3.1. Growth Behavior of Small Phosphorus–Doped Boron Clusters
The low-lying isomers of phosphorus-doped boron P
2B
n0/−/+clusters are shown in
Figure 1,
Figure 2 and
Figure 3. All have real vibrational frequencies. Based on the high-precision CCSD(T) energies, five low-lying isomers for each P
2B
n0/−/+ cluster are enumerated and shown in the main text. In the following discussion and analysis, unless otherwise stated, we will mainly use the CCSD(T) energies.
3.1.1. Low-Lying Isomers of Neutral P2Bn (n = 3–7) Clusters
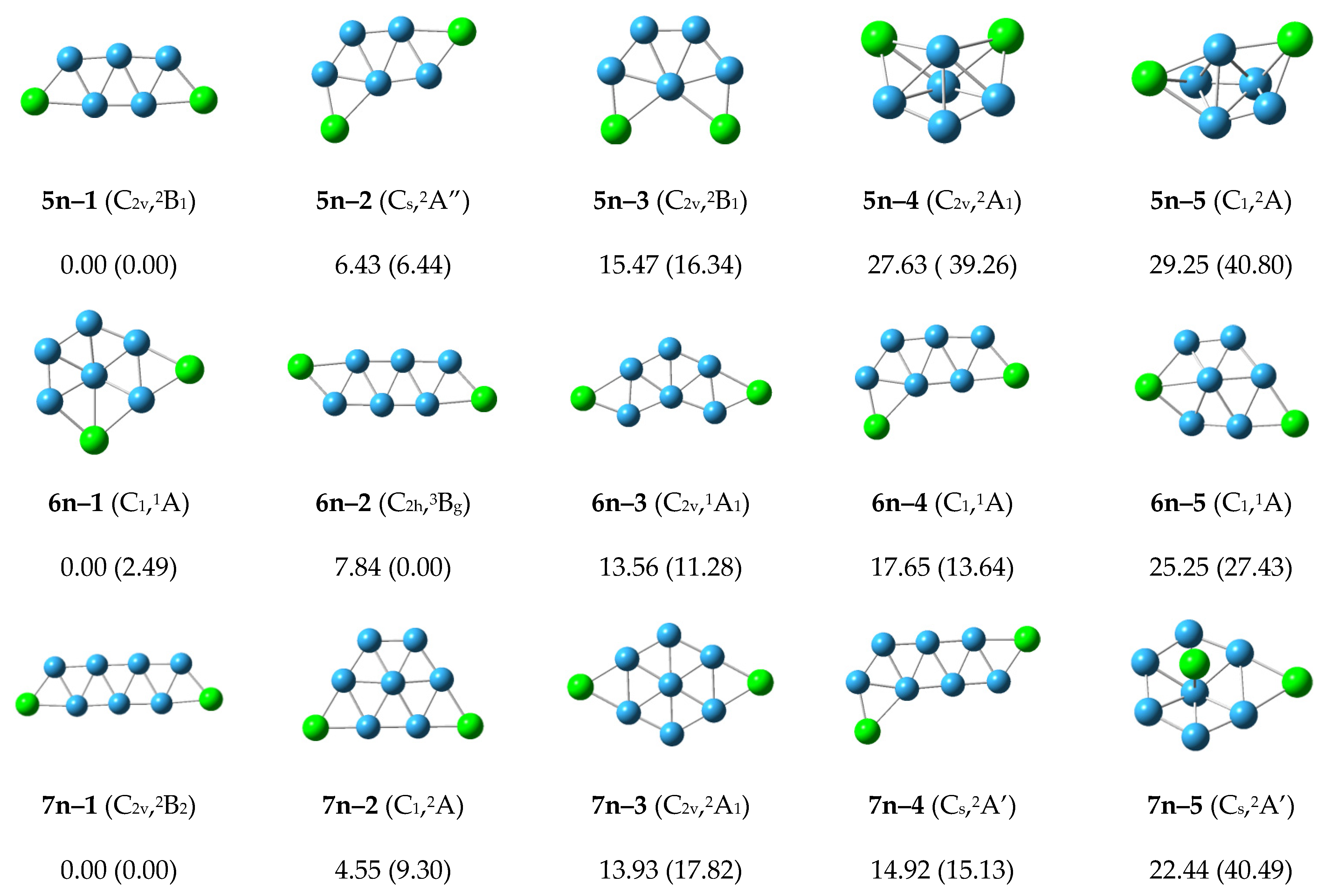
In
Figure 1, five low-lying isomers for each neutral P
2B
n cluster are ordered starting with the lowest energy configurations. For the neutral P
2B
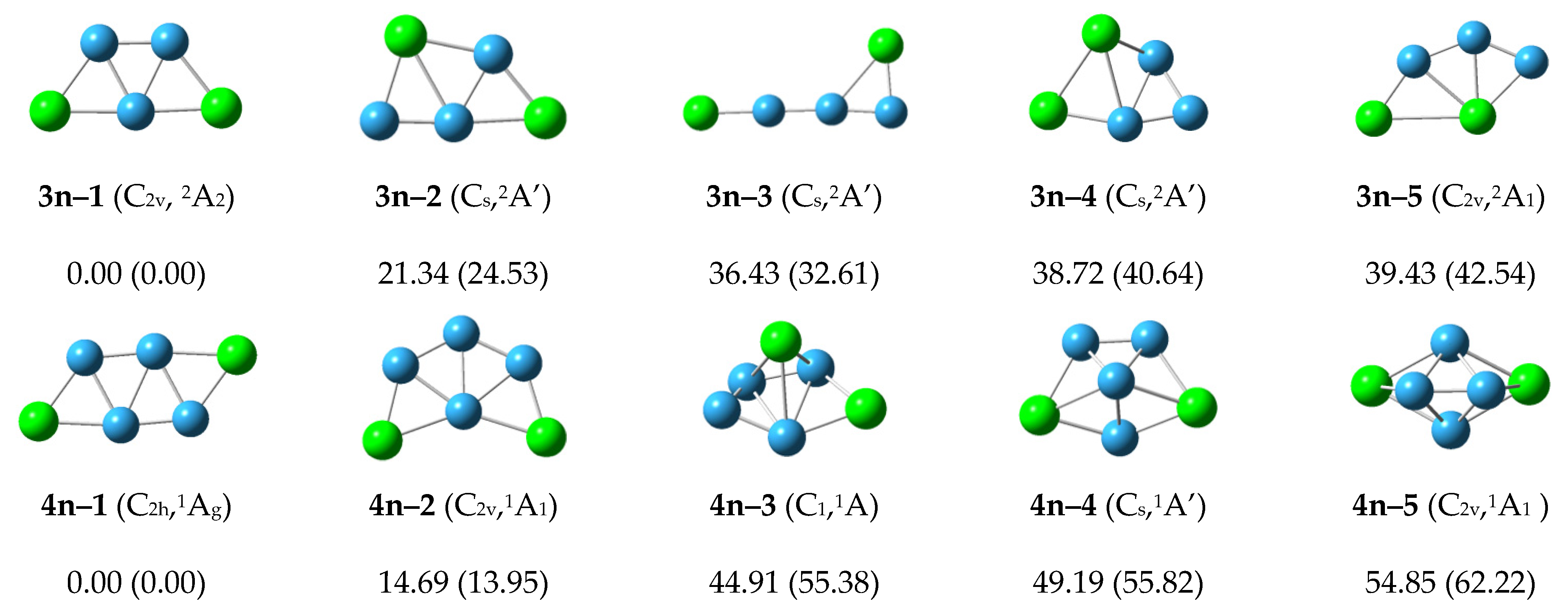
3 cluster, the lowest isomer structure 3n–1 is flat, and has a stable equilateral boron triangle. The two P atoms are located on the periphery. The relative energies of the 3n–2 to 3n–5 structures are more than 20 kcal/mol, demonstrating a sufficient gap away from the lowest energy state.
Figure 1.
The optimized structures and relative energies (kcal/mol) of neutral P2Bn clusters. The relative order of different isomers is determined using the CCSD(T)/6–311+G(2df) energies. The energies in parentheses are the B3LYP/6–311+G(d) results.
Figure 1.
The optimized structures and relative energies (kcal/mol) of neutral P2Bn clusters. The relative order of different isomers is determined using the CCSD(T)/6–311+G(2df) energies. The energies in parentheses are the B3LYP/6–311+G(d) results.
For the neutral P2B4 cluster, the lowest energy structure 4n–1 has C2h symmetry. Its four boron atoms form a stable rhombus. The structure is flat, and the four boron atoms form two stable equilateral triangles. The two P atoms are located in the corners of the raft–like structure. The structure 4n–2 has relative energy 14.7 kcal/mol and C2v symmetry. Four boron atoms also form a stable rhombus, and two phosphorus atoms are added on the periphery. The relative energies of 4n–3 and other higher energy structures exceed 20 kcal/mol. 4n–3 starts to show 3D structure. In general, the 4n–1 structure is the most stable structure of the neutral P2B4 cluster.
For the neutral P2B5 cluster, the lowest energy structure 5n–1 has the symmetry of C2v with five boron atoms forming a W-shape pentagon, or the five boron atoms forming three stable equilateral triangles. Lower lying energy isomers 5n–2 and 5n–3 have 6.43 and 15.47 kcal/mol with five boron atoms preferentially forming a pentagon, and the remaining two phosphorus atoms are located at different positions on the periphery. The energy of the other structures is higher than 20 kcal/mol. The 5n–1 structure is the most stable structure of the P2B5 cluster.
For the neutral P2B6 cluster, the structure 6n–1 at the CCSD(T)/6–311+G(2df) level has the lowest energy. The two-dimensional planar structure 6n–2 at the B3LYP/6–311+G(d) level has the lowest energy. The structure of 6n–2 is arranged in a zigzag pattern. Other higher energy structures are shown in the Figure. Structures 6n–1 and 6n–2 may compete with each other to become the most stable structure of P2B6 cluster.
For the neutral P2B7 cluster, the lowest energy structure 7n–1 with C2v symmetry has a zigzag arrangement in which two phosphorus atoms are located at both ends. The higher energy 7n–2, 7n–3, and 7n–4 structures are planar or planar-like configurations. The 7n–1 structure is the most stable structure of the neutral P2B7 cluster.
In general, for the neutral P2Bn (n = 3–7) structures, the most stable structures present planar or planar-like configurations, and are arranged in a zigzag pattern with two phosphorus atoms located at the two ends.
3.1.2. Low-Lying Isomers of Anionic P2Bn− (n = 3–7) Clusters
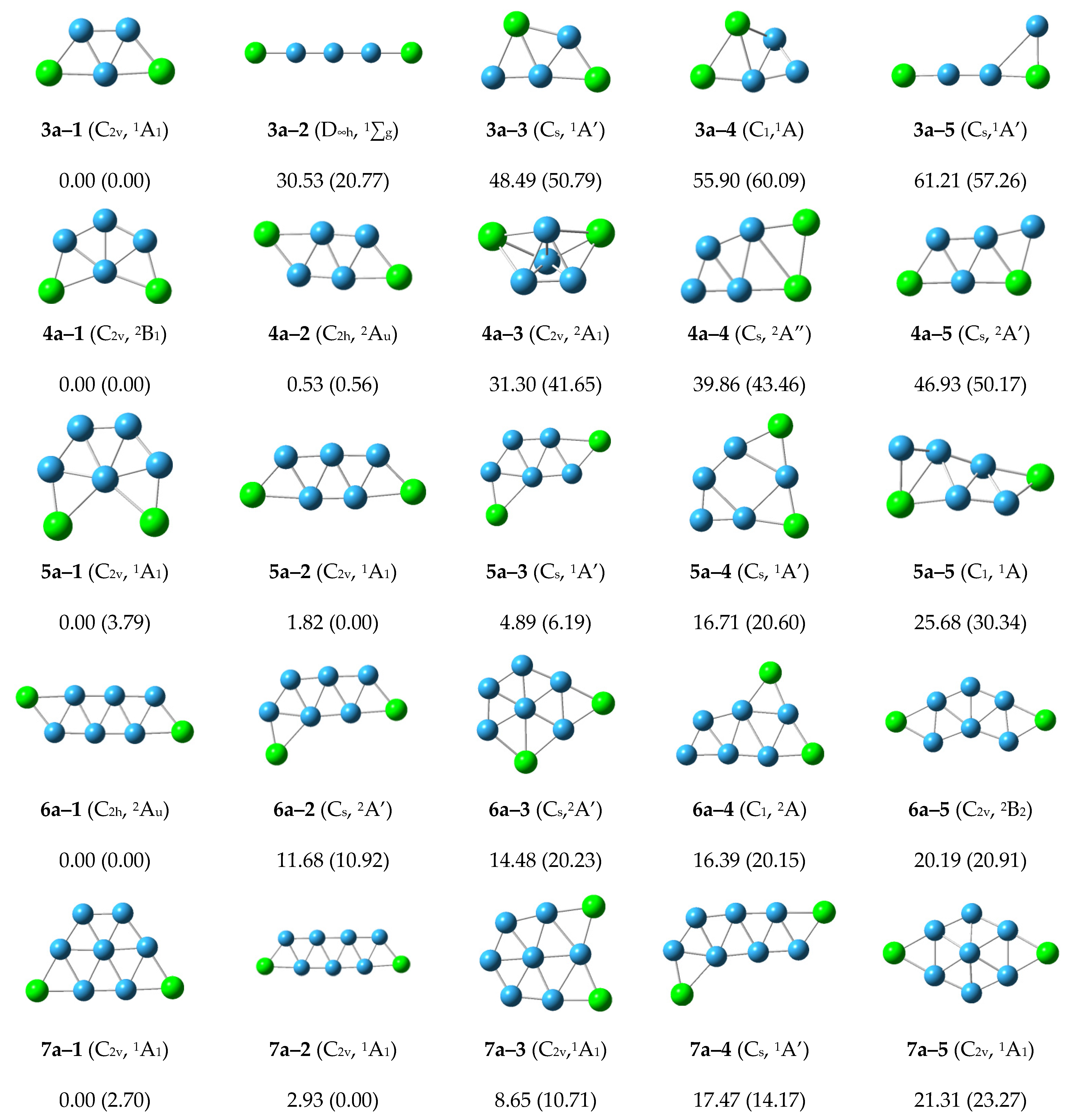
The low-lying isomers for anionic P
2B
n− clusters are shown in
Figure 2. For P
2B
3, the lowest energy structure 3a–1 with planar
C2v symmetry is similar to the structure 3n–1 of the neutral P
2B
3 cluster. The energy of the other structures are more than 20 kcal/mol. We can conclude that the structure 3a–1 is the most stable anionic P
2B
3− cluster.
