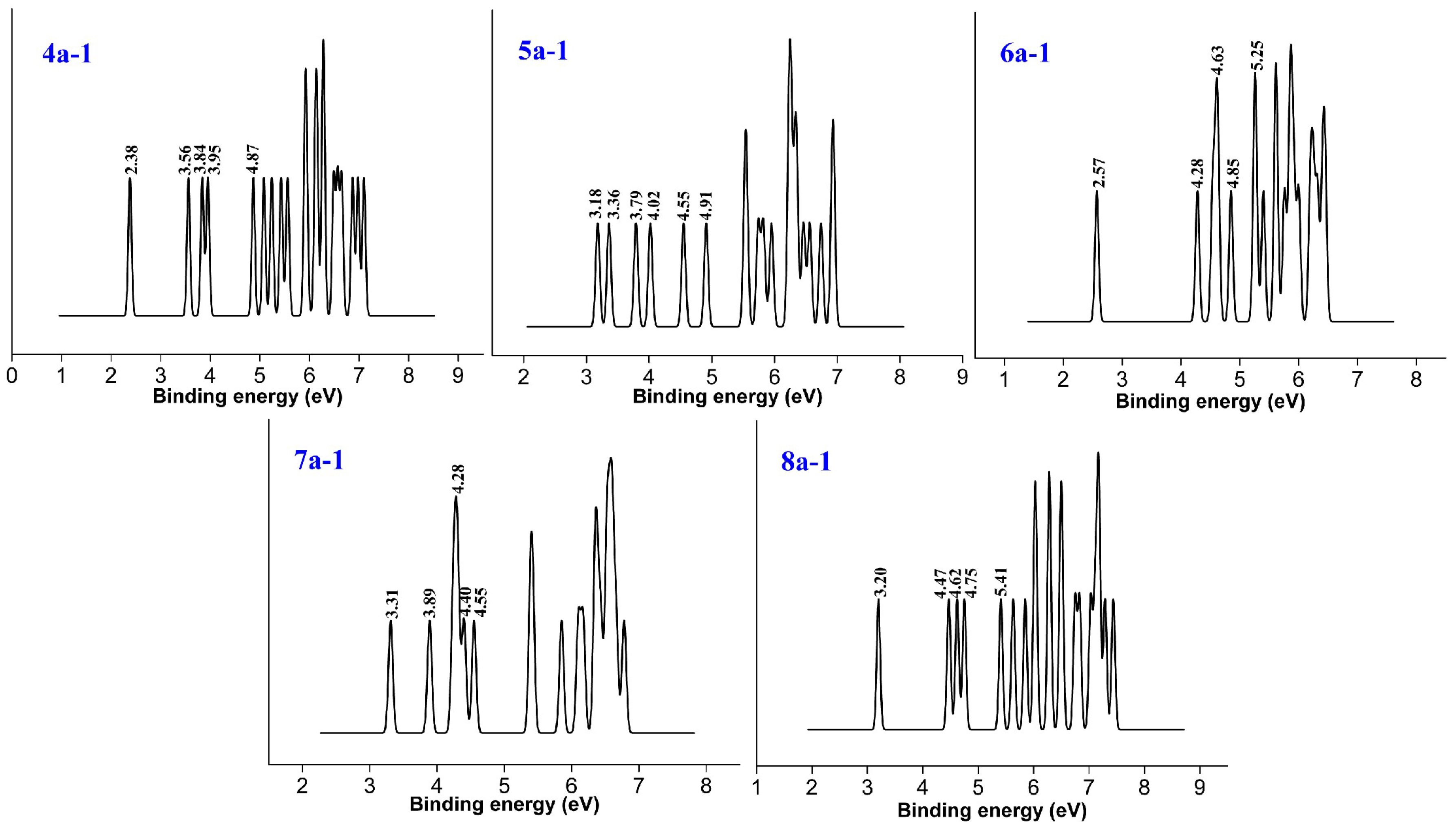
3.1.2. Structure of Anionic Clusters
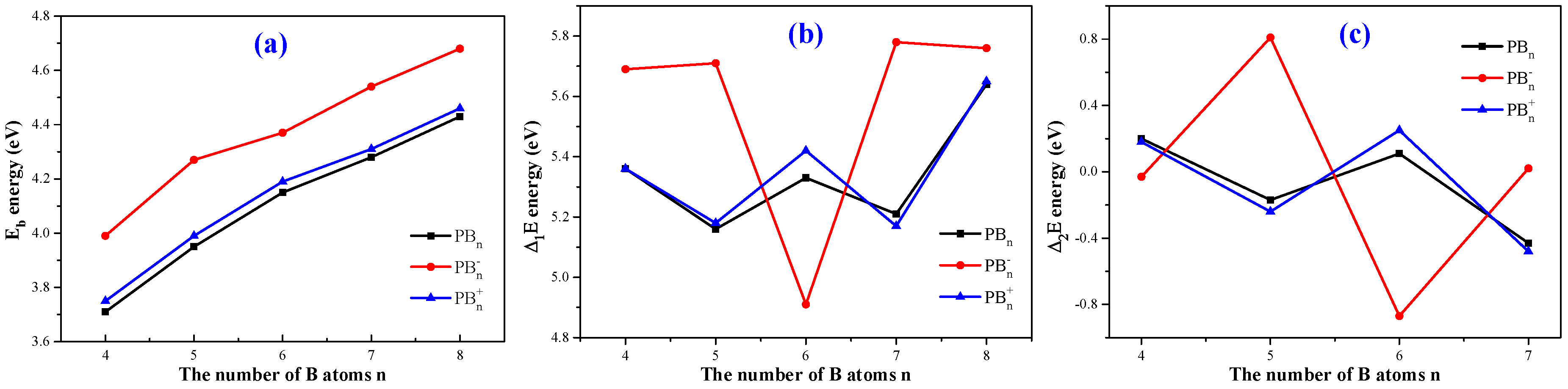
3.2. Relative Stabilities
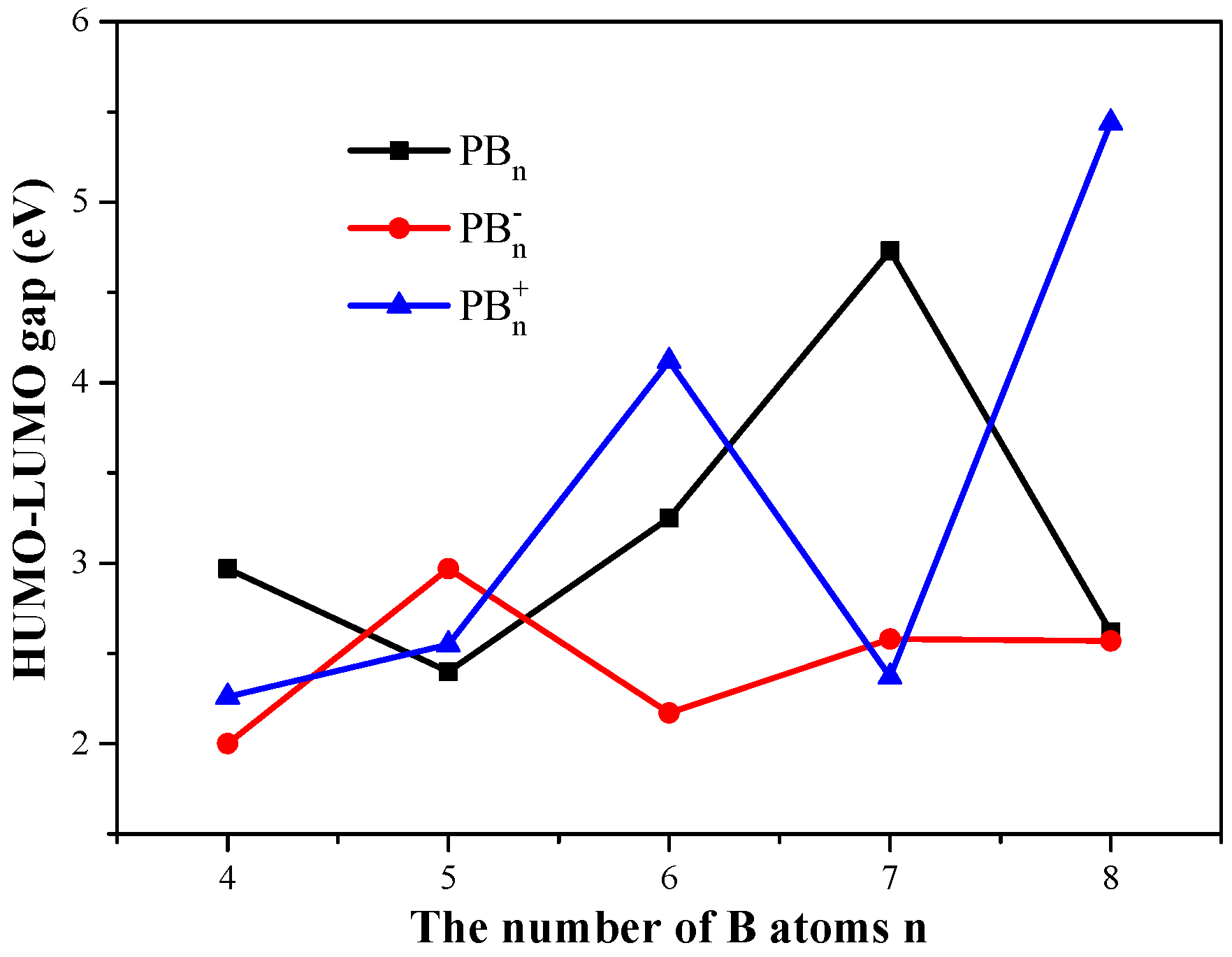
3.3. HOMO–LUMO Gaps
3.4. The Ionization Potential (IP) and Electron Affinity (EA)
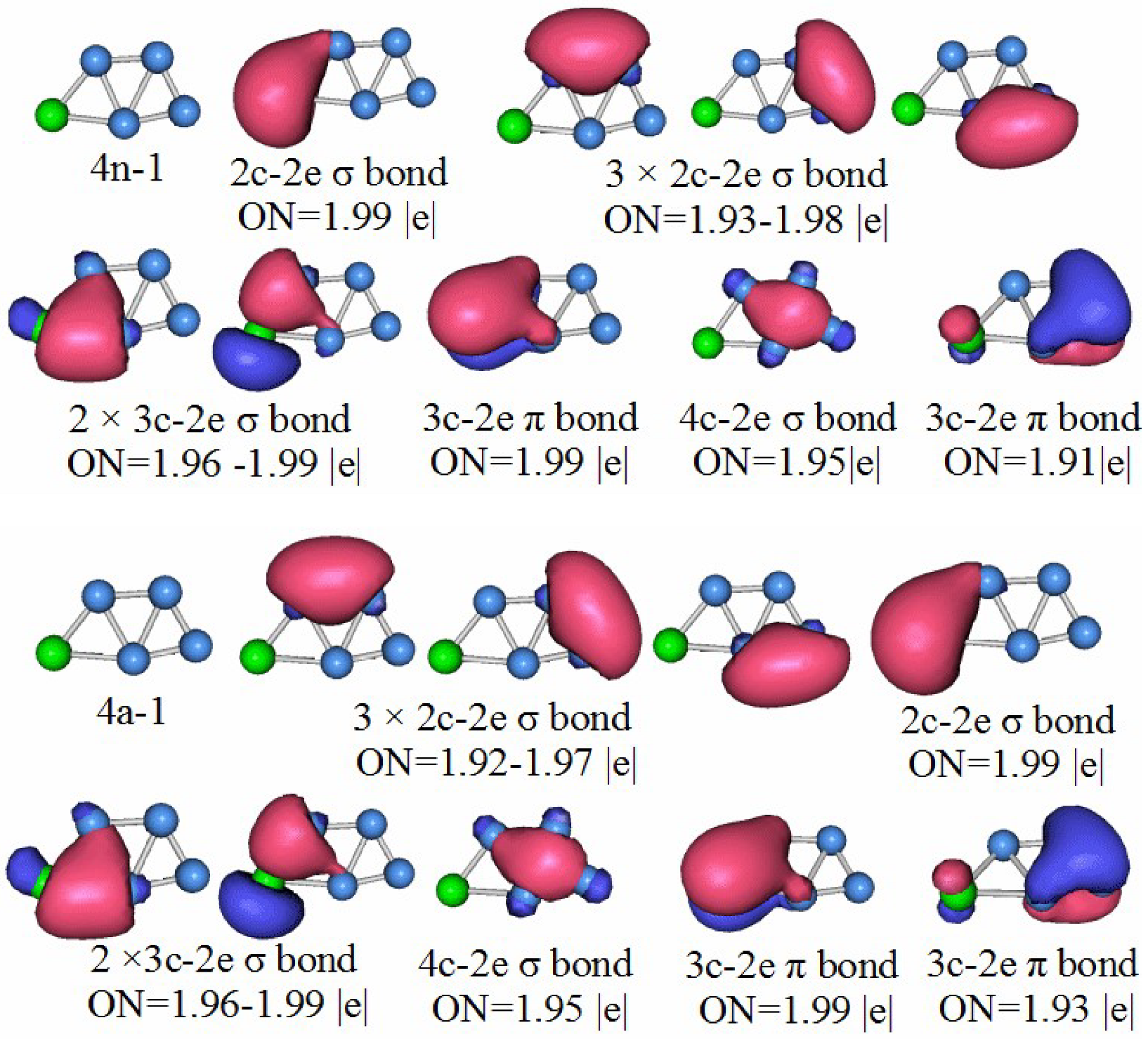
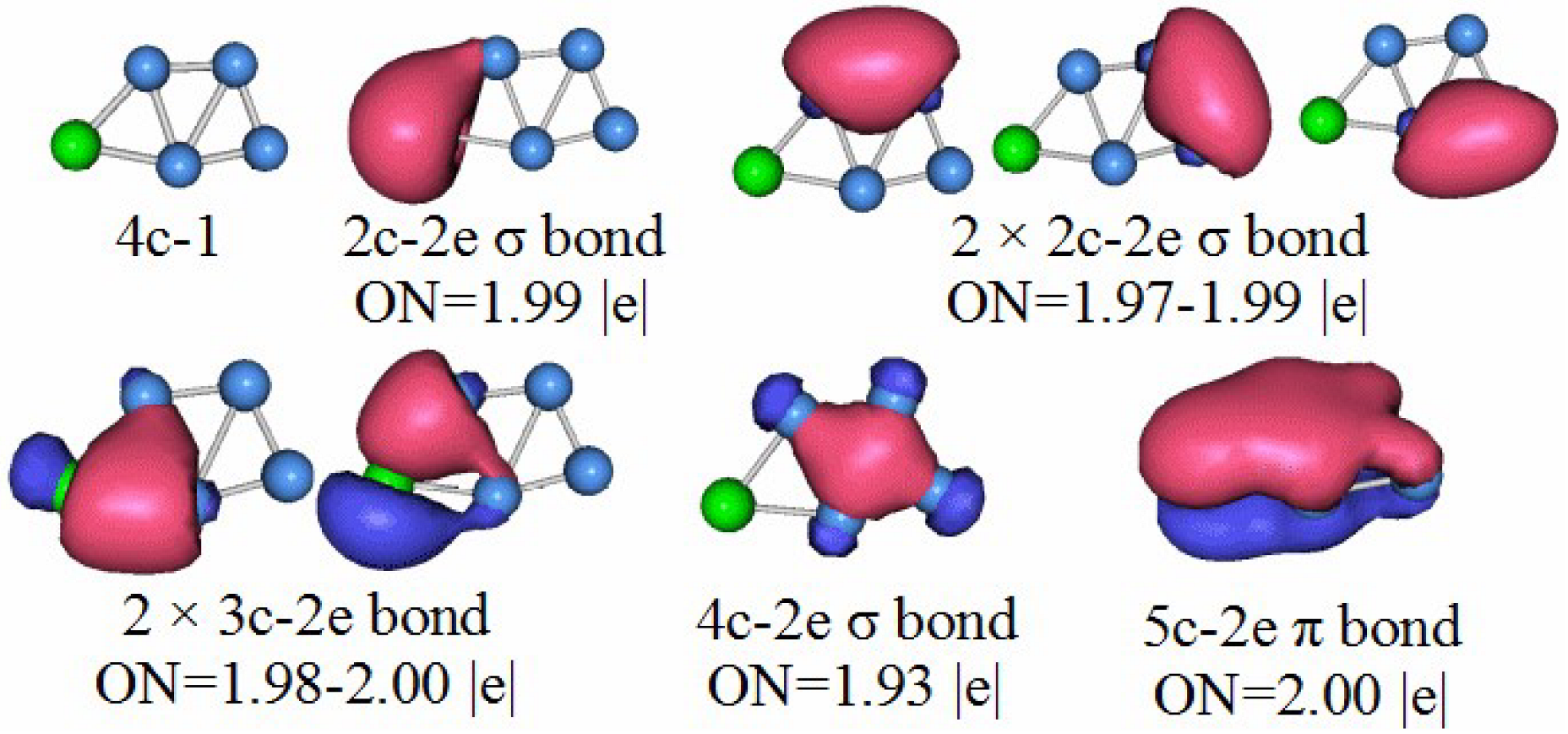
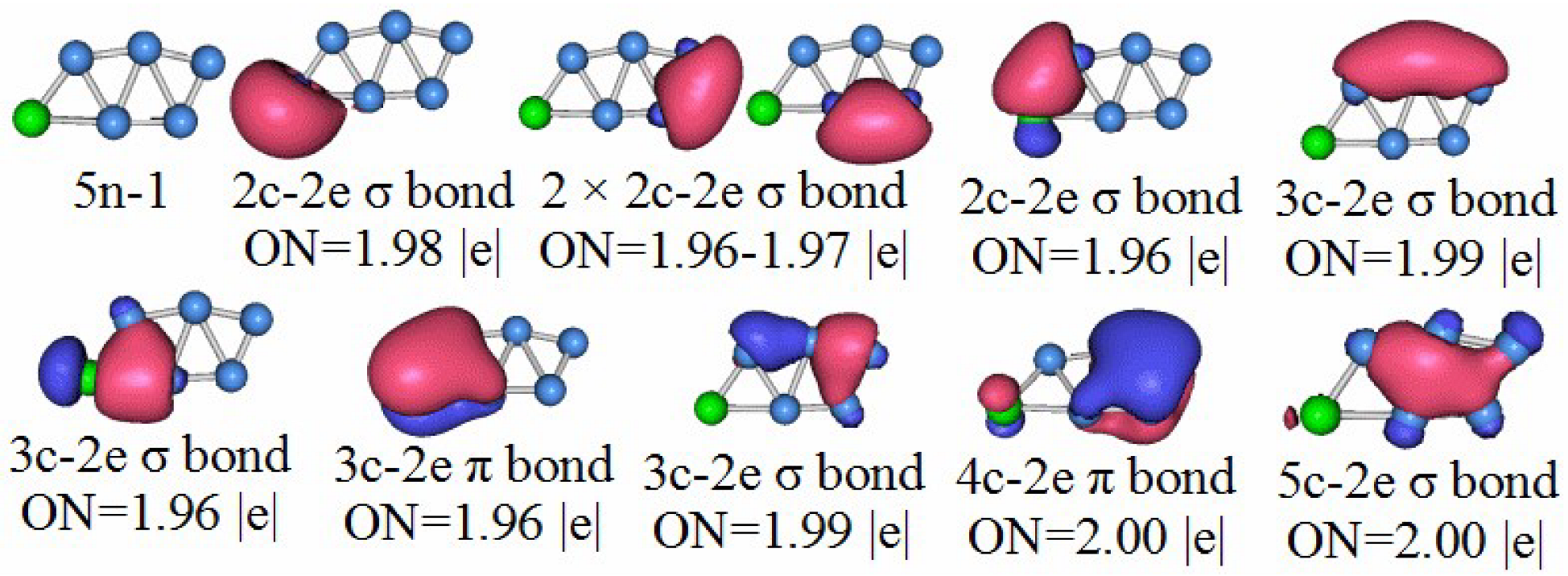
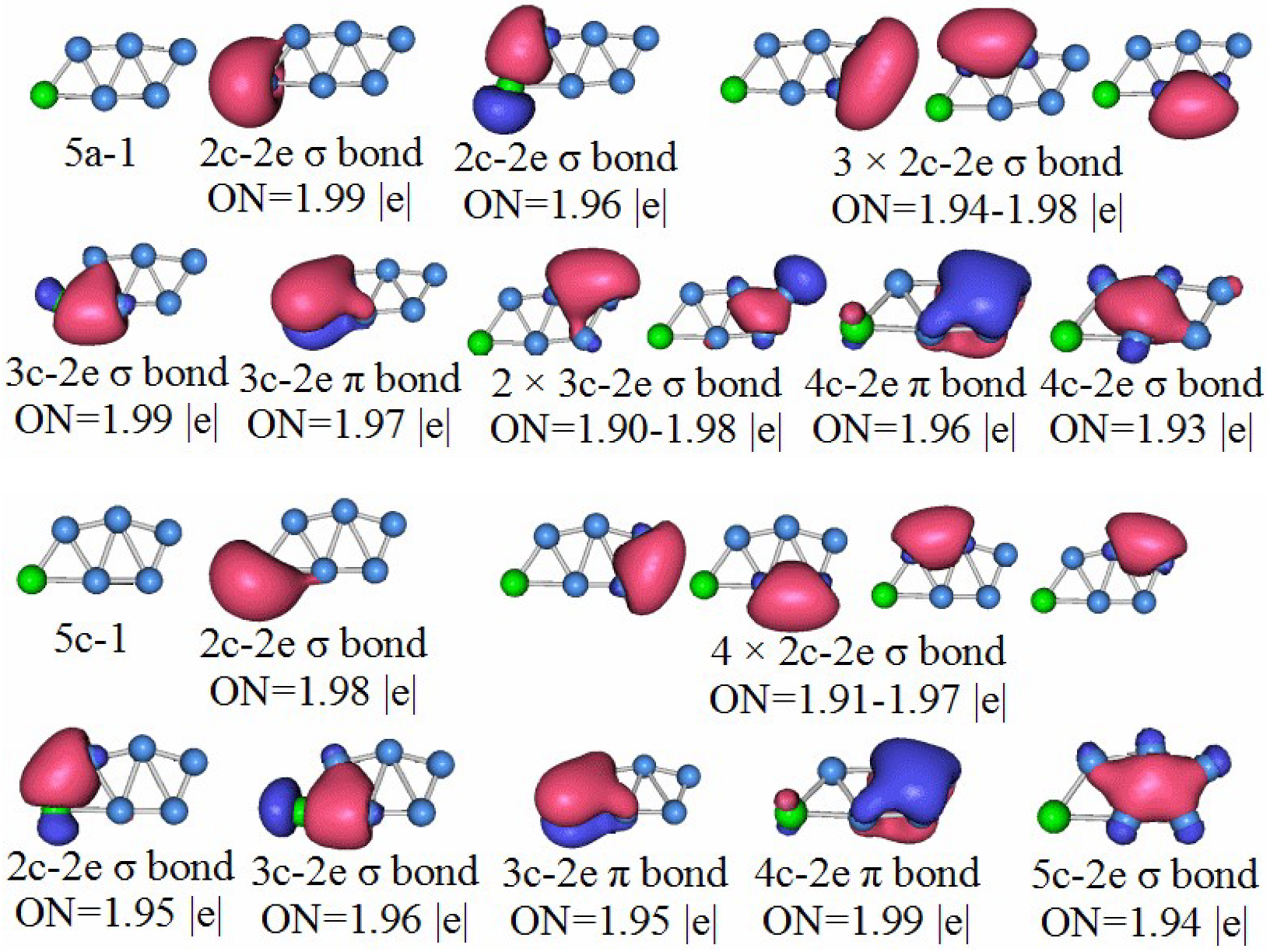
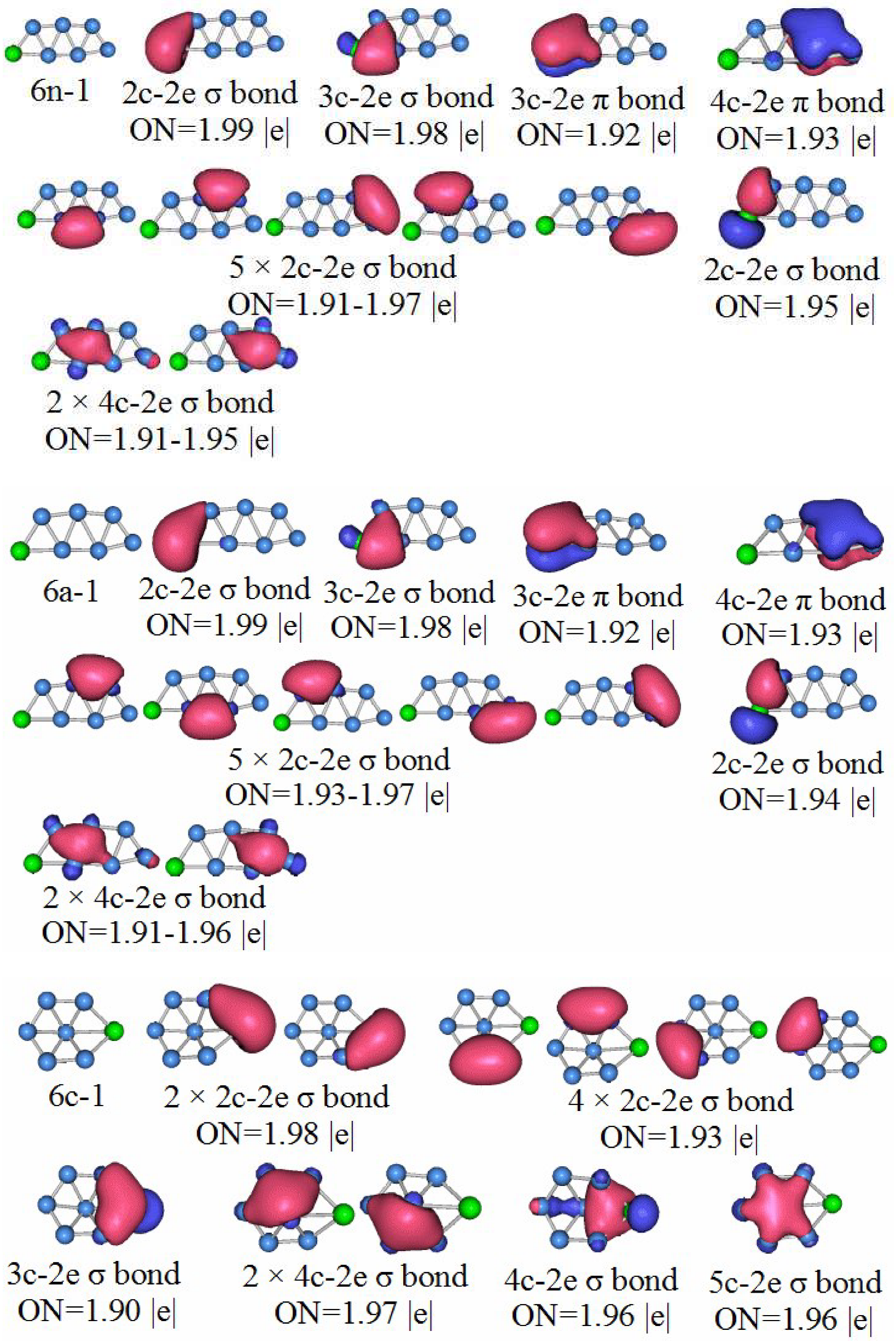
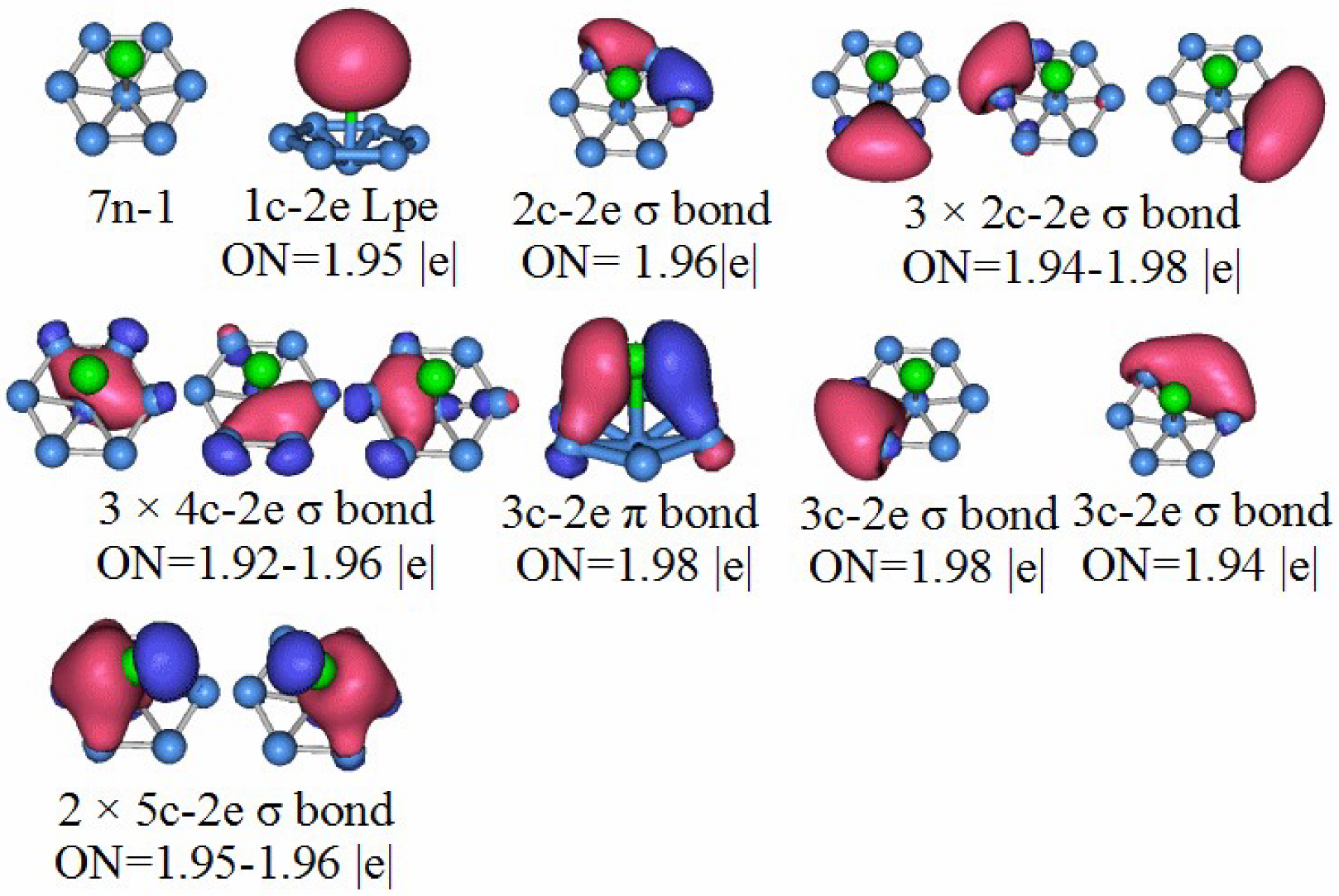
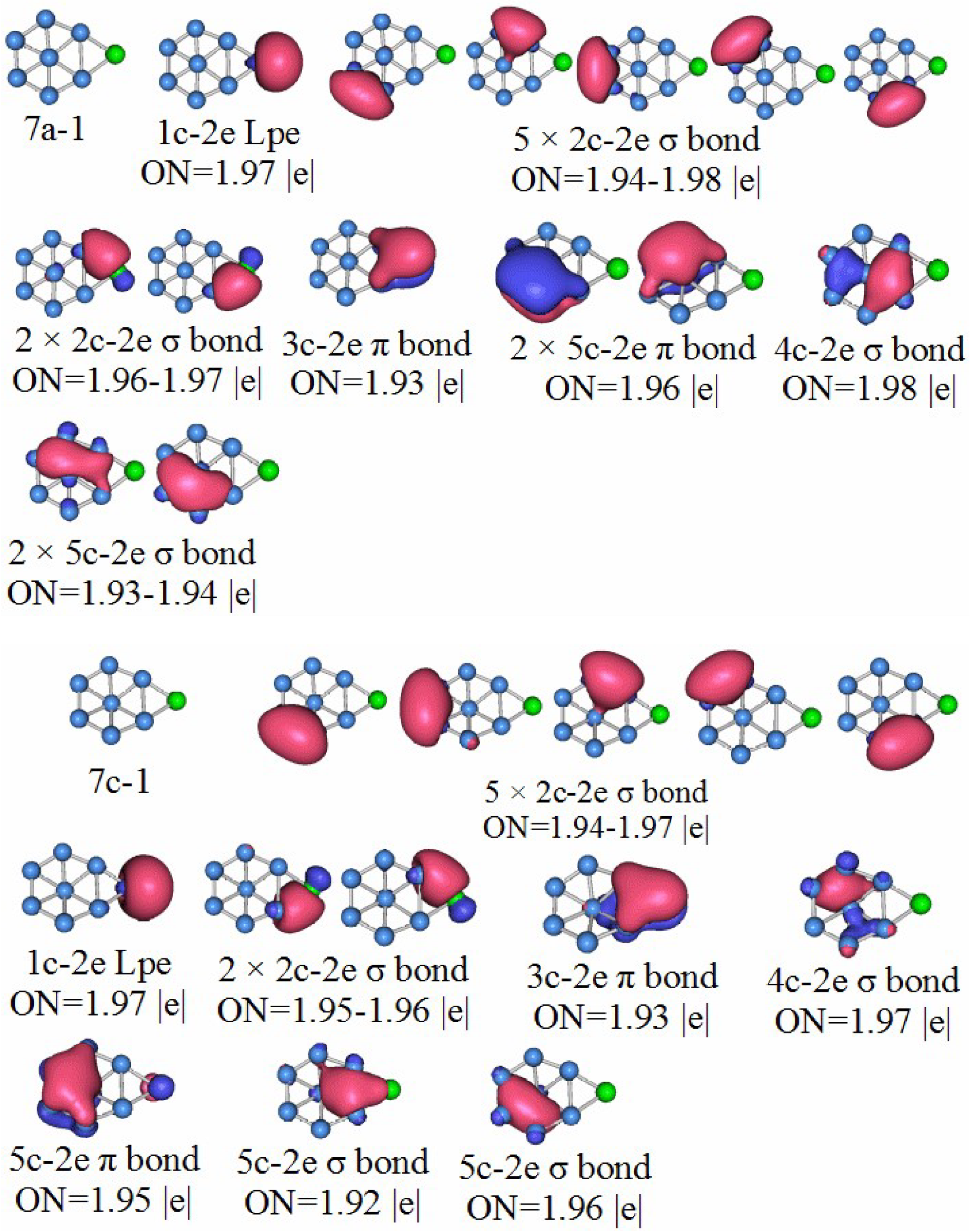
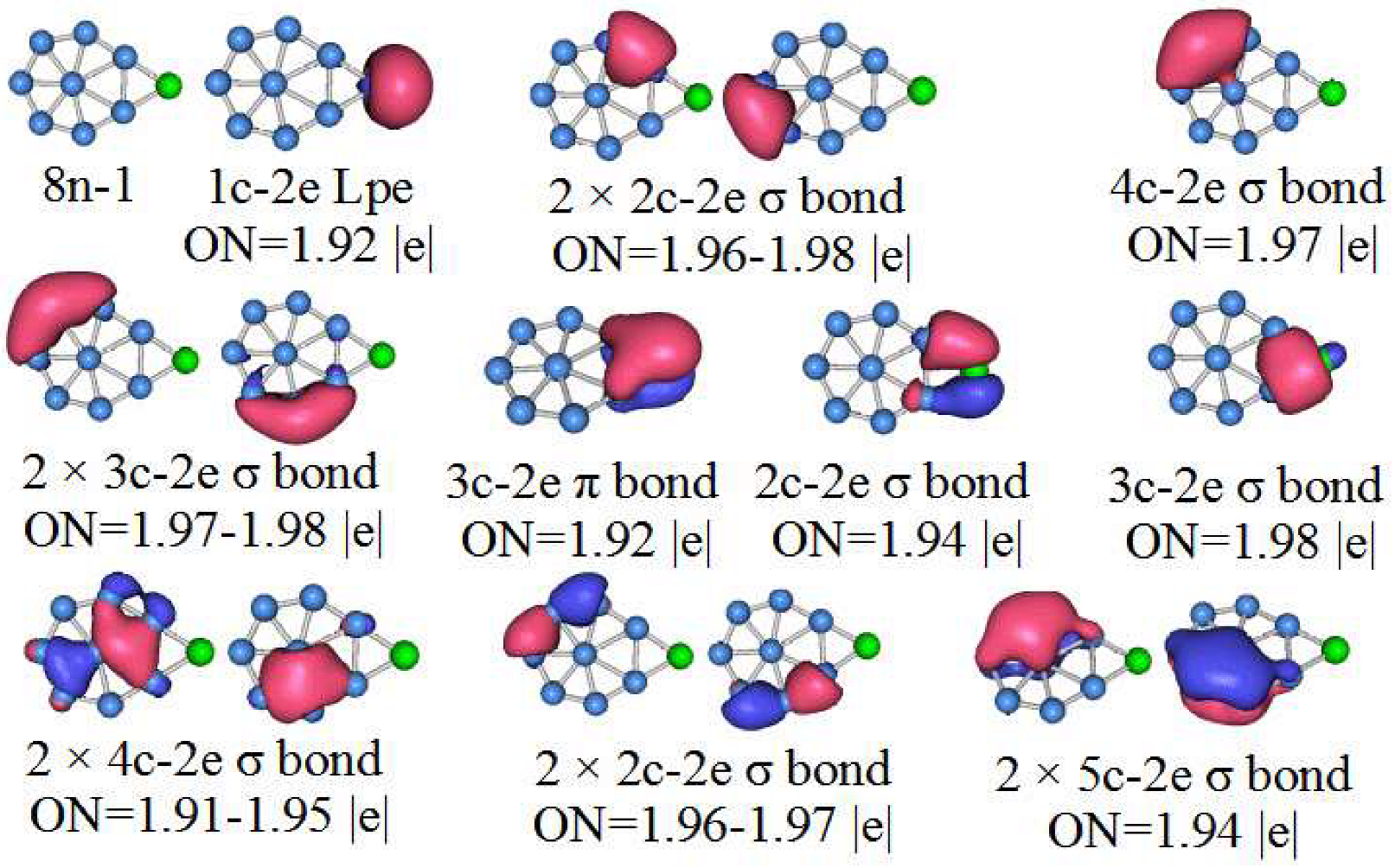
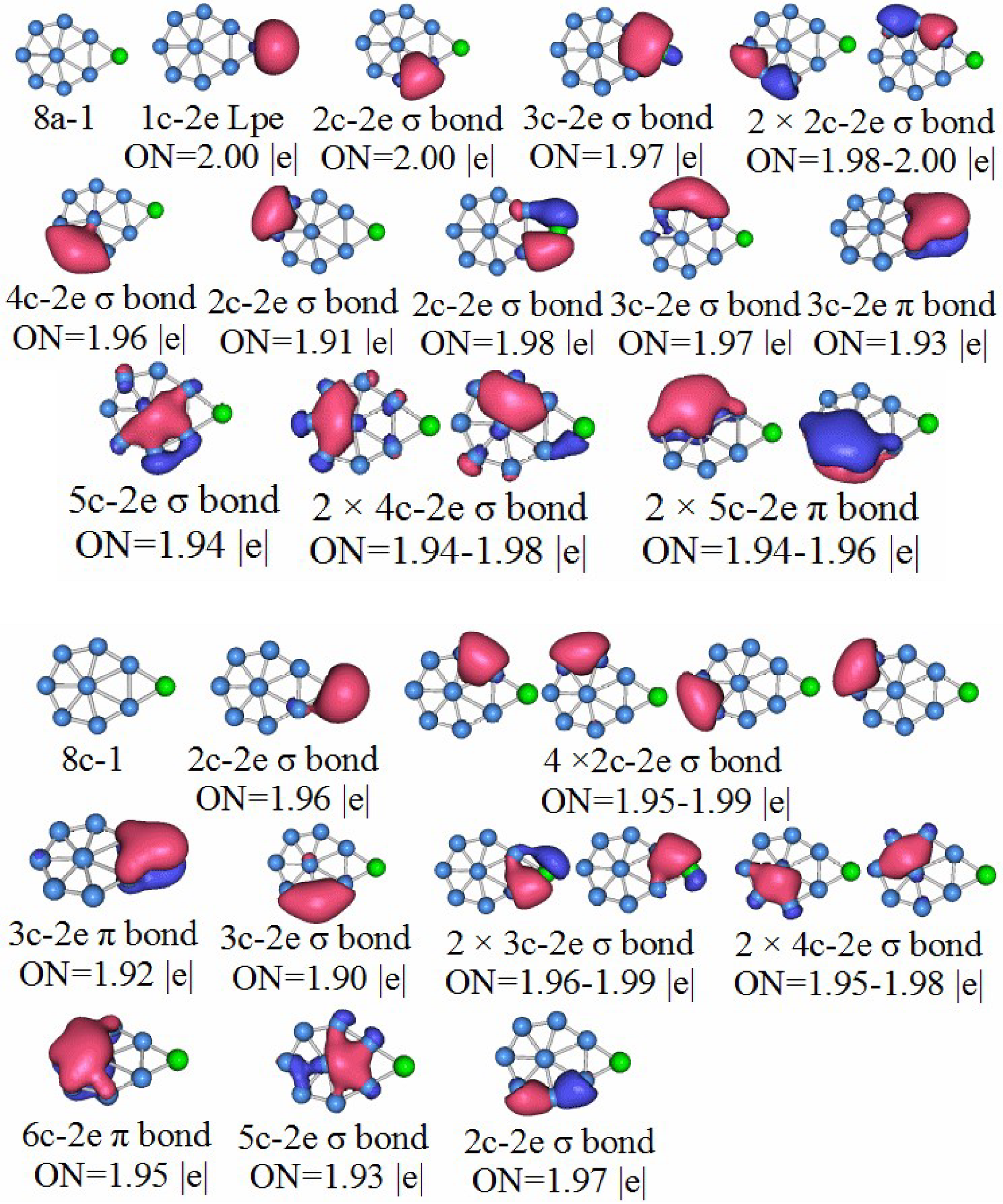
3.5. Chemical Bonding Analysis
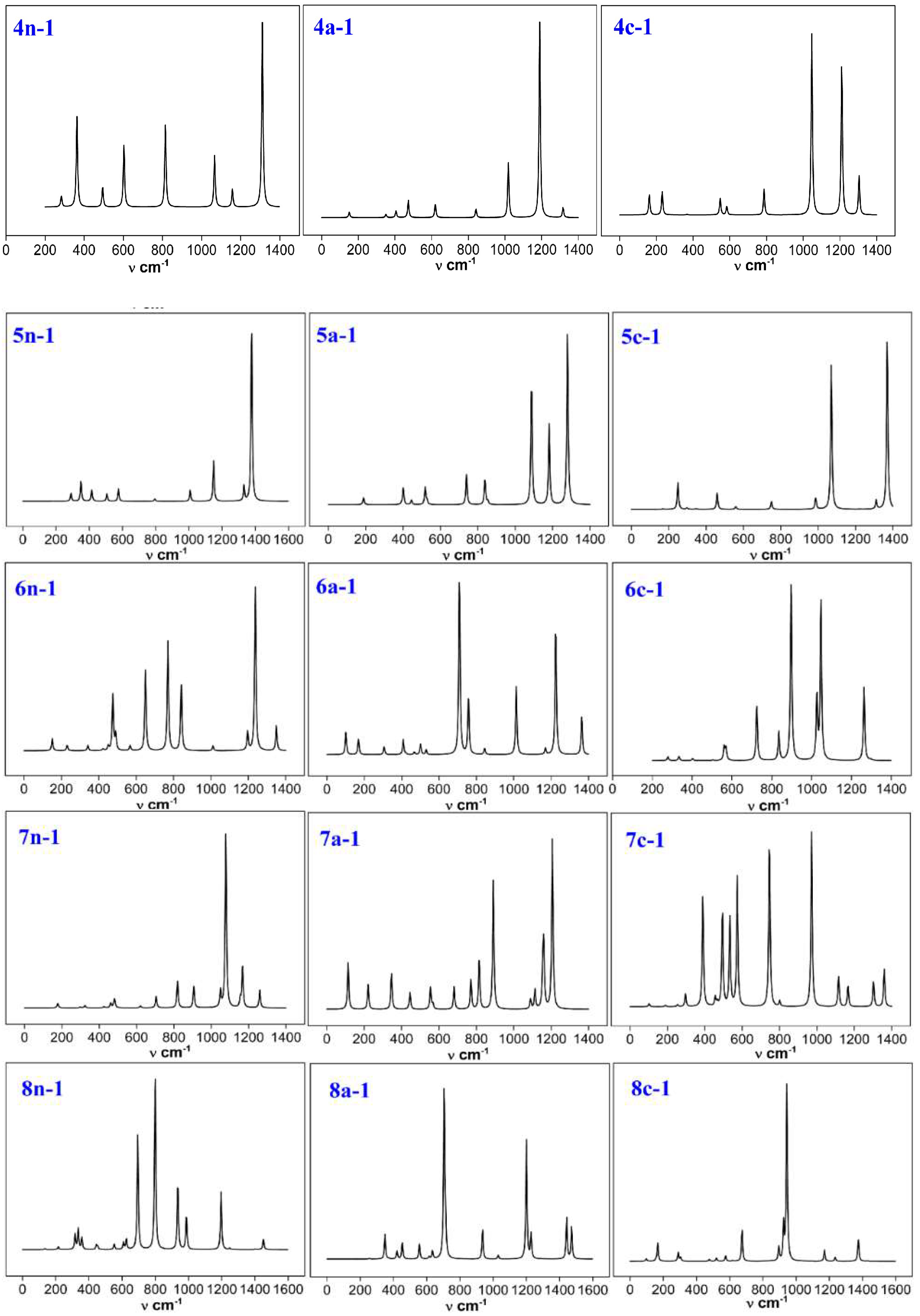
3.6. Infrared Spectroscopy
3.7. Simulated Photoelectron Spectrum
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kiran, B.; Gopa Kumar, G.; Nguyen, M.T.; Kandalam, A.K.; Jena, P. Origin of the Unusual Stability of B12 and B13+ Clusters. Inorg. Chem. 2009, 48, 9965. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, A.P.; Piazza, Z.A.; Romanescu, C.; Li, W.-L.; Boldyrev, A.I.; Wang, L.-S. B22− and B23−: All-Boron Analogues of Anthracene and Phenanthrene. J. Am. Chem. Soc. 2012, 134, 18065. [Google Scholar] [CrossRef] [PubMed]
- Piazza, Z.A.; Popov, I.A.; Li, W.-L.; Pal, R.; Zeng, X.C.; Boldyrev, A.I.; Wang, L.-S. A photoelectron spectroscopy and ab initio study of the structures and chemical bonding of the B25−cluster. J. Chem. Phys. 2014, 141, 034303. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-L.; Pal, R.; Piazza, Z.A.; Zeng, X.C.; Wang, L.-S. B27−: Appearance of the smallest planar boron cluster containing a hexagonal vacancy. J. Chem. Phys. 2015, 142, 204305. [Google Scholar] [CrossRef] [PubMed]
