
Anodic Activity of Hydrated and Anhydrous Iron (II) Oxalate in Li-Ion Batteries



Abstract
:1. Introduction
2. Computational Method
3. Results and Discussion
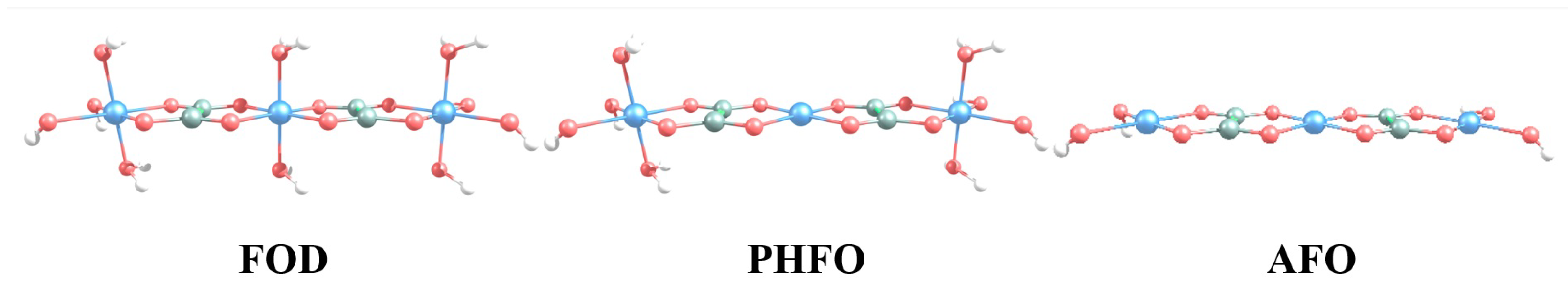
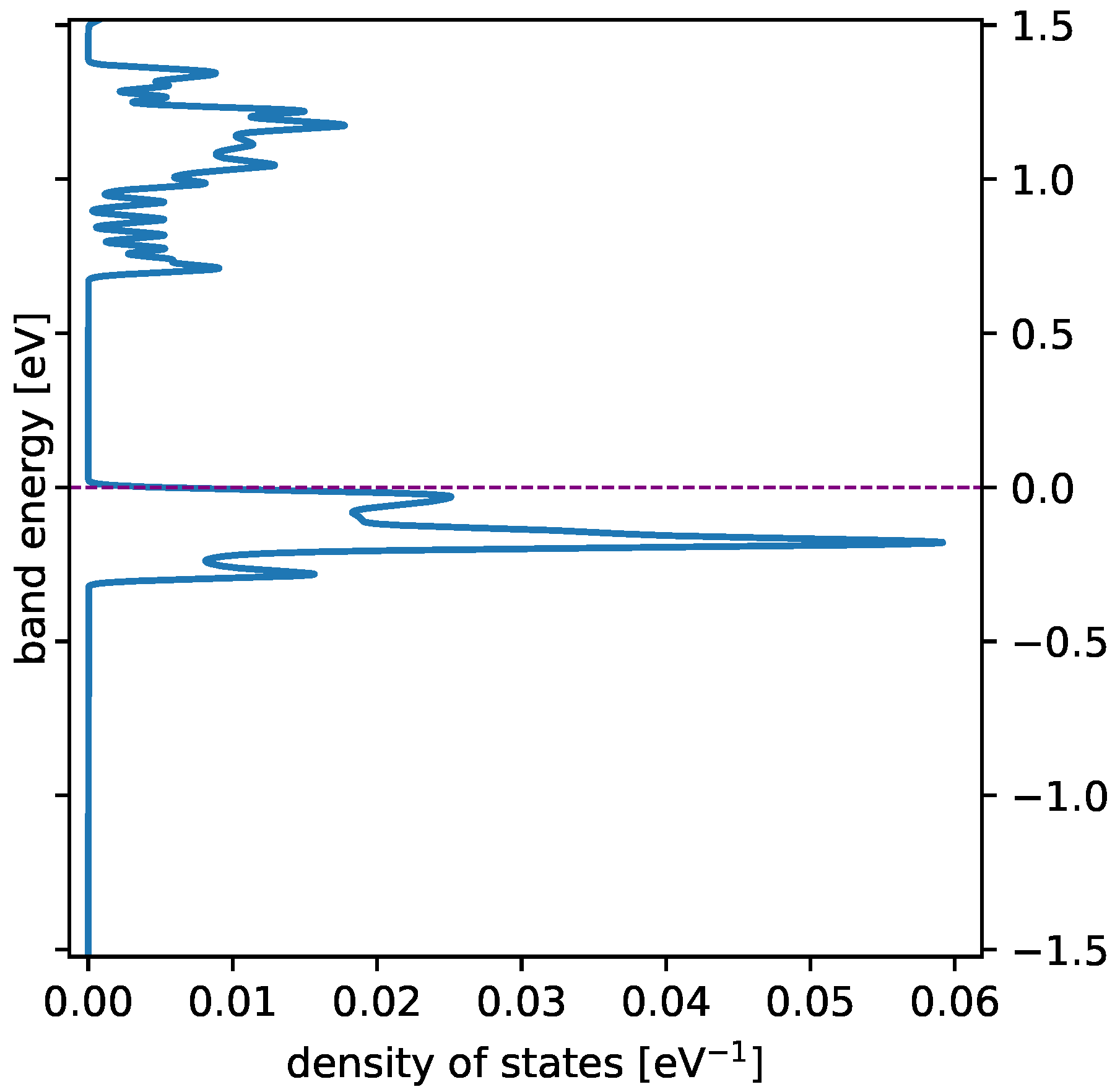
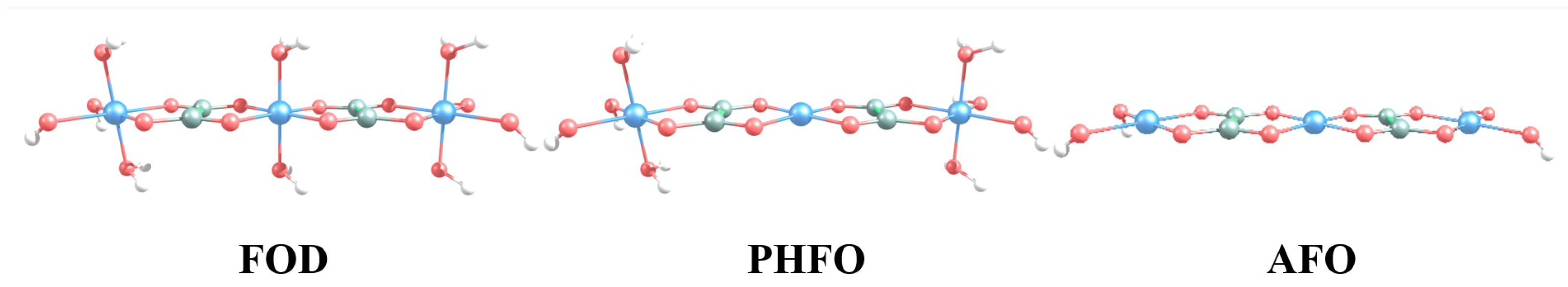
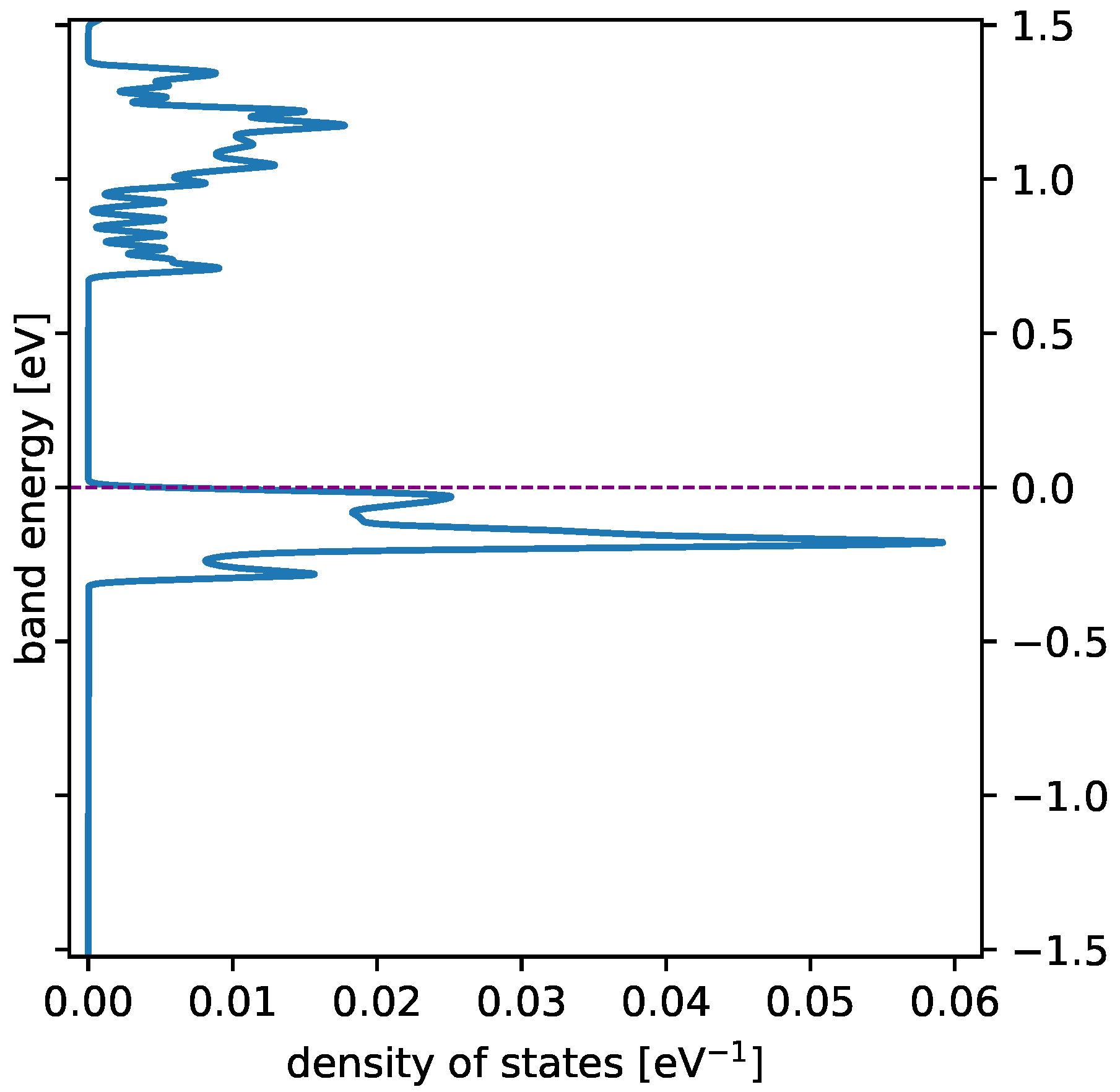
3.1. Spin State and Electronic Band Structure
3.2. Intercalation Mechanism
3.3. Electrochemical Potential
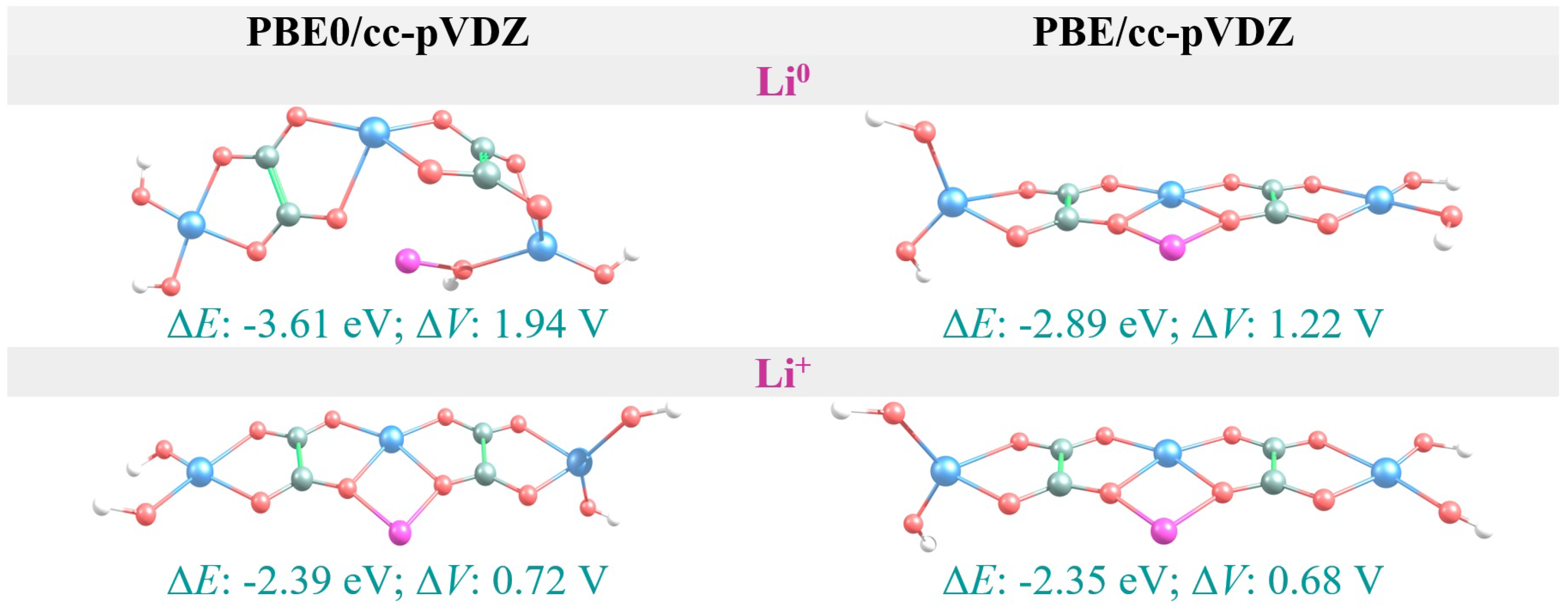
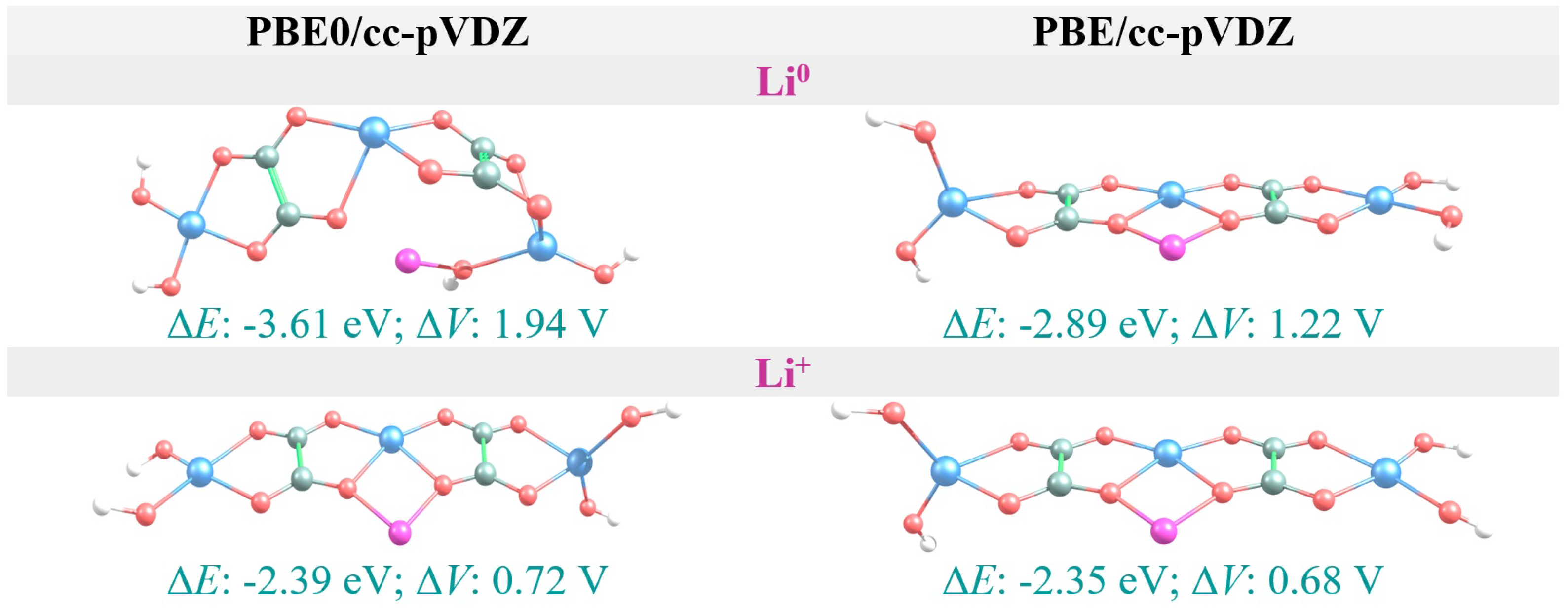
3.4. Li vs. Li Intercalation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Echigo, T.; Kimata, M. Single-crystal X-ray diffraction and spectroscopic studies on humboldtine and lindbergite: Weak Jahn–Teller effect of Fe2+ ion. Phys. Chem. Miner. 2008, 35, 467–475. [Google Scholar] [CrossRef]
- Vehmaanperä, P.; Gong, B.; Sit, P.H.L.; Salmimies, R.; Barbiellini, B.; Häkkinen, A. Formation of humboldtine during the dissolution of hematite in oxalic acid–density functional theory (DFT) calculations and experimental verification. Clays Clay Miner. 2021. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, L.; Li, M.; Wang, M.; Zhou, X.; Cheng, R.; Zhou, Y.; Shi, J. α-Ferrous oxalate dihydrate: A simple coordination polymer featuring photocatalytic and photo-initiated Fenton oxidations. Sci. China Mater. 2016, 59, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Liang, Y.; Yang, J.; Yang, G.; Xu, R.; Xie, X. α-Ferrous oxalate dihydrate: An Fe-based one-dimensional metal organic framework with extraordinary photocatalytic and Fenton activities. Catal. Sci. Technol. 2018, 8, 6057–6061. [Google Scholar] [CrossRef]