Figure 2.
Optimized global minimum and low-lying isomers for anionic P2Bn− clusters. The relative energies at the CCSD(T)/6–311+G(2df) level are shown below each structure. The B3LYP/6–311+G(d) energies are shown in parentheses.
Figure 2.
Optimized global minimum and low-lying isomers for anionic P2Bn− clusters. The relative energies at the CCSD(T)/6–311+G(2df) level are shown below each structure. The B3LYP/6–311+G(d) energies are shown in parentheses.
For the anionic P2B4− cluster, the structure 4a–1 with C2v symmetry has the lowest energy and is similar to the structure 4n–2 of the neutral P2B4 cluster. The structure 4a–2 is 0.53 kcal/mol higher than the energy of structure 4a–1, and is similar to the structure 4n–1 of the neutral P2B4 cluster. The relative energies of the other structures are more than 20 kcal/mol. We can conclude that the structure 4a–1 and 4a–2 may compete with each other to become the most stable anionic P2B4− cluster.
For the anionic P2B5− cluster, the structures 5a–1 and 5a–2 with C2v symmetry have lower energy, and are similar to the structures 5n–3 and 5n–1 of the neutral P2B5 cluster, respectively. The structure 5a–3 is similar to the structure 5n–2, and the other structures have higher energy. The structures 5a–1 and 5a–2 may compete with each other to become the most stable anionic P2B5− cluster.
The structure 6a–1 with the lowest energy and C2h symmetry grows in zigzag shape, and is similar to the structure 6n–2. The structures 6a–2 and 6a–3 with higher energy are similar to structures 6n–4 and 6n–1, respectively. The other structures are higher energy and the structure 6a–1 is the most stable anionic P2B6− cluster.
The structure 7a–1 has the lowest energy and is similar to the structure 7n–2. The structure 7a–2 growing in a zigzag shape is similar to the structure 7n–1. The other structures are higher energy, and the structures 7a–1 and 7a–2 may compete with each other to become the most stable anionic P2B7− cluster.
In general, it can be seen from
Figure 2 that any three atoms can preferentially form stable triangles and grow in a planar network.
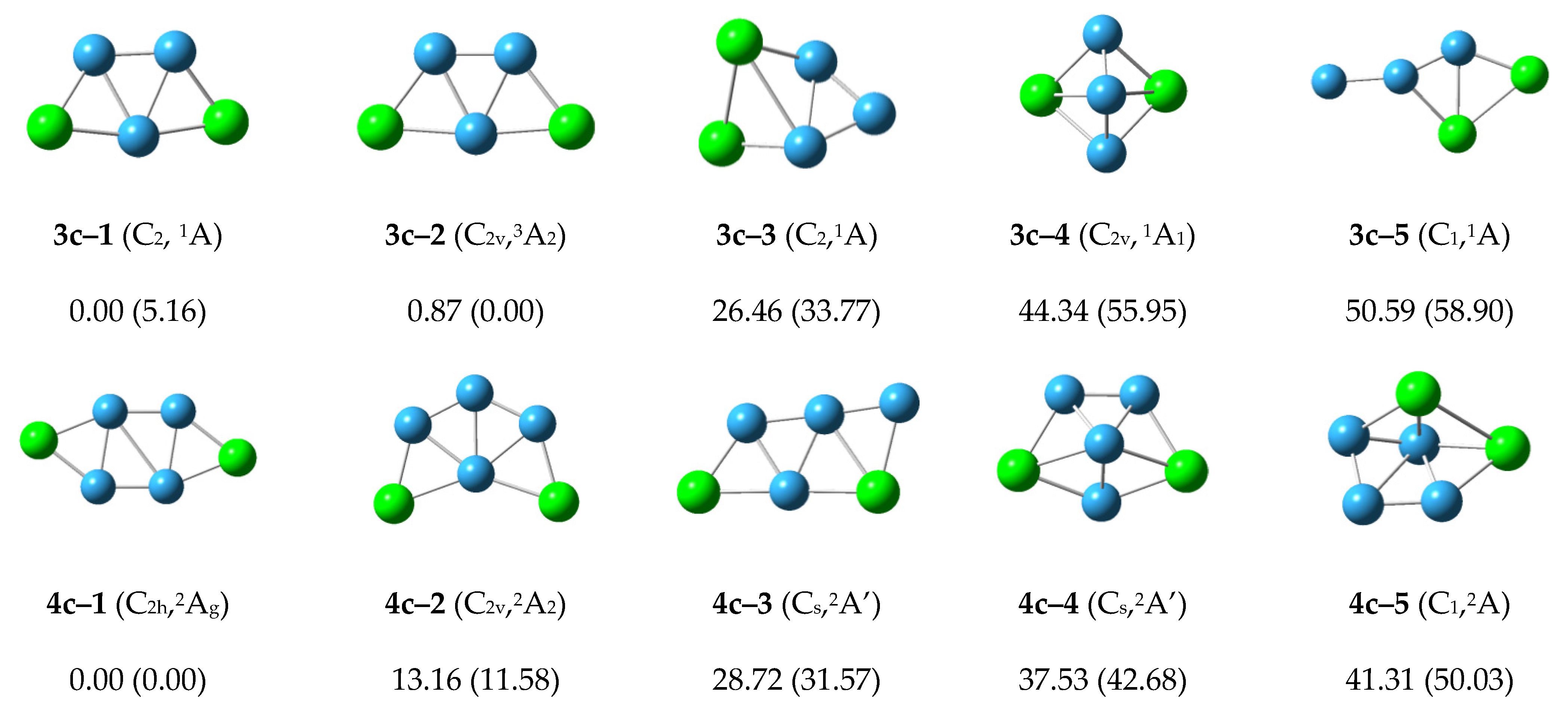
3.1.3. Low-Lying Isomers of Cationic P2Bn+ (n = 3–7) Clusters
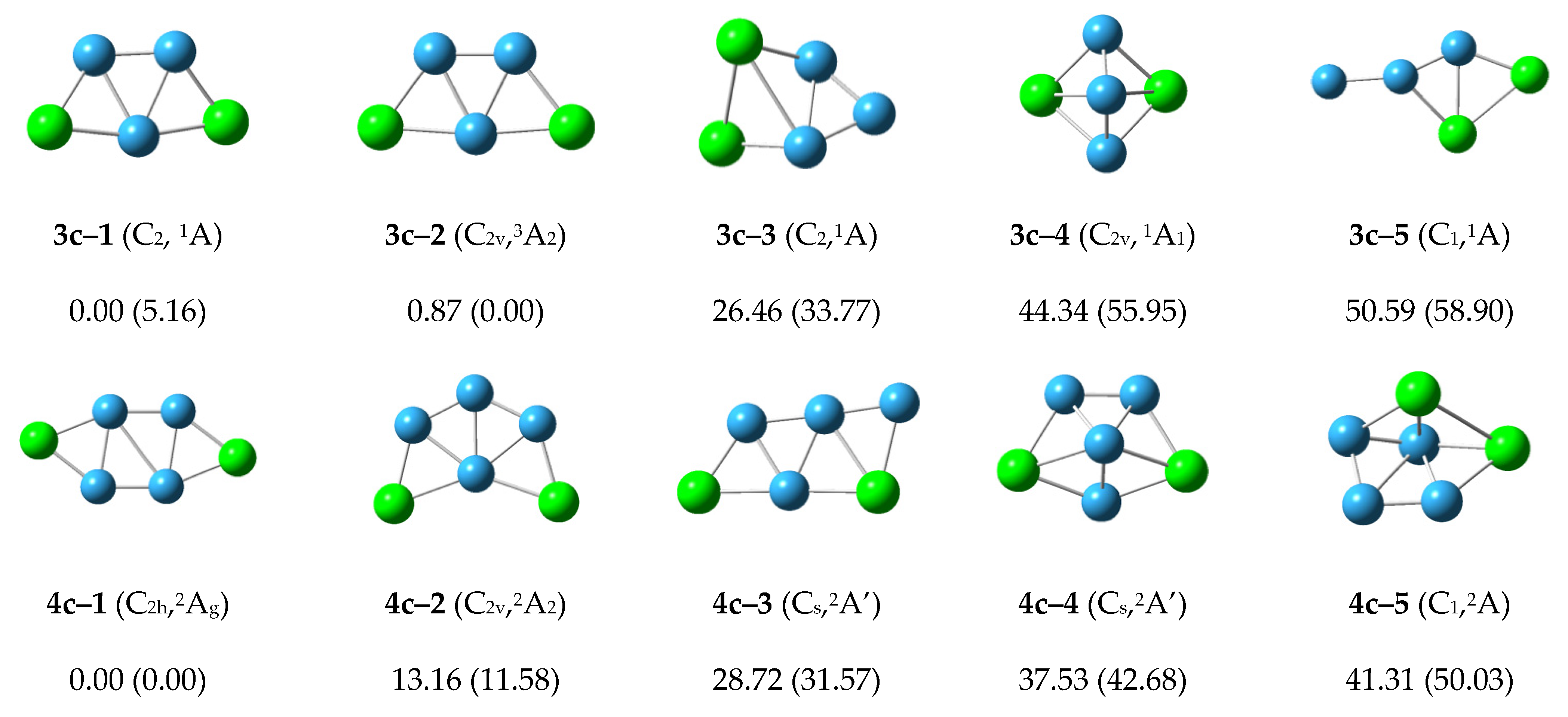
The low-lying isomers for each cationic P
2B
n+ cluster are shown in
Figure 3. For the cationic P
2B
3+ cluster, the lowest energy structure 3c–1 with
C2 symmetry is similar to the structure 3n–1 and 3a–1. The second structure 3c–2 is 0.87 kcal/mol higher than structure 3c–1 with
C2v symmetry and
3A
2 electronic state. The relative energy of other structures is more than 20 kcal/mol. We can conclude that the structures 3c–1 and 3c–2 may compete with each other to become the most stable structure in the cationic P
2B
3+ cluster.
Figure 3.
Optimized global minimum and low-lying isomers for cationic P2Bn+ clusters. The relative energies at CCSD(T)/6–311+G(2df) level are shown below each structure. The B3LYP/6–311+G(d) energies are shown in parentheses.
Figure 3.
Optimized global minimum and low-lying isomers for cationic P2Bn+ clusters. The relative energies at CCSD(T)/6–311+G(2df) level are shown below each structure. The B3LYP/6–311+G(d) energies are shown in parentheses.
