
The Mechanism of Elizabethkingia miricola Infection of the Black Spotted Frog as Revealed by Multi-Omics Analysis

Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Elizabethkingia miricola Challenge Experiment and Frog Sampling
2.3. RNA Extraction, Library Construction, and Sequencing
2.4. Analysis of Intestinal Microbiota
2.5. Quantitative Real-Time PCR
3. Results
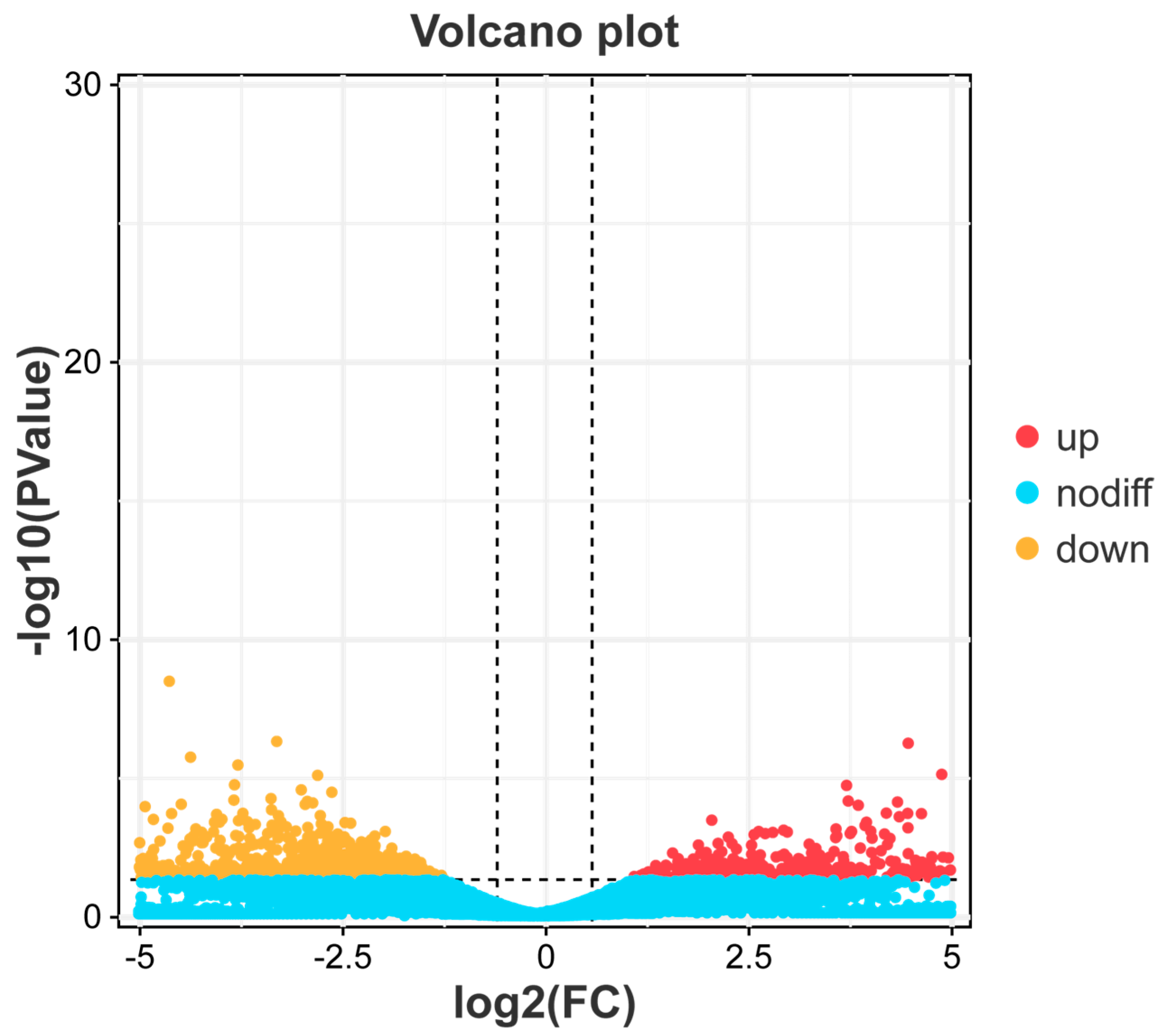