- Deyrieux, R.; Peneloux, A. Studies on some divalent metal oxalates. I. Crystal structure of 2 allotropic forms of dihydrated ferrous oxalate. Bull. Soc. Chim. Fr. 1969, 8, 2675. [Google Scholar]
- Śledzińska, I.; Murasik, A.; Piotrowski, M. Neutron diffraction study of crystal and magnetic structures of α-FeC2O4·2D2O. Phys. B+ C 1986, 138, 315–322. [Google Scholar] [CrossRef]
- Lagier, J.; Pezerat, H. Etude des phases obtenues lors de la preparation doxalates de metaux de transition. Comptes Rendus Hebd. Des Seances Acad. Des Sci. Ser. 1967, 264, 496. [Google Scholar]
- Yamada, T.; Sadakiyo, M.; Kitagawa, H. High proton conductivity of one-dimensional ferrous oxalate dihydrate. J. Am. Chem. Soc. 2009, 131, 3144–3145. [Google Scholar] [CrossRef]
- Liu, Z.J.; Liu, W.; Wang, Y.; Guo, M.L. Preparation of β-ferrous oxalate dihydrate layered nanosheets by mechanochemical method and its visible-light-driven photocatalytic performance. Mater. Lett. 2016, 178, 83–86. [Google Scholar] [CrossRef]
- Aragón, M.J.; León, B.; Perez Vicente, C.; Tirado, J.L. Synthesis and electrochemical reaction with lithium of mesoporous iron oxalate nanoribbons. Inorg. Chem. 2008, 47, 10366–10371. [Google Scholar] [CrossRef]