For the cationic P2B4+ cluster, the structure 4c–1 with C2h symmetry has the lowest energy and is similar to the structures 4n–2 and 4a–2. The structure 4c–2 is 13.16 kcal/mol more than the energy of structure 4c–1, and is similar to the structure 4n–1 and 4a–2. The energy of the other structures is more than 20 kcal/mol, so we can conclude that the structure 4c–1 is the most stable structure in cationic P2B4+ cluster.
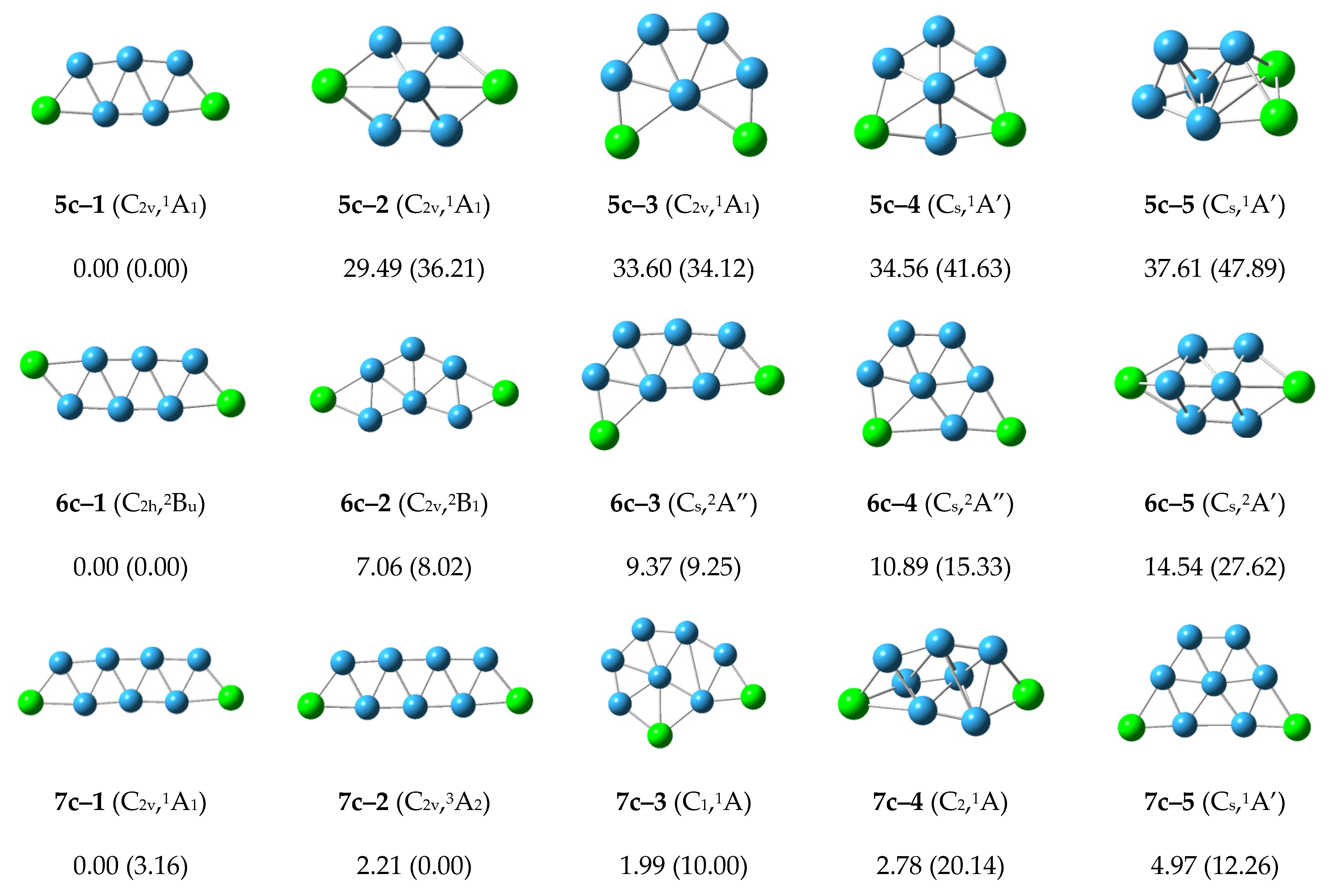
For the cationic P2B5+ cluster, the structure 5c–1 with C2v symmetry has the lowest energy and is similar to the structures 5n–1 and 5a–2. The second lower energy structure 5c–2 and the other structures have higher energy. The structure 5c–1 is the most stable structure in the cationic P2B5+ cluster.
The structure 6c–1 with the lowest energy and C2h symmetry. It grows in a zigzag shape, and is similar to the structures 6n–2 and 6a–1. The structures 6c–2, 6c–3 and 6c–4 with higher energy similar to structures 6n–3, 6n–4 and 6n–1, respectively. The other structures are higher energy, and the structure 6c–1 is the most stable structure in the cationic P2B6+ cluster.
The structures 7c–1 and 7c–3 grow in a zigzag shape, and have lower energy. These are similar to the structures 7n–1 and 7a–2. The structures 7c–2 and 7c–5 grow in a net-like fashion with relative energies of 1.99 and 4.97 kcal/mol. The other structures are higher energy and the structure 7c–1 is the most stable structure in the cationic P2B7+ cluster.
According to
Figure 3, the structures of the cationic P
2B
n+ clusters tend to preferentially show zigzag shapes and grow in a flat network. This may reduce the energy of the structures and make the structures more stable.
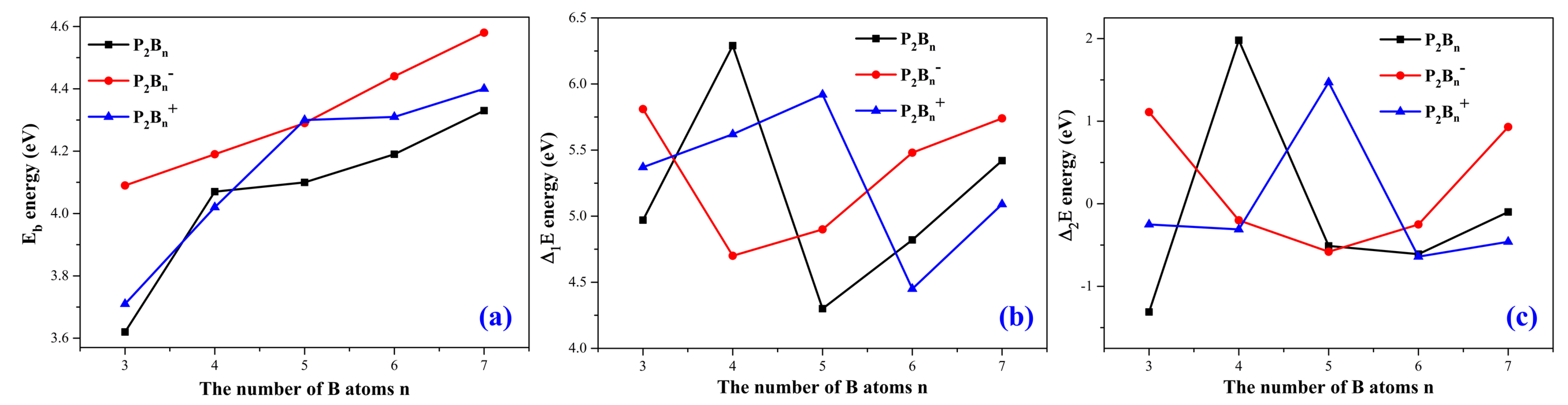
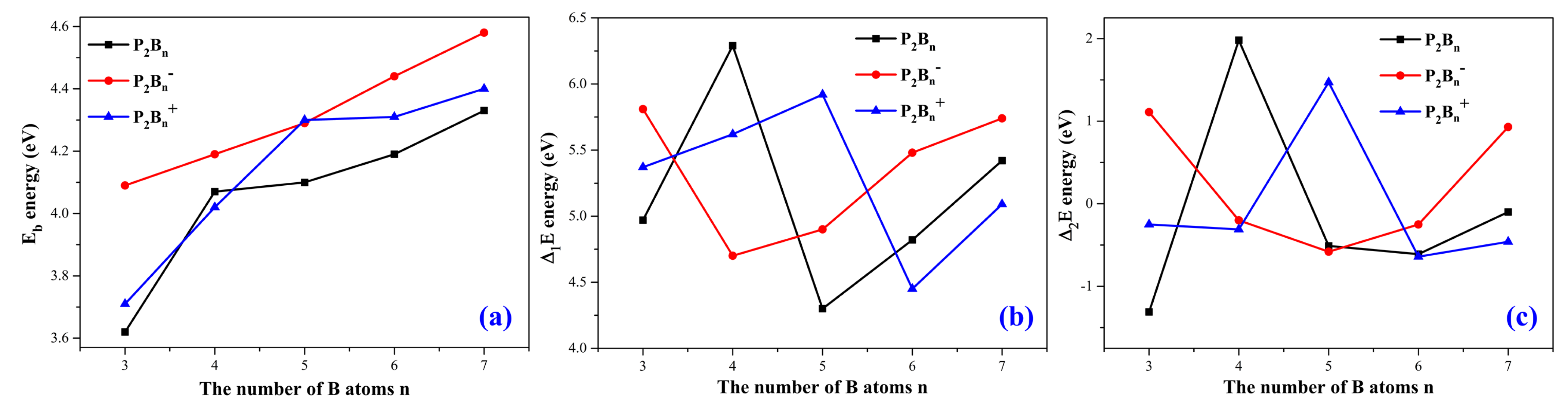
3.2. Relative Stabilities
In order to understand the stability of the structures of the phosphorus–doped boron clusters, we can calculate the average bonding energies, the fragmentation energies, and the difference energies for the clusters. They are represented by E
b, ∆E
1 and ∆E
2, respectively. These energies are calculated using the formulas above in
Table 1.
The average bonding energy (E
b) of the atomic clusters P
2B
n/P
2B
n−/P
2B
n+ are shown in
Figure 4a. All three curves show steady increase as the number of boron atoms increases. Therefore, an increase in the number of boron atoms can lead to higher stability of the structures.
The fragmentation energies (∆
1E) of the atomic clusters P
2B
n/P
2B
n−/P
2B
n+ are shown in
Figure 4b. For the neutral P
2B
n clusters, P
2B
4 is the maximum, and P
2B
5 is the minimum. For the anionic P
2B
n− clusters, as n increases, the value of ∆
1E first decreases, and then gradually increases. For the cationic P
2B
n+ clusters, as n increases, the value of ∆
1E first rises, then gradually decreases, and finally rises.
The second–order energy differences (∆
2E) for these clusters P
2B
n/P
2B
n−/P
2B
n+ are shown in
Figure 4c. We see that this has a small range of ±2 eV.