3.1. Analysis of Differentially Expressed Genes
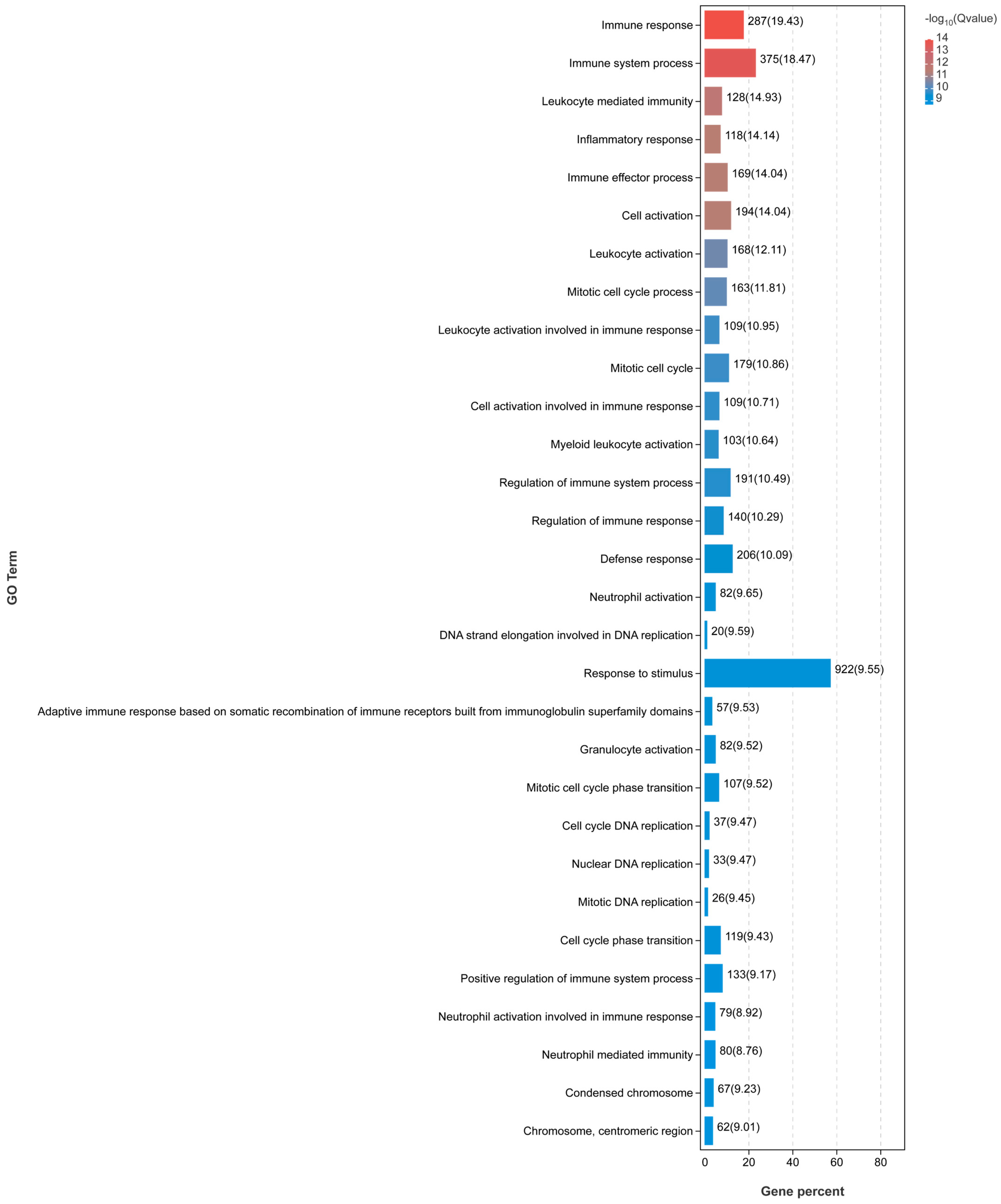
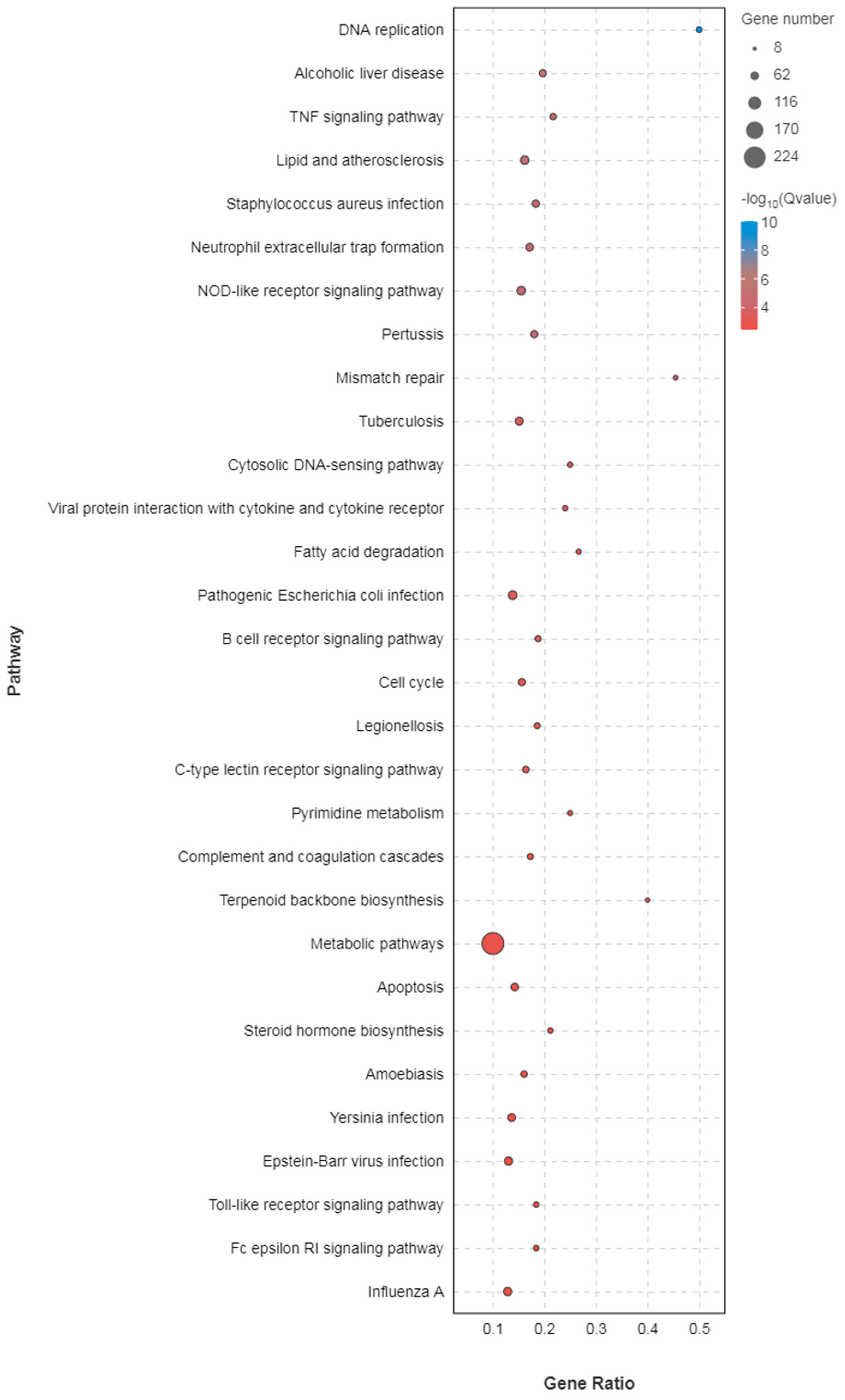