- Li, H.R.; Jian, T.; Li, W.L.; Miao, C.Q.; Wang, Y.J.; Chen, Q.; Luo, X.M.; Wang, K.; Zhai, H.J.; Li, S.D. Competition between quasi-planar and cage-like structures in the B29− cluster: Photoelectron spectroscopy and ab initio calculations. Phys. Chem. Chem. Phys. 2016, 18, 29147–29155. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-L.; Zhao, Y.-F.; Hu, H.-S.; Li, J.; Wang, L.-S. [B30]−: A Quasiplanar Chiral Boron Cluster. Angew. Chem. Int. Ed. 2014, 53, 5540. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-L.; Chen, Q.; Tian, W.-J.; Bai, H.; Zhao, Y.-F.; Hu, H.-S.; Li, J.; Zhai, H.-J.; Li, S.-D.; Wang, L.-S. The B35 Cluster with a Double-Hexagonal Vacancy: A New and More Flexible Structural Motif for Borophene. J. Am. Chem. Soc. 2014, 136, 12257. [Google Scholar] [CrossRef]
- Piazza, Z.A.; Hu, H.-S.; Li, W.-L.; Zhao, Y.-F.; Li, J.; Wang, L.-S. Planar hexagonal B36 as a potential basis for extended single-atom layer boron sheets. Nat. Commun. 2014, 5, 3113. [Google Scholar] [CrossRef]
- Chen, Q.; Wei, G.F.; Tian, W.J.; Bai, H.; Liu, Z.P.; Zhai, H.J.; Li, S.D. Quasi-planar aromatic B36 and B36− clusters: All-boron analogues of coronene. Phys. Chem. Chem. Phys. 2014, 16, 18282. [Google Scholar] [CrossRef]
- Romanescu, C.; Harding, D.J.; Fielicke, A.; Wang, L.-S. Probing the structures of neutral boron clusters using infrared/vacuum ultraviolet two color ionization: B11, B16, and B17. J. Chem. Phys. 2012, 137, 014317. [Google Scholar] [CrossRef]