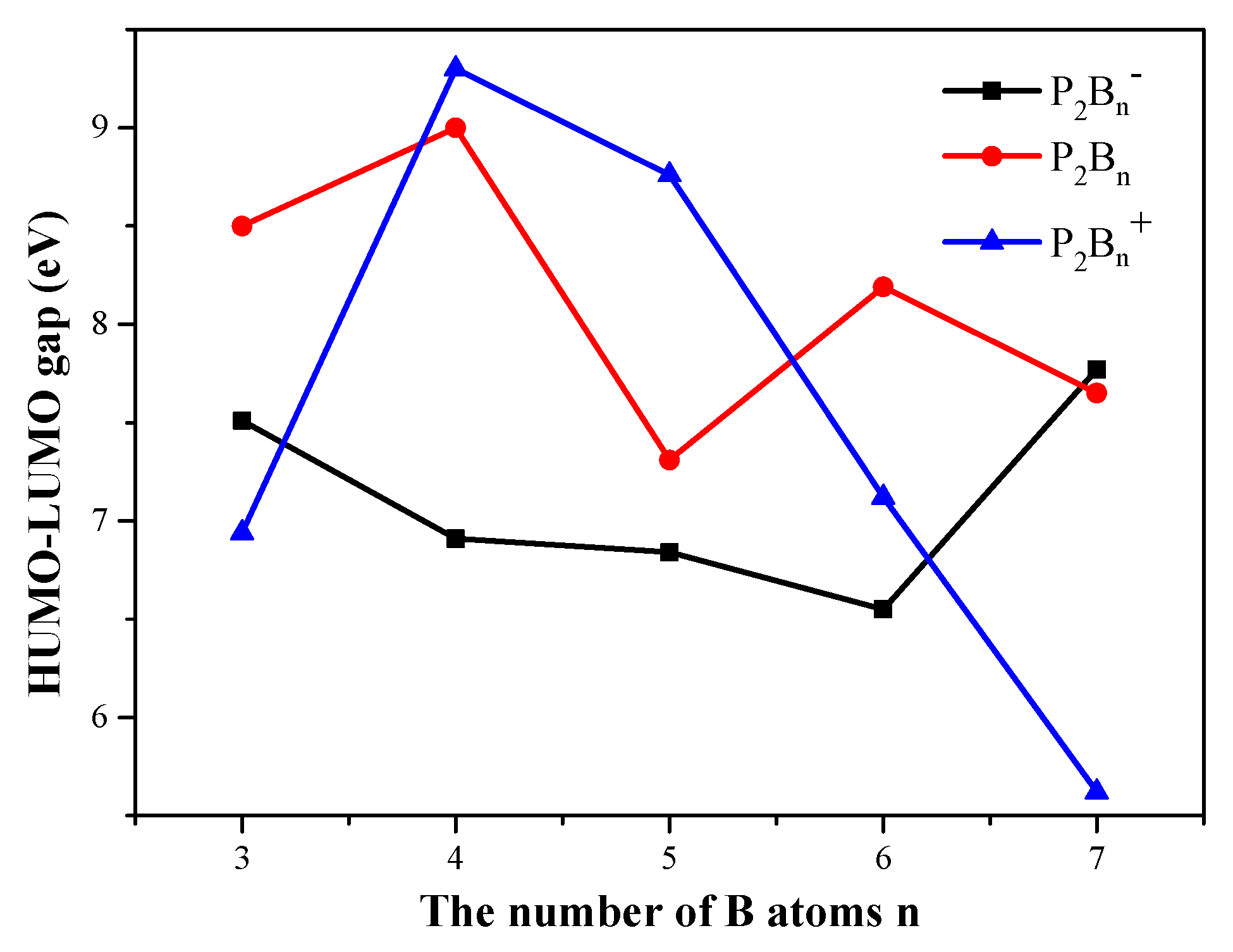
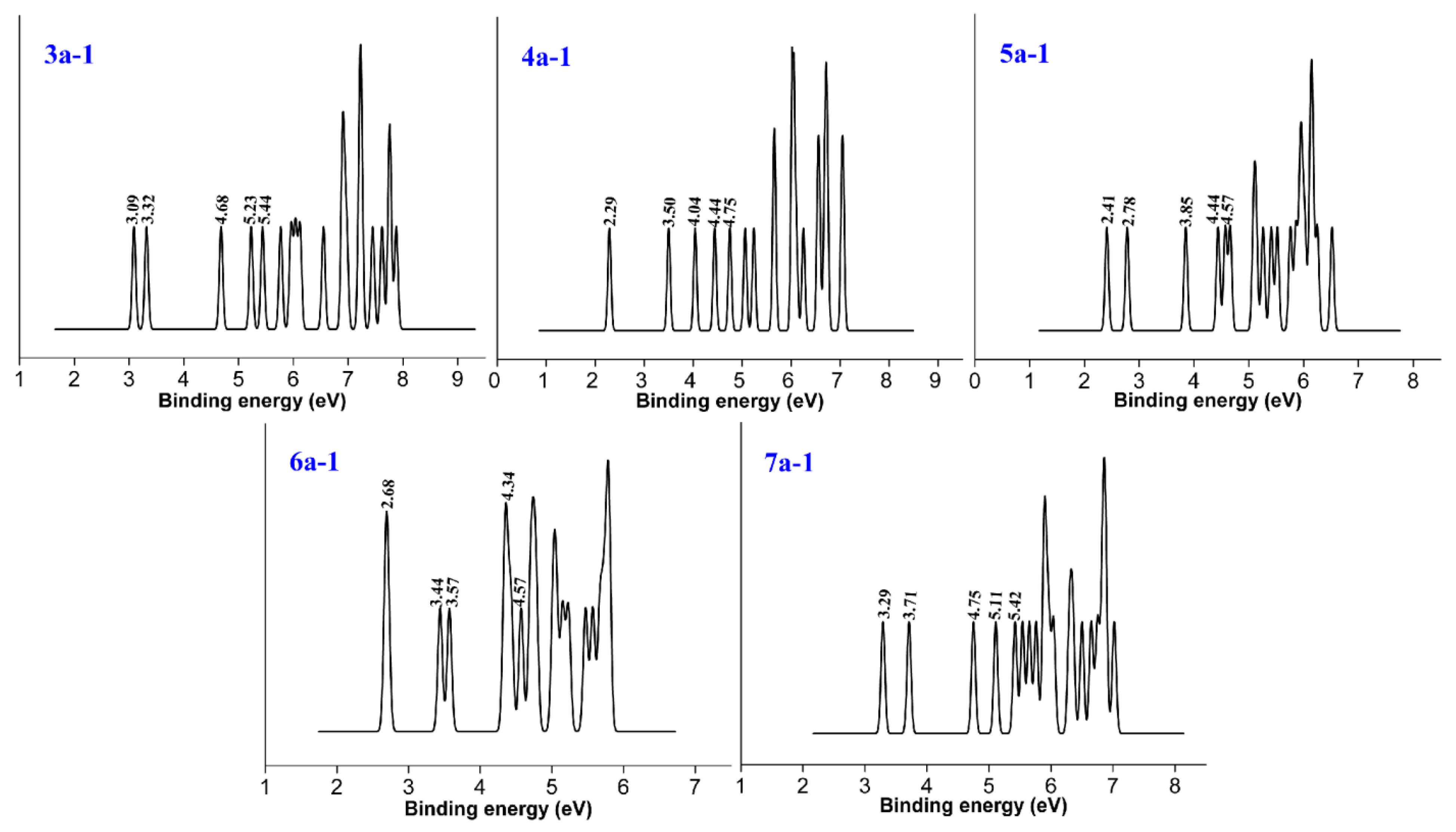
3.3. HOMO–LUMO Gap
HOMO and LUMO are collectively referred to as frontier orbital theory, the energy difference between HOMO and LUMO is called the energy band gap. This energy difference is called the HOMO–LUMO energy level, and used to measure whether a molecule is easily excited. The smaller the gap, the easier for the molecule to be excited.
As shown in
Figure 5, for the anionic P
2B
n− clusters, as the number of boron atoms increases, the HUMO–LUMO gaps show a downward trend, but when the number of boron atoms is seven, the value increases rapidly. The P
2B
3− and P
2B
7− clusters have higher stability than the other clusters. For the neutral P
2B
n clusters, we observe an odd–even oscillation rule. Therefore, we conjecture that when the number of boron atoms is even, more energy needs to be provided when electron transition occurs. For the HOMO–LUMO gap curve of cationic P
2B
n+ clusters in
Figure 5, the curve first rises and then continues to fall. The cationic P
2B
4+ cluster has the maximum, and the P
2B