3.2. Liver Transcriptome Function Analysis
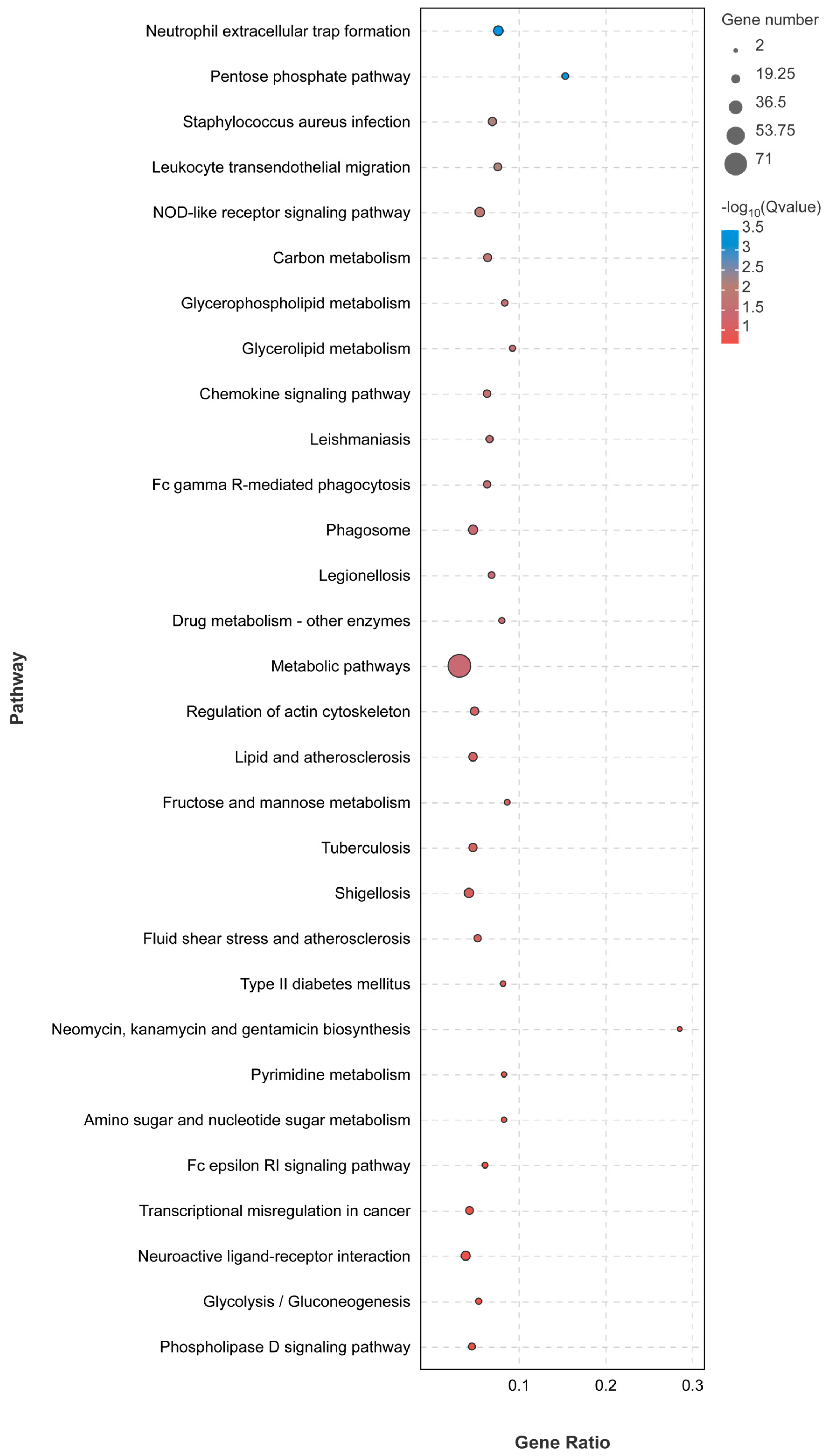
3.3. Kidney Transcriptome Function Analysis