- Zhang, K.; Li, Y.; Wang, Y.; Zhao, J.; Chen, X.; Dai, Y.; Yao, Y. Enhanced electrochemical properties of iron oxalate with more stable Li+ ions diffusion channels by controlling polymorphic structure. Chem. Eng. J. 2020, 384, 123281. [Google Scholar] [CrossRef]
- Ojczyk, W.; Marzec, J.; Świerczek, K.; Zając, W.; Molenda, M.; Dziembaj, R.; Molenda, J. Studies of selected synthesis procedures of the conducting LiFePO4-based composite cathode materials for Li-ion batteries. J. Power Sources 2007, 173, 700–706. [Google Scholar] [CrossRef]
- Li, M.; Wang, W.; Yang, M.; Lv, F.; Cao, L.; Tang, Y.; Sun, R.; Lu, Z. Large-scale fabrication of porous carbon-decorated iron oxide microcuboids from Fe–MOF as high-performance anode materials for lithium-ion batteries. RSC Adv. 2015, 5, 7356–7362. [Google Scholar] [CrossRef]
- Wang, C.; Wang, R.; Peng, Y.; Chen, J.; Chen, Z.; Yin, H.; Li, J. Nb-incorporated Fe (oxy) hydroxide derived from structural transformation for efficient oxygen evolution electrocatalysis. J. Mater. Chem. A 2020, 8, 24598–24607. [Google Scholar] [CrossRef]
- Müller, H.; Bourcet, L.; Hanfland, M. Iron (II) oxalate dihydrate—Humboldtine: Synthesis, spectroscopic and structural properties of a versatile precursor for high pressure research. Minerals 2021, 11, 113. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16 Revision a. 03. 2016; Gaussian Inc.: Wallingford, CT, USA, 2016; Volume 2. [Google Scholar]