7+ cluster has the minimum value and stability. In general, the gap values are large, ranging between 5.5 and 9.5 eV.
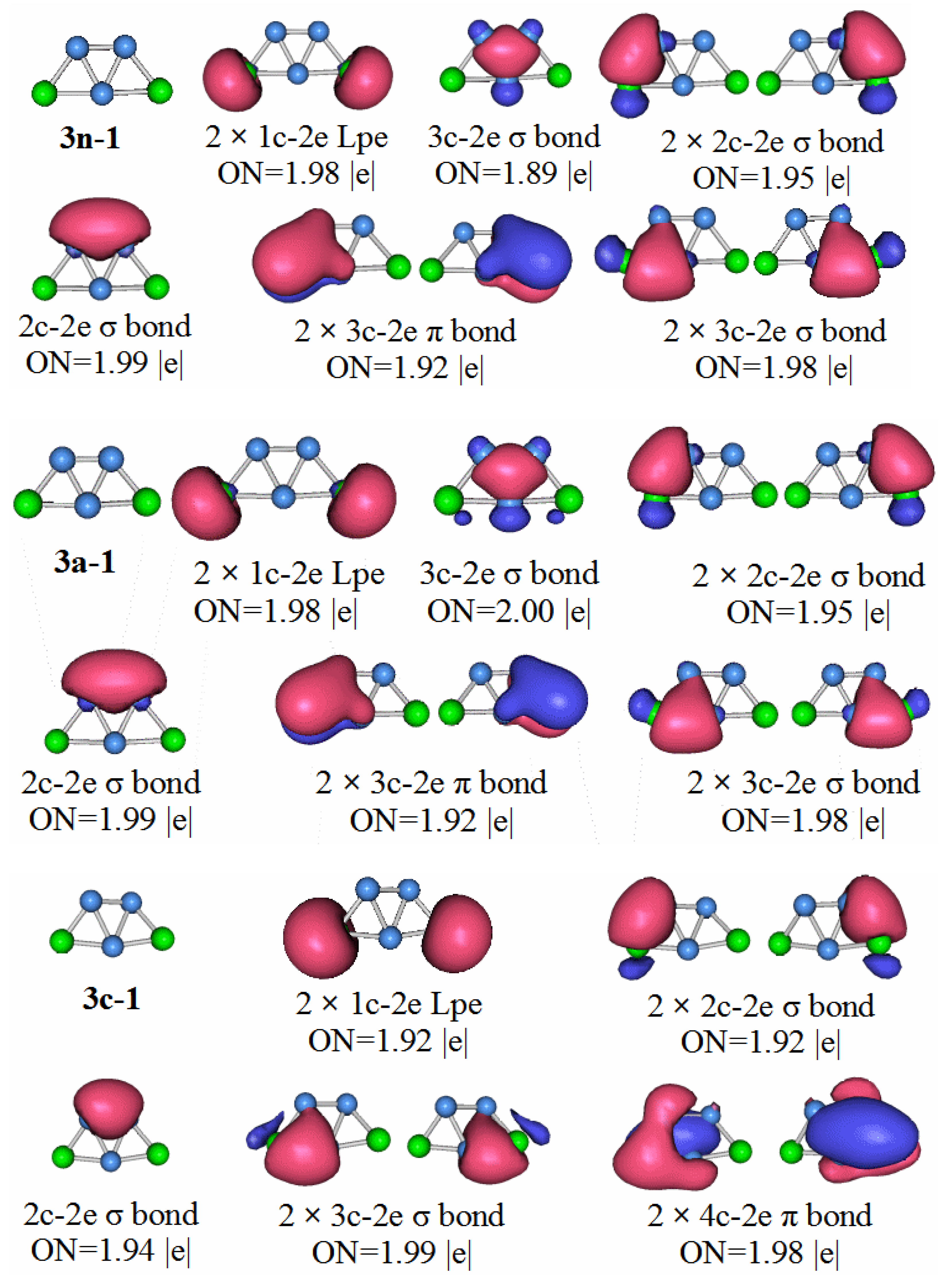
3.5. Chemical Bonding Analysis
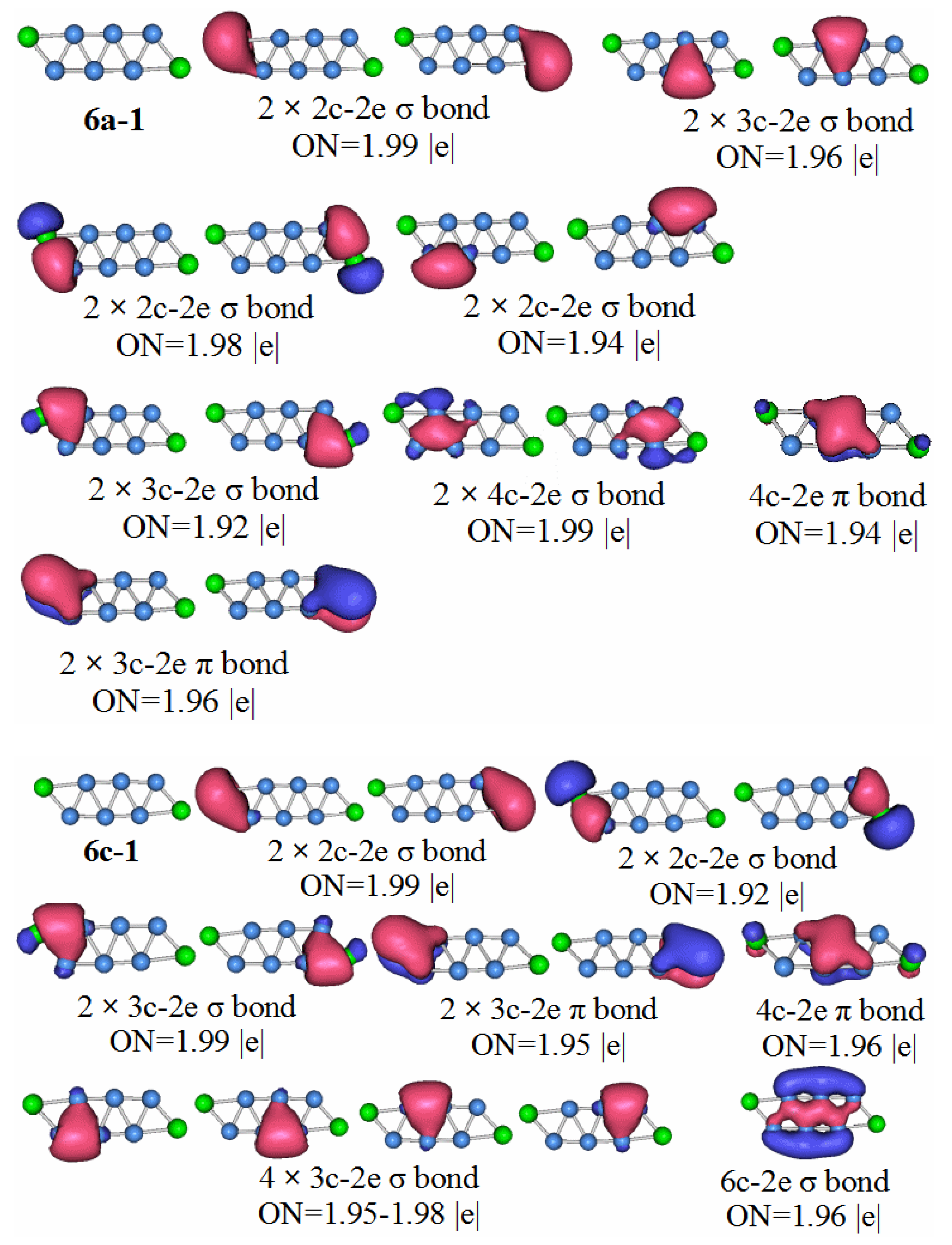
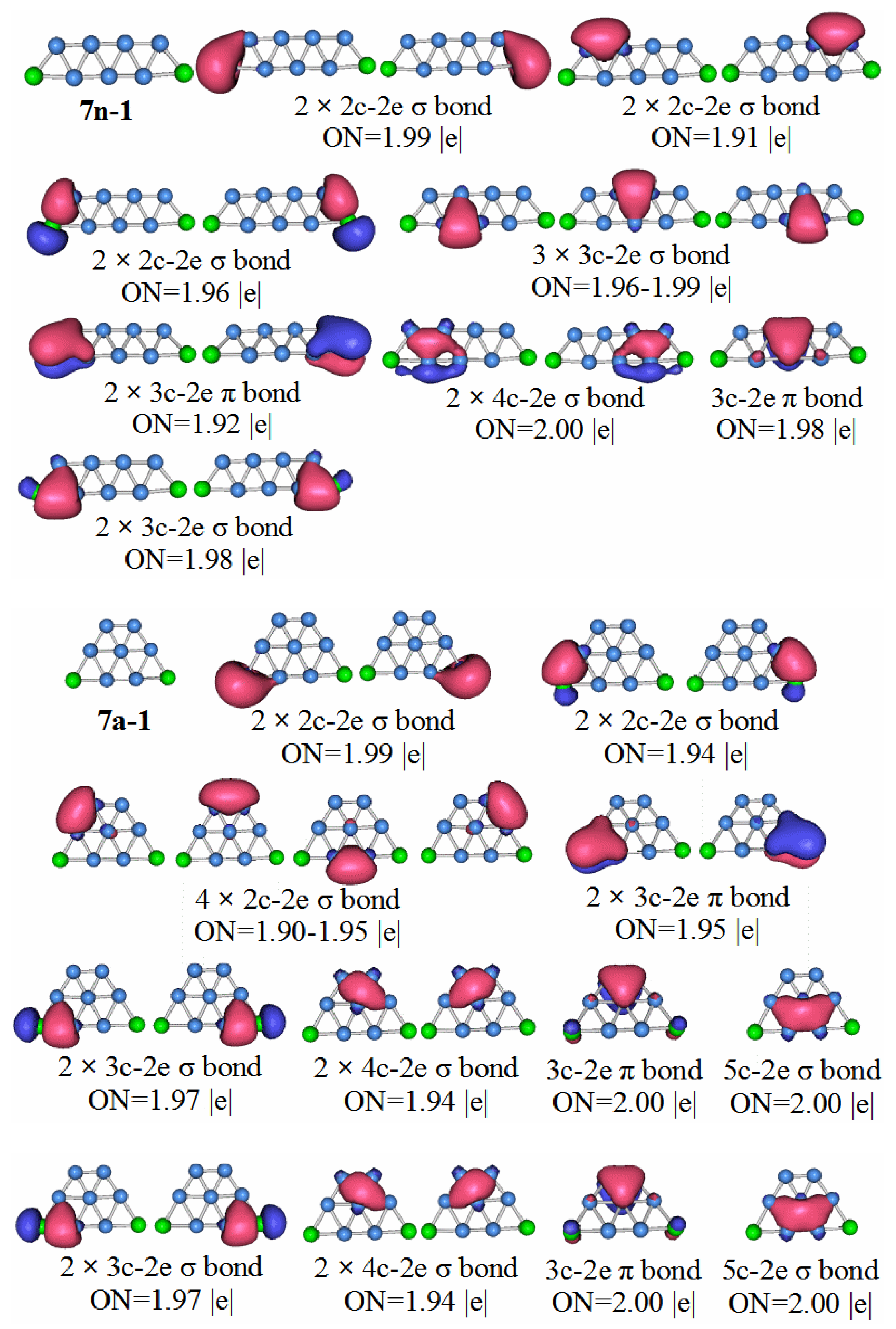
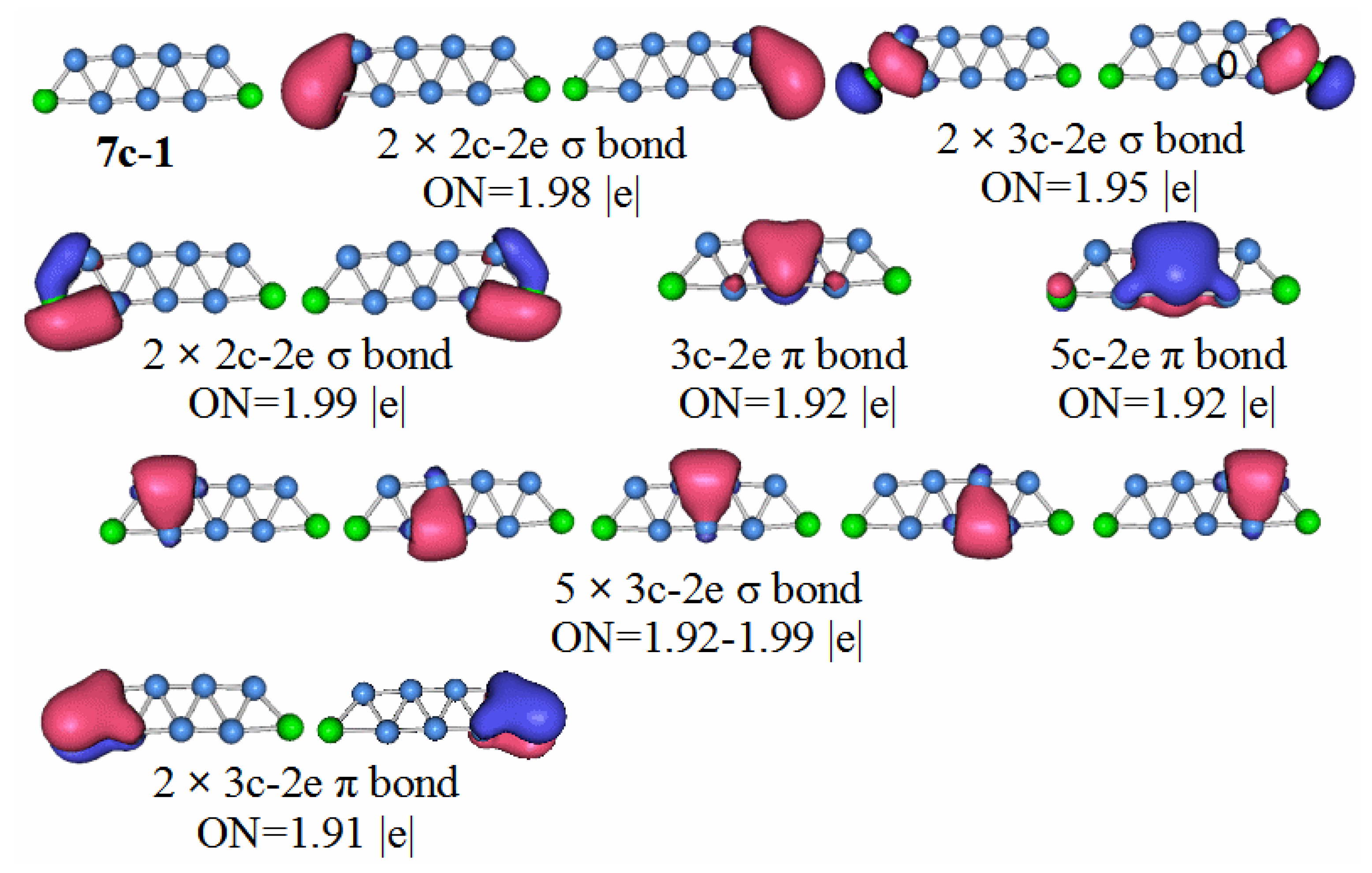
In order to have a deeper understanding of the stable structure of these clusters, chemical bond analysis is important. Below is a chemical bond analysis of the stable structure of these clusters using AdNDP code. Although the figure shows the bond formation of neutral molecules, anions, and cations, we only analyze the bond formation of neutral molecules here.
For an anion with a unit of negative charge, if its HOMO–LUMO gap is larger, it will have higher stability when it loses an electron and becomes a neutral molecule. Our previous analysis of the HOMO–LUMO gaps of cluster anion molecules shows that the HOMO–LUMO gap value of P2B3− is larger than that of the other phosphorus-boron cluster molecules. Therefore, P2B3 neutral molecules should have high stability, and thus stronger chemical bonds.