- Jian, T.; Chen, X.; Li, S.-D.; Boldyrev, A.I.; Li, J.; Wang, L.-S. Probing the structures and bonding of size-selected boron and doped-boron clusters. Chem. Soc. Rev. 2019, 48, 3550. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Kiran, B.; Li, J.; Wang, L.-S. Hydrocarbon analogues of boron clusters—Planarity, aromaticity and antiaromaticity. Nat. Mater. 2003, 2, 827. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Wang, J.; Zhang, X.; Zhu, L. Hydrogen Storage on Metal-Coated B80 Buckyballs with Density Functional Theory. J. Phys. Chem. C 2009, 113, 7052. [Google Scholar] [CrossRef]
- Qi, B.; Wu, C.; Li, X.; Wang, D.; Sun, L.; Chen, B.; Liu, W.; Zhang, H.; Zhou, X. Self-Assembled Magnetic Gold Catalysts from Dual-Functional Boron Clusters. ChemCatChem 2015, 10, 2285–2290. [Google Scholar] [CrossRef]
- Tu, D.; Cai, S.; Fernandez, C.; Ma, H.; Wang, X.; Wang, H.; Ma, C.; Yan, H.; Lu, C.; An, Z. Boron-Cluster-Enhanced Ultralong Organic Phosphorescence. Angew. Chem. Int. Ed. 2019, 58, 9129–9133. [Google Scholar] [CrossRef]
- Warneke, J.; Hou, G.-L.; Aprà, E.; Jenne, C.; Yang, Z.; Qin, Z.; Kowalski, K.; Wang, X.-B.; Xantheas, S.S. Electronic Structure and Stability of [B12X12]2− (X = F–At): A Combined Photoelectron Spectroscopic and Theoretical Study. J. Am. Chem. Soc. 2017, 139, 14749–14756. [Google Scholar] [CrossRef]