- Beck, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Koleżyński, A.; Handke, B.; Drożdż-Cieśla, E. Crystal structure, electronic structure, and bonding properties of anhydrous nickel oxalate. J. Therm. Anal. Calorim. 2013, 113, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Colmenero, F.; Timón, V. Extreme negative mechanical phenomena in the zinc and cadmium anhydrous metal oxalates and lead oxalate dihydrate. J. Mater. Sci. 2020, 55, 218–236. [Google Scholar] [CrossRef]
- Colmenero, F. Silver oxalate: Mechanical properties and extreme negative mechanical phenomena. Adv. Theory Simul. 2019, 2, 1900040. [Google Scholar] [CrossRef]
- South, C.J.; Roy, L.E. Insights into the thermal decomposition of plutonium (IV) oxalate–a DFT study of the intermediate structures. J. Nucl. Mater. 2021, 549, 152864. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.K.; Rathke, B.; Kiefer, J.; Materny, A. Molecular structure and interactions in the ionic liquid 1-ethyl-3-methylimidazolium trifluoromethanesulfonate. J. Phys. Chem. A 2016, 120, 6274–6286. [Google Scholar] [CrossRef]
- Repisky, M.; Komorovsky, S.; Kadek, M.; Konecny, L.; Ekström, U.; Malkin, E.; Kaupp, M.; Ruud, K.; Malkina, O.L.; Malkin, V.G. ReSpect: Relativistic spectroscopy DFT program package. J. Chem. Phys. 2020, 152, 184101. [Google Scholar] [CrossRef] [PubMed]
- Komorovskỳ, S.; Repiskỳ, M.; Malkina, O.L.; Malkin, V.G.; Malkin Ondík, I.; Kaupp, M. A fully relativistic method for calculation of nuclear magnetic shielding tensors with a restricted magnetically balanced basis in the framework of the matrix Dirac–Kohn–Sham equation. J. Chem. Phys. 2008, 128, 104101. [Google Scholar] [CrossRef]