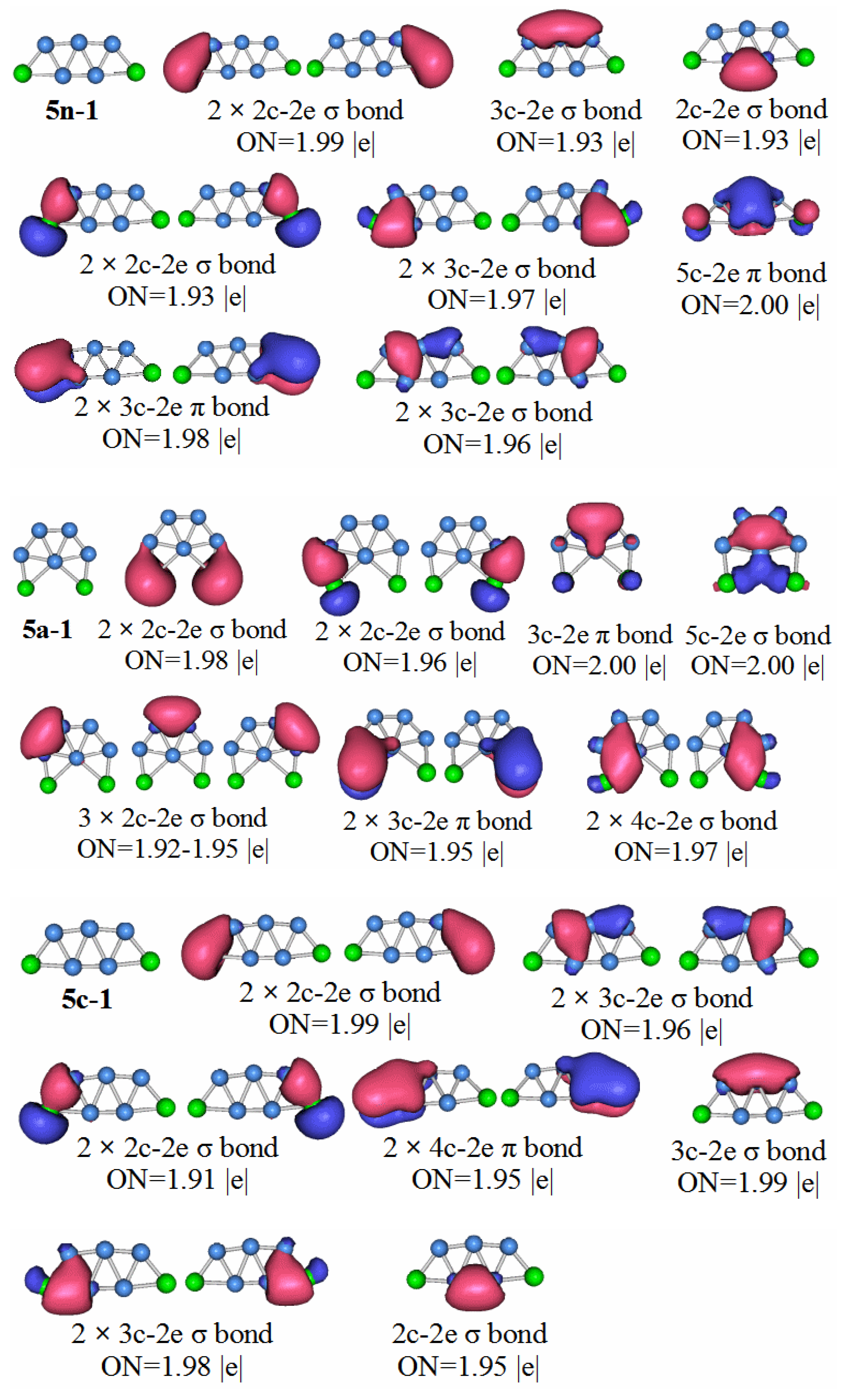
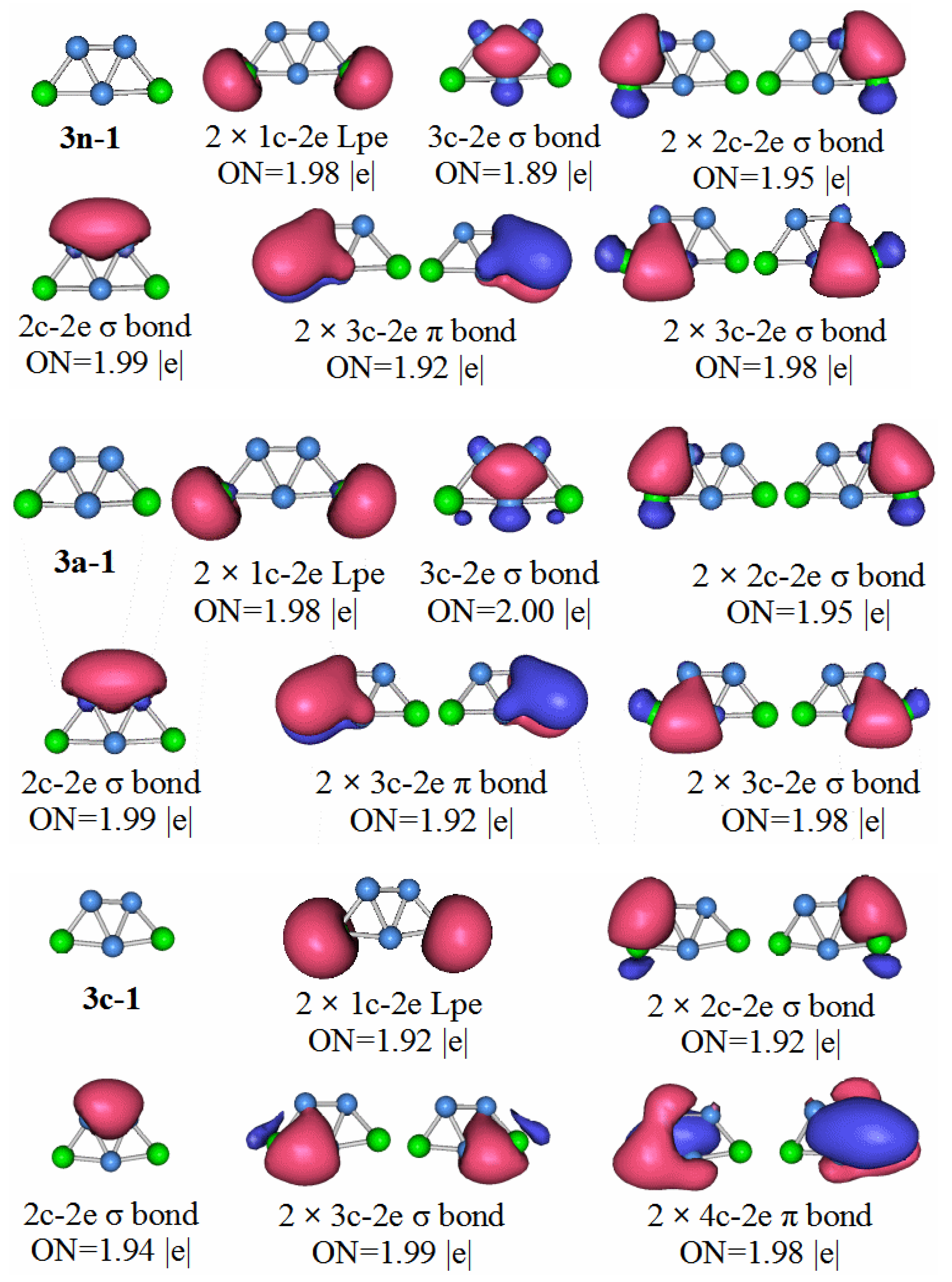
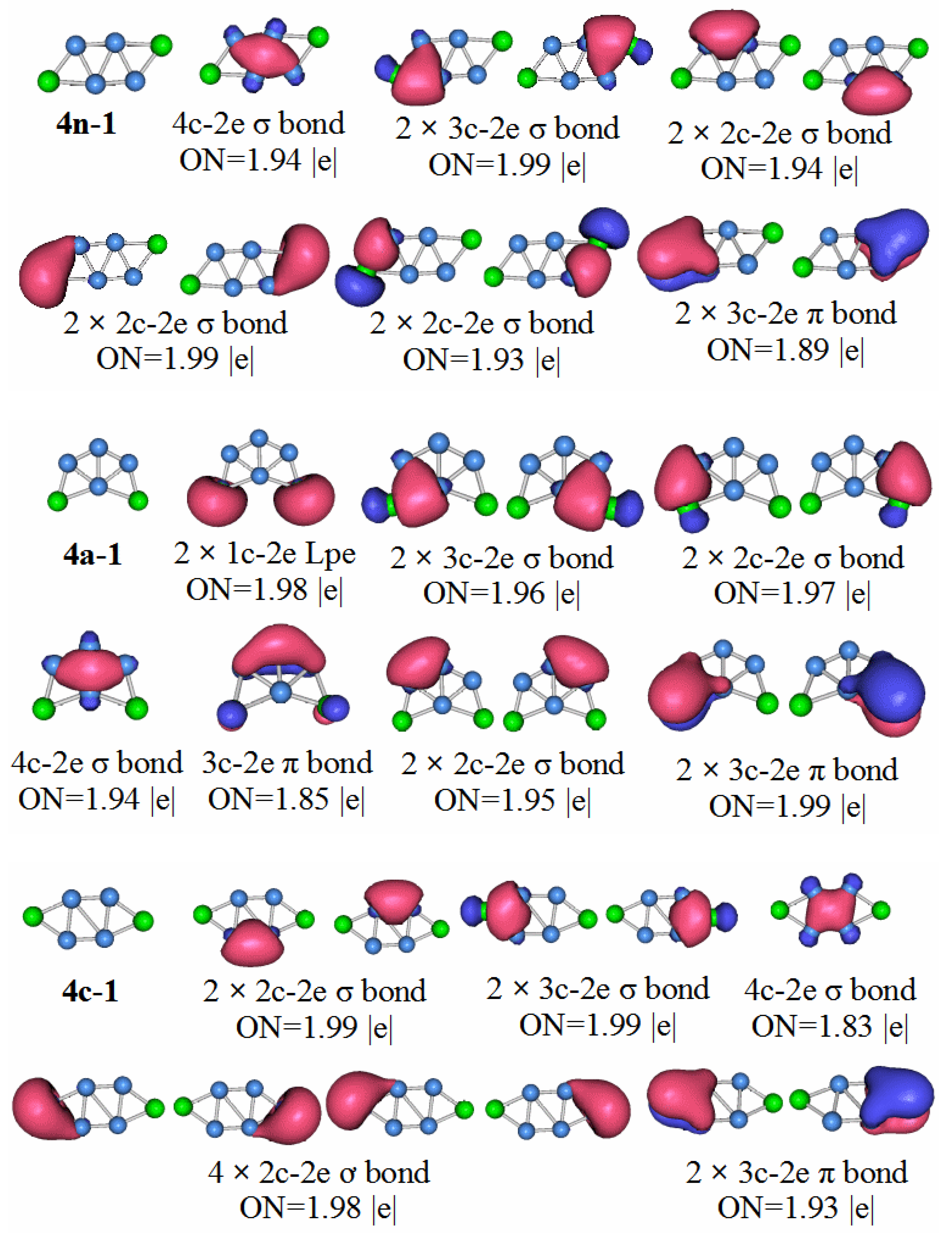
P2B3: According to the AdNDP analysis of the 3n–1 structure in
Figure 7, it can be seen that the structure has three 2c–2e chemical bonds, one of which is the B–B bond. The other two are P–B bonds. The structure also has two 3c–2e B–P–B σ bonds, and one 3c–2e B–B–B σ bond, as well as two 3c–2e B–P–B π bonds. Finally, there are two lone pairs. It can be seen that the chemical bond formed by the P
2B
3 molecule has six delocalized sigma electrons, which can be regarded as having sigma electron aromaticity, and two pairs of delocalized π electrons can be regarded as having π electron aromaticity, making the P
2B
3 molecule more stable.
Compared with the P2B3 anion, the HOMO–LUMO gap value of P2B4− is smaller. For neutral molecules, P2B4 molecules are slightly less stable than P2B3 molecules. The results of the chemical bond analysis of P2B4 molecules also support this view.
P2B4: Looking at the structure of 4n–1 in
Figure 8, it is found that the P–B chemical bonds of the two 2c–2e bonds has ON = 1.93, and the other P–B bond of the two 2c–2e bonds has ON = 1.99. Two 2c–2e B–B bonds have ON = 1.94. Two 3c–2e P–B bonds have ON = 1.99, 3c–2e P–B π bonds with two ON = 1.89, and 4c–2e B–B bonds with ON = 1.94. The P
2B
4 molecule has two pairs of sigma electrons connected. This can be regarded as having sigma electron antiaromaticity, which is not conducive to the stability of P
2B
4. The two pairs of π delocalized π electrons can be regarded as having π electron aromaticity, so the P
2B
4 molecule is stable. This stability is slightly weaker than the P
2B
3 molecule.