- Warneke, J.; Konieczka, S.Z.; Hou, G.-L.; Aprà, E.; Kerpen, C.; Keppner, F.; Schäfer, T.C.; Deckert, M.; Yang, Z.; Bylaska, E.J.; et al. Properties of perhalogenated {closo-B10} and {closo-B11} multiply charged anions and a critical comparison with {closo-B12} in the gas and the condensed phase. Phys. Chem. Chem. Phys. 2019, 21, 5903–5915. [Google Scholar] [CrossRef]
- Mayer, M.; van Lessen, V.; Rohdenburg, M.; Hou, G.L.; Yang, Z.; Exner, R.M.; Apra, E.; Azov, V.A.; Grabowsky, S.; Xantheas, S.S.; et al. Rational design of an argon-binding superelectrophilic anion. Proc. Natl. Acad. Sci. USA 2019, 116, 8167–8172. [Google Scholar] [CrossRef]
- Rohdenburg, M.; Yang, Z.; Su, P.; Bernhardt, E.; Yuan, Q.; Apra, E.; Grabowsky, S.; Laskin, J.; Jenne, C.; Wang, X.-B.; et al. Properties of gaseous closo-[B6X6]2− dianions (X = Cl, Br, I). Phys. Chem. Chem. Phys. 2020, 22, 17713–17724. [Google Scholar] [CrossRef]
- Apra, E.; Warneke, J.; Xantheas, S.S.; Wang, X.B. A benchmark photoelectron spectroscopic and theoretical study of the electronic stability of [B12H12]2−. J. Chem. Phys. 2019, 150, 164306. [Google Scholar] [CrossRef]
- Czekner, J.; Cheung, L.F.; Johnson, E.L.; Fortenberry, R.C.; Wang, L.-S. A high-resolution photoelectron imaging and theoretical study of CP− and C2P−. J. Chem. Phys. 2018, 148, 044301. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Li, Q.; Song, B.; Yang, L.; Ganz, E. The Evolution of Geometric Structures, Electronic Properties, and Chemical Bonding of Small Phosphorus-Boron Clusters. Condens. Matter 2022, 7, 36. [Google Scholar] [CrossRef]
- Saha, P.; Rahane, A.B.; Kumar, V.; Sukumar, N. Analysis of the electron density features of small boron clusters and the effects of doping with C, P, Al, Si, and Zn: Magic B7P and B8Si clusters. Phys. Scr. 2016, 91, 053005. [Google Scholar] [CrossRef]