- Repiskỳ, M.; Komorovskỳ, S.; Malkina, O.L.; Malkin, V.G. Restricted magnetically balanced basis applied for relativistic calculations of indirect nuclear spin–spin coupling tensors in the matrix Dirac–Kohn–Sham framework. Chem. Phys. 2009, 356, 236–242. [Google Scholar] [CrossRef]
- Repiskỳ, M.; Komorovskỳ, S.; Malkin, E.; Malkina, O.L.; Malkin, V.G. Relativistic four-component calculations of electronic g-tensors in the matrix Dirac–Kohn–Sham framework. Chem. Phys. Lett. 2010, 488, 94–97. [Google Scholar] [CrossRef]
- Kadek, M.; Konecny, L.; Gao, B.; Repisky, M.; Ruud, K. X-ray absorption resonances near L 2, 3-edges from real-time propagation of the Dirac–Kohn–Sham density matrix. Phys. Chem. Chem. Phys. 2015, 17, 22566–22570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konecny, L.; Kadek, M.; Komorovsky, S.; Ruud, K.; Repisky, M. Resolution-of-identity accelerated relativistic two-and four-component electron dynamics approach to chiroptical spectroscopies. J. Chem. Phys. 2018, 149, 204104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konecny, L.; Repisky, M.; Ruud, K.; Komorovsky, S. Relativistic four-component linear damped response TDDFT for electronic absorption and circular dichroism calculations. J. Chem. Phys. 2019, 151, 194112. [Google Scholar] [CrossRef] [Green Version]