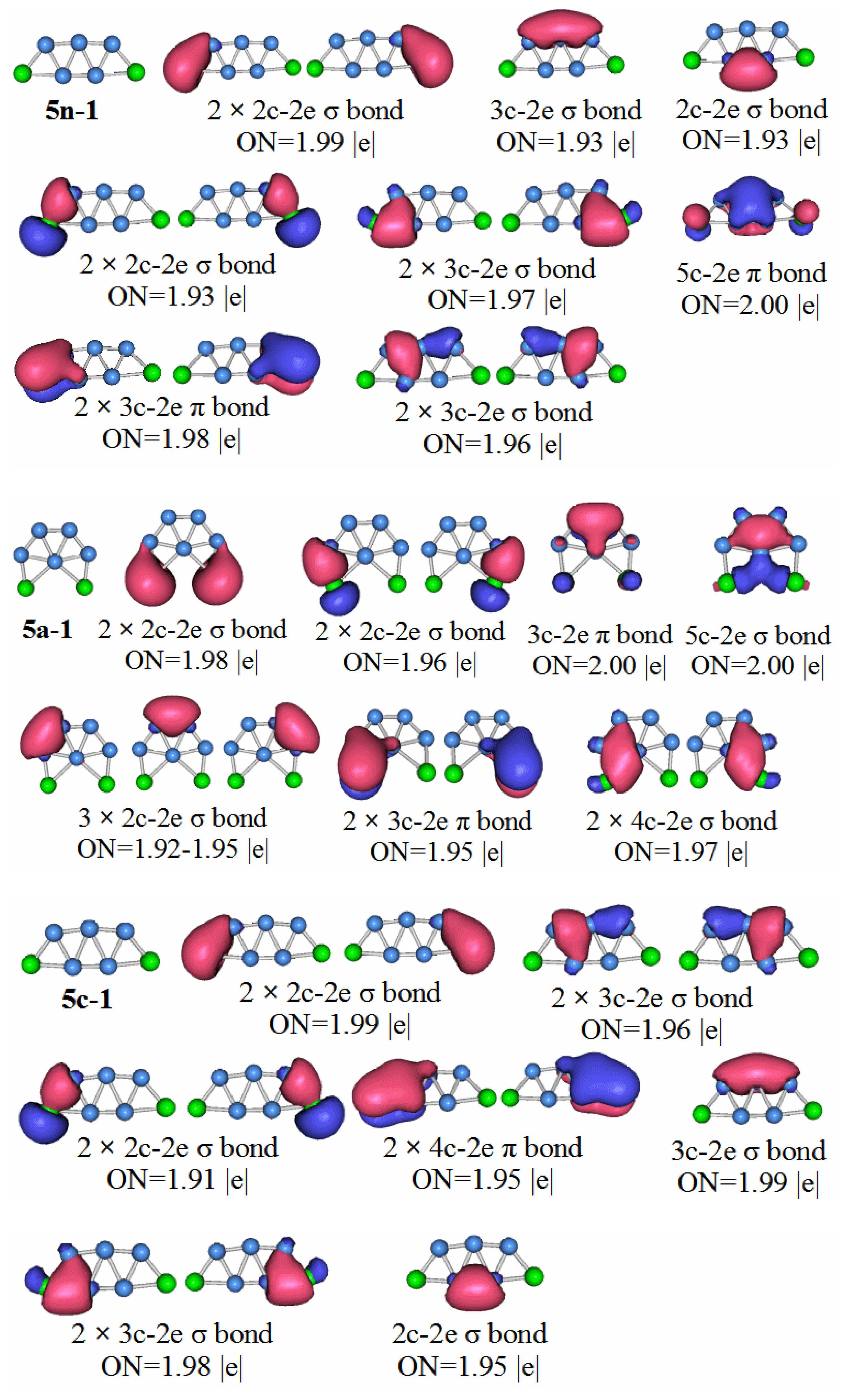
Compared with the previous HOMO–LUMO gap of P2B3− and P2B4−, the value of P2B5− is smaller. It shows that in terms of stability, the P2B5 molecule is weaker than the previous two types of molecules. Through the chemical bond analysis of the P2B5 molecule, we have also found the corresponding basis.
P2B5: Figure 9 shows the AdNDP analysis of the stable structure of the P
2B
5 neutral cluster. The results of the analysis are as follows, the structure contains five 2c–2e chemical bonds containing a B–B bond, two P–B bonds with ON = 1.99, and two P–B bonds of ON = 1.93. There are also seven 3c–2e chemical bonds, and two 3c–2e π bonds. In addition, a 5c–2e chemical bond with ON = 2.00. We can regard the 3c–2e sigma bonds with ON values equal to 1.97 and 1.96 as two pairs of 4c–4e sigma bonds. This means that the P
2B
5 molecules have two pairs of anti-aromatic delocalized sigma bonds, which is not conducive to the stability of the molecule. Although the P
2B
5 molecule has a large aromatic delocalized π bond, it still makes the stability of the molecule lower than that of P
2B
3 and P
2B
4.
Compared with the other clusters, the HOMO–LUMO gap value of the P2B6 anion is the smallest. It is not difficult to conclude that the stability of neutral molecules of P2B6 is the worst. The symmetry of the neutral molecules of P2B6 is the C1 point group. Let us look at the chemical bond analysis of the P2B6 molecule.
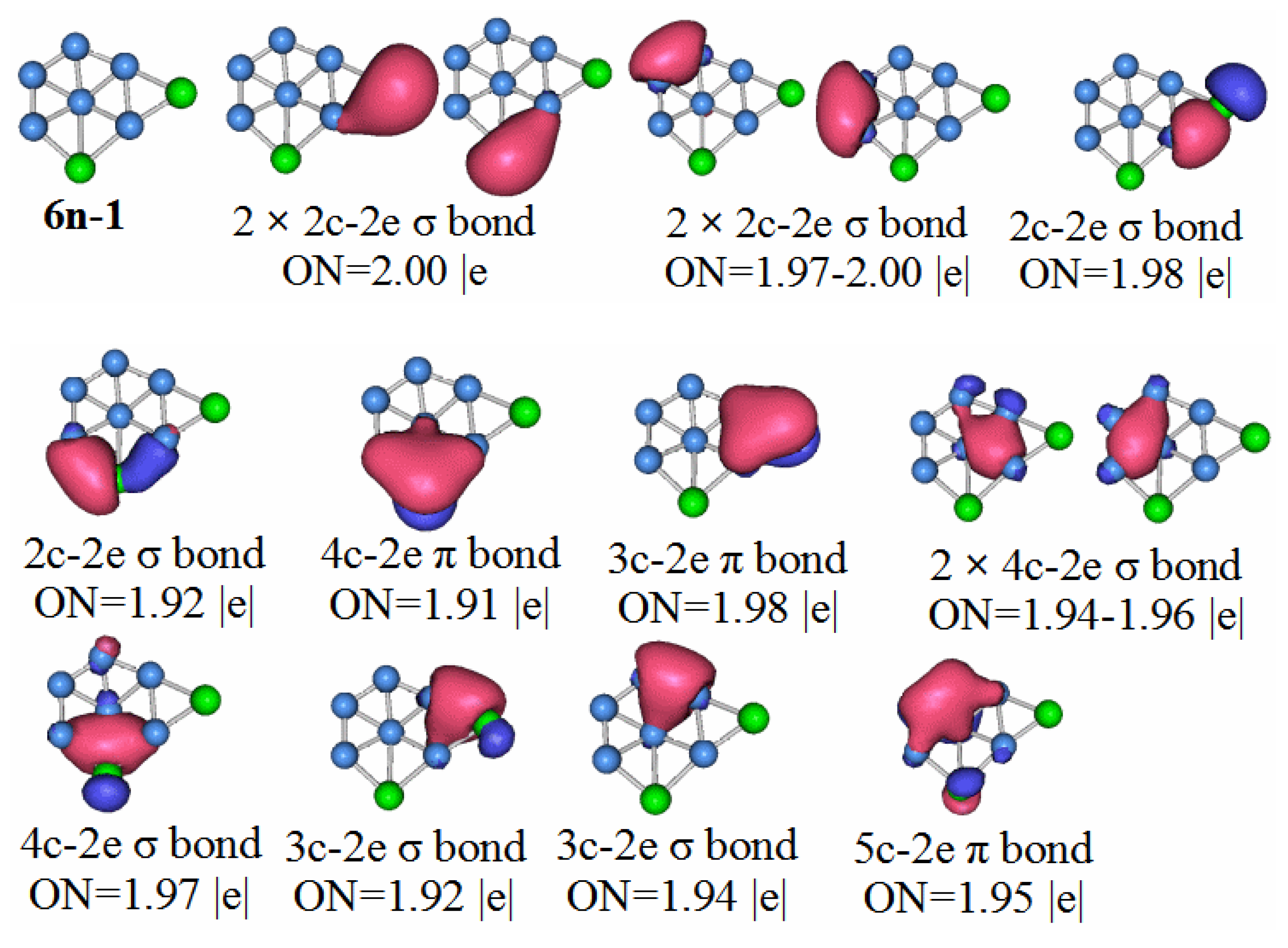
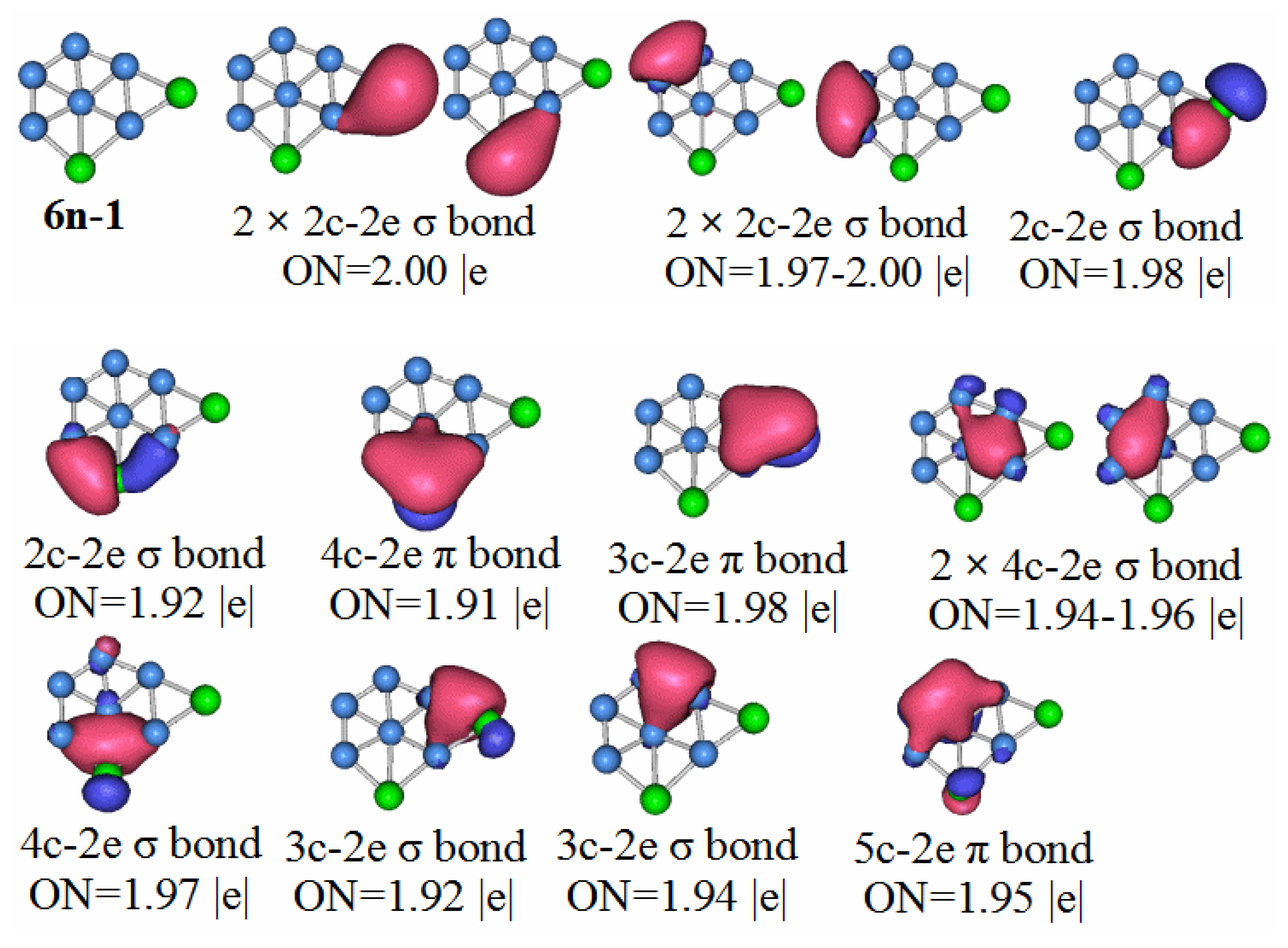
P2B6: According to the AdNDP analysis of 6n–1 in
Figure 10, the bonding of the most stable structure of the P
2B
6 neutral cluster is as follows. The structure has six 2c–2e chemical bonds, five 3c–2e chemical bonds, two 4c–2e chemical bonds, and one 5c–2e chemical bond. Only two of the 2c–2e bonds are B–B. The rest are P–B. Three of the 3c–2e chemical bonds are σ bonds, two are π bonds, and there is a single π bond of 5c–2e. We can regard the delocalized 4c–2e and 5c–2e π bonds as antiaromatic delocalized π bonds. For the delocalized sigma bonds formed by P
2B
6 molecules, the ON values are 1.97 for the 4c–2e sigma bond, 1.92 for the 3c–2e sigma, and 1.94–1.96 for the sigma bonds. We can regard these as two sets of antiaromatic delocalized sigma bonds. This is the main reason that makes the P
2B
6 molecules weaker than other the other molecules under study.