- Jin, S.; Chen, B.; Kuang, X.; Lu, C.; Sun, W.; Xia, X.; Gutsev, G.L. Structural and electronic properties of medium-sized aluminum-doped boron clusters AlB n and their anions. J. Phys. Chem. C 2019, 123, 6276–6283. [Google Scholar] [CrossRef]
- Barroso, J.; Pan, S.; Merino, G. Structural transformations in boron clusters induced by metal doping. Chem. Soc. Rev. 2022, 51, 1098–1123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Guo, J.C.; Zhai, H.J. Why nanoscale tank treads move? Structures, chemical bonding, and molecular dynamics of a doped boron cluster B 10 C. Nanoscale 2017, 9, 9310–9316. [Google Scholar] [CrossRef]
- Tai, T.B.; Kadłubański, P.; Roszak, S.; Majumdar, D.; Leszczynski, J.; Nguyen, M.T. Electronic Structures and Thermochemical Properties of the Small Silicon-Doped Boron Clusters BnSi (n = 1–7) and Their Anions. ChemPhysChem 2011, 12, 2948–2958. [Google Scholar] [CrossRef]
- Gu, J.B.; Yang, X.D.; Wang, H.Q.; Li, H.F. Structural, electronic, and magnetic properties of boron cluster anions doped with aluminum: BnAl−(2 ≤ n ≤ 9). Chin. Phys. B 2012, 21, 043102. [Google Scholar] [CrossRef]
- Anis, I.; Dar, M.S.; Rather, G.M.; Dar, M.A. Exploring the structure and electronic properties of germanium doped boron clusters using density functional theory based global optimization method. New J. Chem. 2022, 46, 6244–6254. [Google Scholar] [CrossRef]