- Kadek, M.; Repisky, M.; Ruud, K. All-electron fully relativistic Kohn-Sham theory for solids based on the Dirac-Coulomb Hamiltonian and Gaussian-type functions. Phys. Rev. B 2019, 99, 205103. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Zhang, L.; Cheng, R.; Wang, M.; Li, M.; Zhou, Y.; Shi, J. Construction of graphitic C3N4-based intramolecular donor–acceptor conjugated copolymers for photocatalytic hydrogen evolution. ACS Catal. 2015, 5, 5008–5015. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Combelles, C.; Yahia, M.B.; Pedesseau, L.; Doublet, M.L. Design of electrode materials for lithium-ion batteries: The example of metal- organic frameworks. J. Phys. Chem. C 2010, 114, 9518–9527. [Google Scholar] [CrossRef]
- Keshavarz, F.; Kadek, M.; Barbiellini, B.; Bansil, A. Electrochemical Potential of the Metal Organic Framework MIL-101 (Fe) as Cathode Material in Li-Ion Batteries. Condens. Matter 2021, 6, 22. [Google Scholar] [CrossRef]
- Tang, Q.; Zhou, Z.; Shen, P. Are MXenes promising anode materials for Li ion batteries? Computational studies on electronic properties and Li storage capability of Ti3C2 and Ti3C2X2 (X= F, OH) monolayer. J. Am. Chem. Soc. 2012, 134, 16909–16916. [Google Scholar] [CrossRef]
- Lv, X.; Li, F.; Gong, J.; Gu, J.; Lin, S.; Chen, Z. Metallic FeSe monolayer as an anode material for Li and non-Li ion batteries: A DFT study. Phys. Chem. Chem. Phys. 2020, 22, 8902–8912. [Google Scholar] [CrossRef]