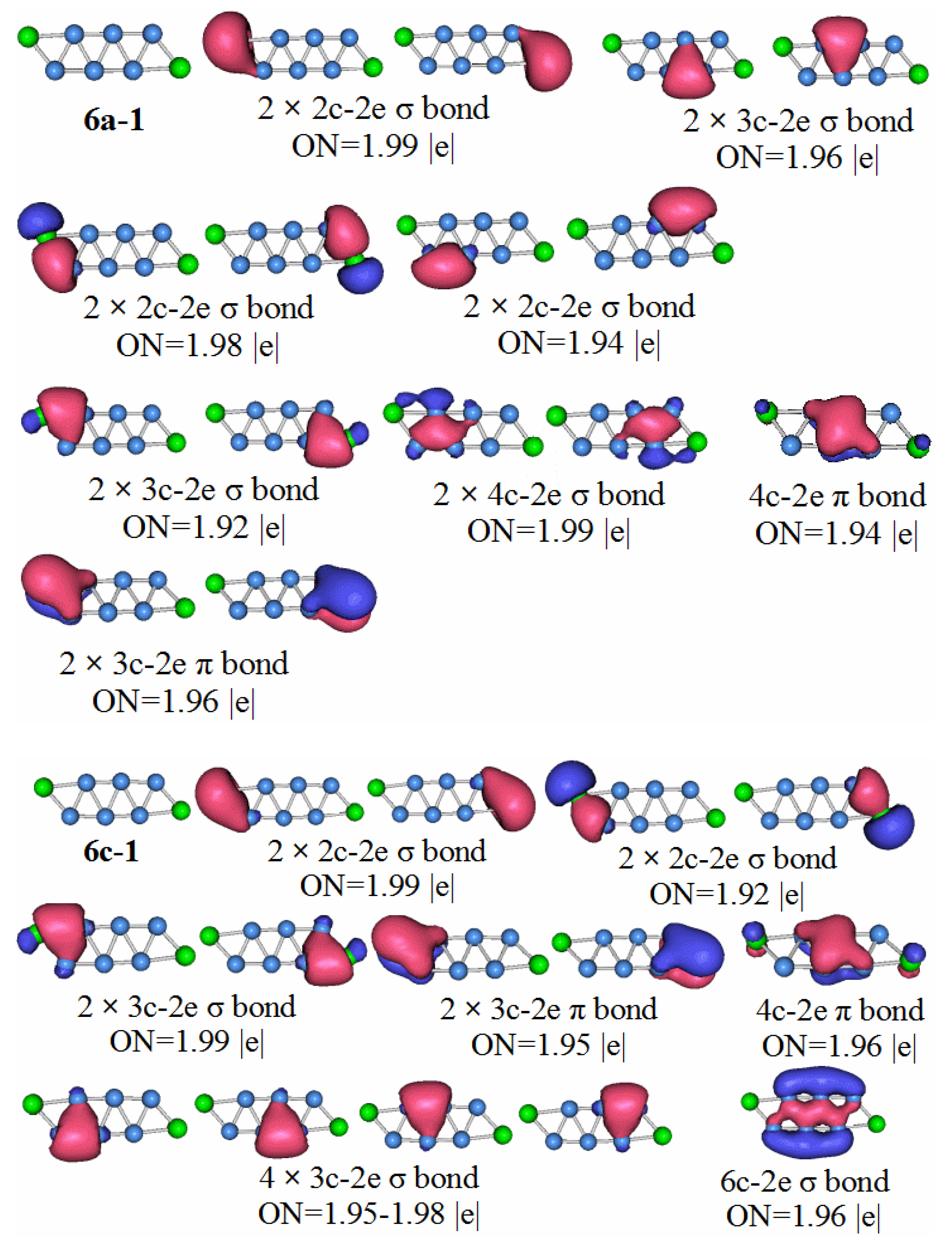
The HOMO–LUMO gap value of the P2B7 anion is the largest of the molecules in this paper. Therefore, the P2B7 neutral molecule should be the most stable. Of course, the results of the chemical bond analysis of the neutral P2B7 molecule also confirmed this.
P2B7: For the 7n–1 structure in
Figure 11, we see six 2c–2e chemical bonds, and nine 3c–2e chemical bonds. Two 2c–2e chemical bonds are B–B bonds, and the other four are P–B bonds. Five of the chemical bonds are 3c–2e σ bonds of B–B, two P–B σ bonds, and two P–B π bonds. We can divide the delocalized sigma electrons into two categories. The first category contains the 3c–2e sigma bond with ON = 1.98, the 4c–2e sigma bond with ON = 2.00, and the 3c–2e sigma bonds with ON range of 1.96 to 1.99 (we can consider these to be like a large 9c-10e aromatic delocalized sigma bond). The second type, the 3c–2e delocalized sigma bond with ON value between 1.96 and 1.99 is regarded as a pair of small aromatic sigma bonds. The π bonds in the P
2B
7 molecule can all be regarded as aromatic delocalized π bonds, which is also in line with the HOMO–LUMO gap value and high molecular stability.
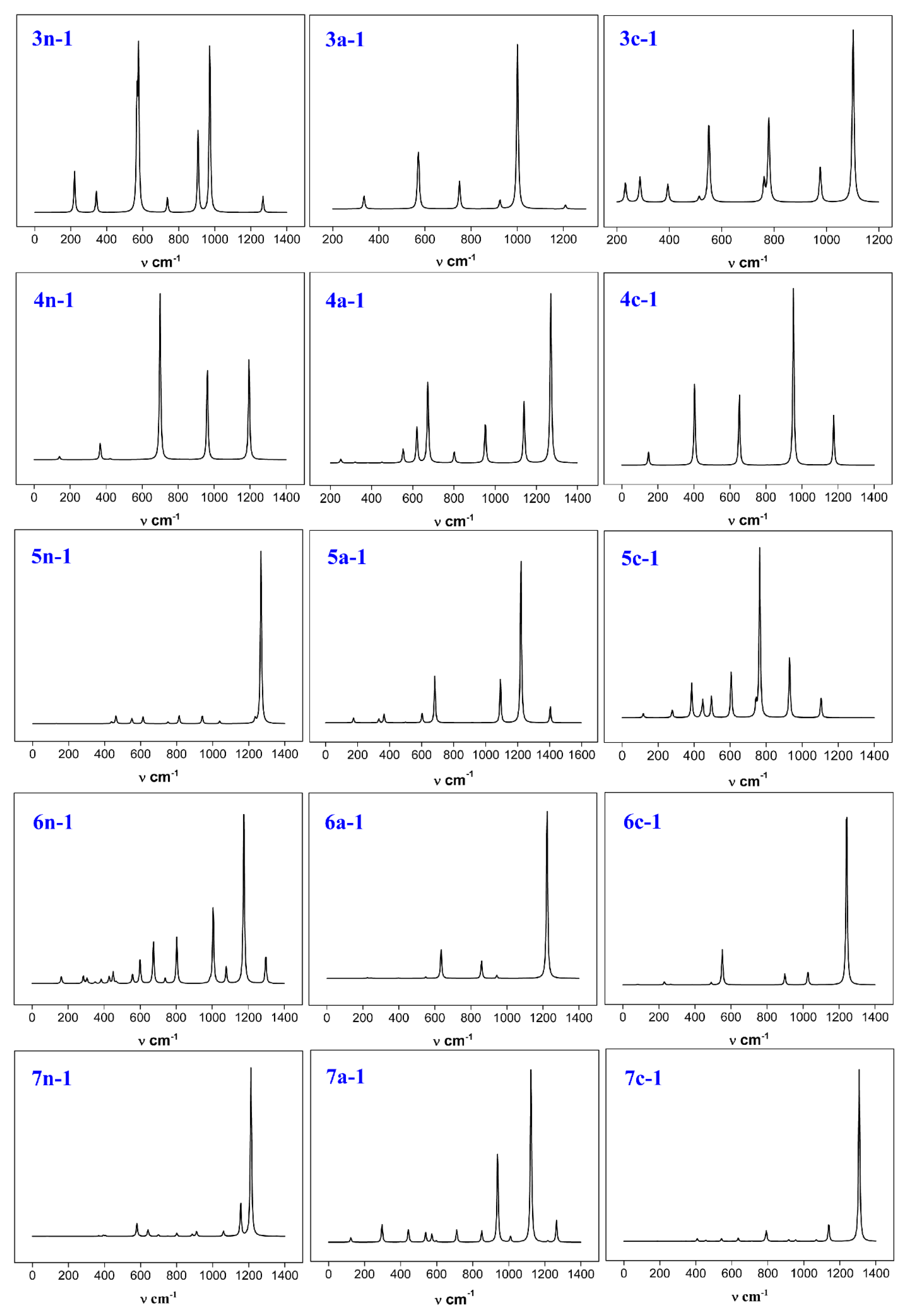
3.6. Infrared Spectroscopy
In order to contribute to potential future synthesis of these systems experimentally, we simulated the infrared spectrum of the stable structure of these boron–doped boron clusters. This will allow the experimenters to compare and identify these materials using the experimental infrared spectrum. The infrared spectrum of each stable structure is shown in
Figure 12.
We briefly introduce a few major peaks for each structure. The 3n–1 structure has three main absorption peaks at 576.29 cm−1, 907.44 cm−1, and 972.41 cm−1. 576.29 cm−1 is the asymmetric stretching vibration absorption peak of the central boron atom and the two peripheral boron atoms. 907.44 cm−1 is the symmetric stretching vibration absorption peak of the central boron atom and the two peripheral boron atoms. 972.41 cm−1 is an asymmetric stretching vibration absorption peak formed by the central boron atom and two phosphorus atoms. For the 3a-1 structure, 335.77 cm−1 is the in-plane bending vibration absorption peak of the peripheral phosphorus-boron bond, and 749.73 cm−1 is the asymmetric stretching vibration absorption peak of the two phosphorus-boron bonds formed by the phosphorus atom. 1001.16 cm−1 is an asymmetric stretching vibration absorption peak formed by a central boron atom and two phosphorus atoms. The 3c–1 structure has a peak at 550.96 cm−1, which is the out-of-plane bending vibration absorption peak of the boron-boron bond formed by the central boron atom.
The 4n-1 structure has four main absorption peaks at 387.79 cm−1, 700.08 cm−1, 962.17 cm−1, and 1193.82 cm−1, respectively. 387.79 cm−1 is the in-plane bending vibration absorption peak of the phosphorus-boron bond. 700.08 cm−1 is the asymmetric stretching vibration absorption peak of the boron-boron bond. 962.17 cm−1 is a symmetric stretching vibration absorption peak formed by a phosphorus atom and a boron atom. 1193.82 cm−1 is an asymmetric stretching vibration absorption peak formed by a phosphorus atom and a boron atom. The 4a–1 structure has 1140.42 cm−1 which is a symmetric stretching vibration absorption peak formed by three peripheral boron atoms. 1270.17 cm−1 is an asymmetric stretching vibration absorption peak formed by three peripheral boron atoms. The infrared spectral absorption peaks of the 4c–1 structure have been described in the absorption peaks of the above two structures.
The 5n-1 structure has 1268.67 cm−1 which is a main absorption peak. It is an asymmetric stretching vibration absorption peak of three boron atoms arranged in a straight line in the structure. The 5a–1 structure has three main absorption peaks. 682.49 cm−1 is an asymmetric stretching vibration absorption peak of the boron-boron bond. 1092.44 cm−1 is the symmetric stretching vibration absorption peak of the peripheral boron atoms in the structure. 1221.02 cm−1 is the asymmetric stretching vibration absorption peak of the peripheral boron atoms in the structure. The 5c–1 structure has 764.29 cm−1 which is a symmetric stretching vibration absorption peak of a boron-boron bond.
The 6n-1 structure has four main absorption peaks. 673.90 cm−1 is an asymmetric stretching vibration absorption peak of the boron-boron bond. 803.61 cm−1 is the symmetric stretching vibration absorption peak of the boron-boron bond at the center of the structure. 1005.92 cm−1 is an asymmetric stretching vibration absorption peak of the phosphorus-boron bond. 1175.87 cm−1 is the symmetric stretching vibration absorption peak of the phosphorus-boron bond. 1222.82 cm−1 in the 6a-1 structure is a symmetric stretching vibration absorption peak of the phosphorus-boron bond. 1241.85 cm−1 of the 6c-1 structure is also a symmetric stretching vibration absorption peak of the phosphorus-boron bond.
The 7n–1 structure has 1115.80 cm−1, which is an asymmetric stretching vibration absorption peak of phosphorus-boron bond and boron-boron bond. 1212.60 cm−1 is the asymmetric stretching vibration absorption peak of the linear boron-boron bond. The structure of 7a–1 has 938.49 cm−1, which is the symmetry stretching vibration absorption peak of the boron-boron bond of the central boron atom. At 1141.40 cm−1, is an asymmetric stretching vibration absorption peak of a boron-boron bond formed by a central boron atom. At 1265.38 cm−1, there is an asymmetric stretching vibration absorption peak of the peripheral boron-boron bond. The 7c–1 structure has only one main absorption peak, at 1307.65 cm−1, which is an asymmetric stretching vibration absorption peak of a boron-boron bond arranged linearly in the structure.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}