- Mai, D.T.T.; Duong, L.V.; Tai, T.B.; Nguyen, M.T. Electronic structure and thermochemical parameters of the silicon-doped boron clusters B n Si, with n = 8–14, and their anions. J. Phys. Chem. A 2016, 120, 3623–3633. [Google Scholar] [CrossRef]
- Chen, B.; Gutsev, G.L.; Li, D.; Ding, K. Structure and Chemical Bonding in Medium-Size Boron Clusters Doped with Praseodymium. Inorganic Chemistry. Inorg. Chem. 2022, 61, 20, 7890–7896. [Google Scholar] [CrossRef]
- Li, D.; Gao, J.; Cheng, P.; He, J.; Yin, Y.; Hu, Y.; Chen, L.; Cheng, Y.; Zhao, J. 2D boron sheets: Structure, growth, and electronic and thermal transport properties. Adv. Funct. Mater. 2020, 30, 1904349. [Google Scholar] [CrossRef]
- Bao, K.; Goedecker, S.; Koga, K.; Lançon, F.; Neelov, A. Structure of large gold clusters obtained by global optimization using the minima hopping method. Phys. Rev. B 2009, 79, 041405. [Google Scholar] [CrossRef]
- Schönborn, S.E.; Goedecker, S.; Roy, S.; Oganov, A.R. The performance of minima hopping and evolutionary algorithms for cluster structure prediction. J. Chem. Phys. 2009, 130, 144108. [Google Scholar] [CrossRef] [PubMed]
- Goedecker, S. Minima hopping: An efficient search method for the global minimum of the potential energy surface of complex molecular systems. J. Chem. Phys. 2004, 120, 9911. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, Y.-F.; Wang, L.-S.; Li, J. Recent progresses of global minimum searches of nanoclusters with a constrained Basin-Hopping algorithm in the TGMin program. Comput. Theor. Chem. 2017, 1107, 57. [Google Scholar] [CrossRef]