- Zhou, F.; Cococcioni, M.; Marianetti, C.A.; Morgan, D.; Ceder, G. First-principles prediction of redox potentials in transition-metal compounds with LDA+ U. Phys. Rev. B 2004, 70, 235121. [Google Scholar] [CrossRef] [Green Version]
- Callaway, J.; Zou, X.; Bagayoko, D. Total energy of metallic lithium. Phys. Rev. B 1983, 27, 631. [Google Scholar] [CrossRef]
- Qian, L.; Yu, T.; Wei, Z.; Chang, B.; Huang, G.; Wang, Z.; Liu, Y.; Sun, H.; Bai, L.; Huang, W. Lower-voltage plateau Zn-substituted Co3O4 submicron spheres anode for Li-ion half and full batteries. J. Alloys Compd. 2022, 890, 161888. [Google Scholar] [CrossRef]
- Zhang, K.; Li, Y.; Hu, X.; Liang, F.; Wang, L.; Xu, R.; Dai, Y.; Yao, Y. Inhibitive role of crystal water on lithium storage for multilayer FeC2O4· xH2O anode materials. Chem. Eng. J. 2021, 404, 126464. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Structure | E | E | E | E | C | C | C |
|---|---|---|---|---|---|---|---|
| FOD | −6.86 | −1.96 | 4.90 | −4.41 | 1.03784 | 3.23523 | - |
| PHFO | −7.18 | −2.44 | 4.74 | −4.81 | 1.25910 | 3.45593 | - |
| AFO | −8.37 | −3.29 | 5.08 | −5.83 | 1.29025 | 3.86257 | - |
| Li/FOD | −9.66 | −4.50 | 5.17 | −7.08 | 1.16869 | 3.50852 | 0.90070 |
| Li/FOD * | −9.68 | −5.03 | 4.65 | −7.35 | 1.06932 | 3.30792 | 0.92476 |
| Li/PHFO | −9.92 | −5.21 | 4.71 | −7.57 | 1.24936 | 3.44727 | 0.94548 |
| Li/AFO | −11.07 | −6.29 | 4.77 | −8.68 | 1.28599 | 3.89290 | 0.95229 |