- Sergeeva, A.P.; Averkiev, B.B.; Zhai, H.-J.; Boldyrev, A.I.; Wang, L.-S. All-boron analogues of aromatic hydrocarbons: B17− and B18−. J. Chem. Phys. 2011, 134, 224304. [Google Scholar] [CrossRef]
- Dondela, B.; Peszke, J.; Śliwa, W. Semiempirical ZINDO/S, AM1 and ab initio HF/STO-3G and HF/6-31G study of quaternary salts of diazaphenanthrenes with ethyl bromide and 1,3-dibromopropane. J. Mol. Struct. 2005, 753, 154. [Google Scholar] [CrossRef]
- Téllez, C.A.; Hollauer, E.; Giannerini, T.; Pais da Silva, M.I.; Mondragón, M.A.; Rodríguez, J.R.; Castaño, V.M. Fourier transform infrared and Raman spectra: Semi empirical AM1 and PM3; MP2/DZV and DFT/B3LYP-6-31G (d) ab initio calculations for dimethylterephthalate (DMT). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2004, 60, 2171. [Google Scholar] [CrossRef]
- Oakes, R.E.; Renwick Beattie, J.; Moss, B.W.; Bell, S.E.J. Conformations, vibrational frequencies and Raman intensities of short-chain fatty acid methyl esters using DFT with 6-31G (d) and Sadlej pVTZ basis sets. J. Mol. Struct. Theochem 2002, 586, 91. [Google Scholar] [CrossRef]
- Negishi, Y.; Kawamata, H.; Hayase, T.; Gomei, M.; Kishi, R.; Hayakawa, F.; Nakajima, A.; Kaya, K. Photoelectron spectroscopy of germanium-fluorine binary cluster anions: The HOMO-LUMO gap estimation of Gen clusters. Chem. Phys. Lett. 1997, 269, 199. [Google Scholar] [CrossRef]
- Cehovin, A.; Mera, H.; Jensen, J.H.; Stokbro, K.; Pedersen, T.B. Role of the virtual orbitals and HOMO-LUMO gap in mean-field approximations to the conductance of molecular junctions. Phys. Rev. B 2008, 77, 195432. [Google Scholar] [CrossRef]