| Li/FOD | −7.15 | −1.75 | 5.40 | −4.45 | 1.16988 | 3.48206 | 0.89520 |
| Li/PHFO | −6.43 | −1.90 | 4.53 | −4.17 | 1.19598 | 3.46516 | 0.89973 |
| Li/AFO | −6.77 | −3.73 | 3.04 | −5.25 | 1.24761 | 3.53108 | 0.90271 |
| PBE0/cc-pVDZ | PBE/cc-pVDZ | |||
|---|---|---|---|---|
| Li intercalation | ||||
| FDO | −4.40 | 2.73 | −3.96 | 2.29 |
| PHFO | −3.14 | 1.47 | −3.10 | 1.43 |
| AFO | −2.39 | 0.72 | −2.35 | 0.68 |
| Li intercalation | ||||
| FDO | −5.25 | 3.58 | −4.52 | 2.85 |
| PHFO | −4.80 | 3.13 | −3.01 | 1.34 |
| AFO | −3.61 | 1.94 | −2.89 | 1.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keshavarz, F.; Kadek, M.; Barbiellini, B.; Bansil, A. Anodic Activity of Hydrated and Anhydrous Iron (II) Oxalate in Li-Ion Batteries. Condens. Matter 2022, 7, 8. https://doi.org/10.3390/condmat7010008
Keshavarz F, Kadek M, Barbiellini B, Bansil A. Anodic Activity of Hydrated and Anhydrous Iron (II) Oxalate in Li-Ion Batteries. Condensed Matter. 2022; 7(1):8. https://doi.org/10.3390/condmat7010008
Chicago/Turabian StyleKeshavarz, Fatemeh, Marius Kadek, Bernardo Barbiellini, and Arun Bansil. 2022. "Anodic Activity of Hydrated and Anhydrous Iron (II) Oxalate in Li-Ion Batteries" Condensed Matter 7, no. 1: 8. https://doi.org/10.3390/condmat7010008
APA StyleKeshavarz, F., Kadek, M., Barbiellini, B., & Bansil, A. (2022). Anodic Activity of Hydrated and Anhydrous Iron (II) Oxalate in Li-Ion Batteries. Condensed Matter, 7(1), 8. https://doi.org/10.3390/condmat7010008