- Chen, W.J.; Kulichenko, M.; Choi, H.W.; Cavanagh, J.; Yuan, D.F.; Boldyrev, A.I.; Wang, L.S. Photoelectron Spectroscopy of Size-Selected Bismuth-Boron Clusters: BiB n-(n = 6–8). J. Phys. Chem. A 2021, 125, 6751–6760. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PBn | Eb(PBn) = [E(P) + nE(B) − E(PBn)]/(1+n) | (1) |
| Δ1E (PBn) = E(PBn−1) + E(B) − E(PBn) | (2) | |
| Δ2E (PBn) = E(PBn−1) + E(PBn+1) − 2E(PBn) | (3) | |
| Eb() = [E(P−) + nE(B) − E()]/(1+n) | (4) | |
| Δ1E () = E() + E(B) − E() | (5) | |
| Δ2E () = E() + E() − 2E() | (6) | |
| Eb() = [E(P) + (n − 1)E(B) + E(B+) − E()]/(1+n) | (7) | |
| Δ1E () = E() + E(B) − E() | (8) | |
| Δ2E () = E() + E() − 2E() | (9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.-S.; Song, B.; Wen, L.; Yang, L.-M.; Ganz, E.
Elucidation of Structures, Electronic Properties, and Chemical Bonding of Monophosphorus-Substituted Boron Clusters in Neutral, Negative, and Positively Charged PBn/
Li Q-S, Song B, Wen L, Yang L-M, Ganz E.
Elucidation of Structures, Electronic Properties, and Chemical Bonding of Monophosphorus-Substituted Boron Clusters in Neutral, Negative, and Positively Charged PBn/
Li, Qing-Shan, Bingyi Song, Limei Wen, Li-Ming Yang, and Eric Ganz.
2022. "Elucidation of Structures, Electronic Properties, and Chemical Bonding of Monophosphorus-Substituted Boron Clusters in Neutral, Negative, and Positively Charged PBn/
Li, Q.-S., Song, B., Wen, L., Yang, L.-M., & Ganz, E.
(2022). Elucidation of Structures, Electronic Properties, and Chemical Bonding of Monophosphorus-Substituted Boron Clusters in Neutral, Negative, and Positively Charged PBn/