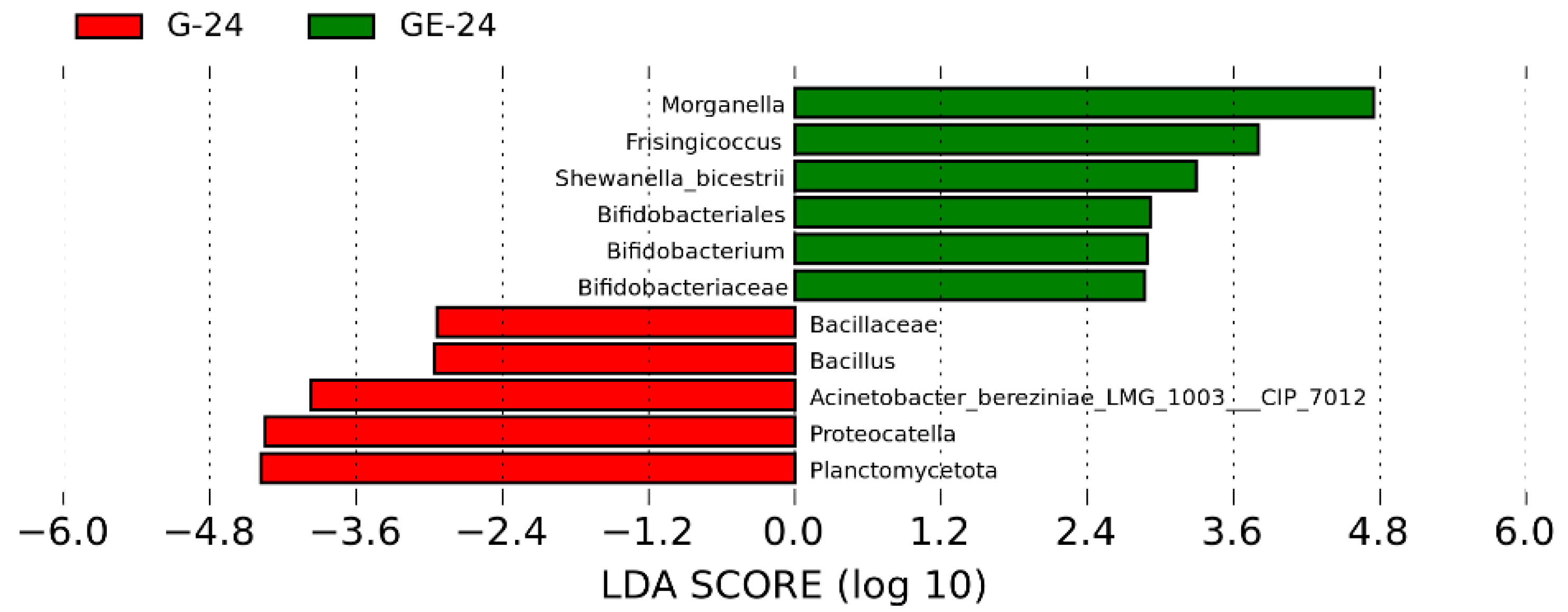
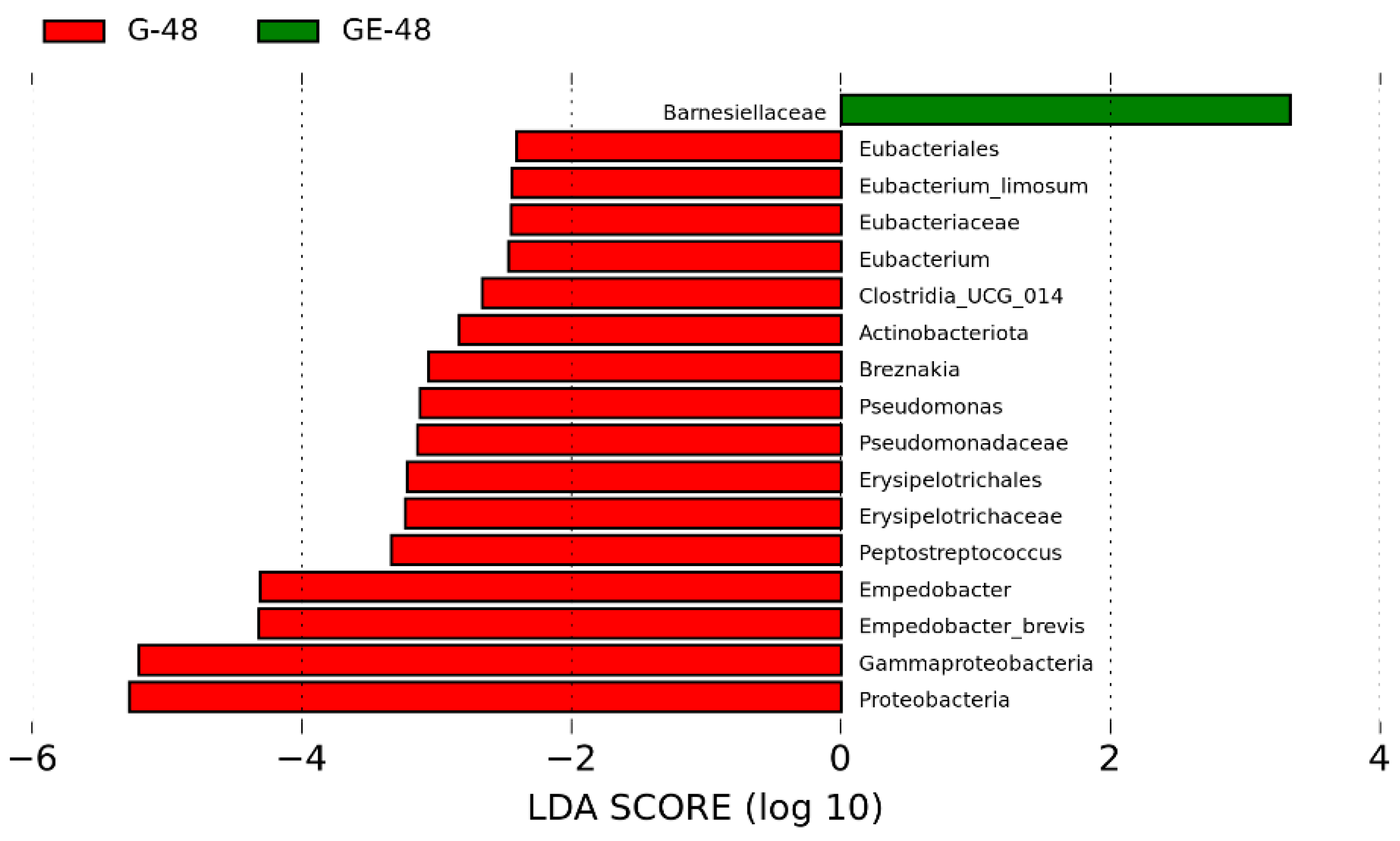
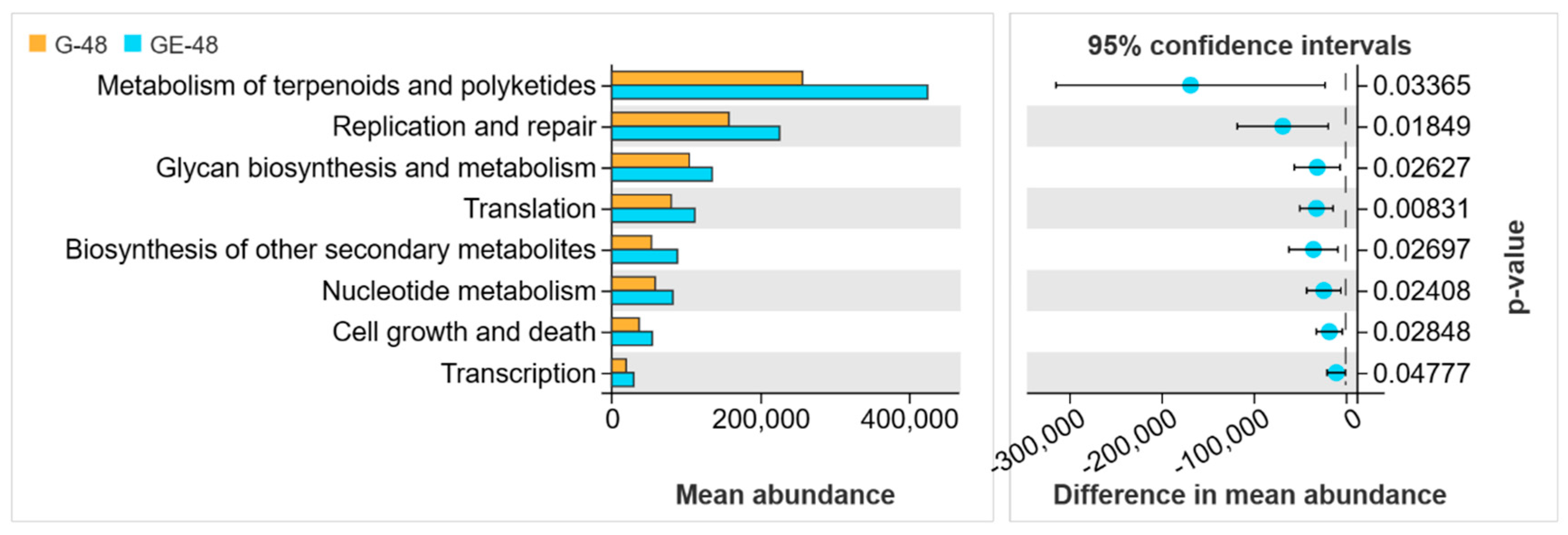
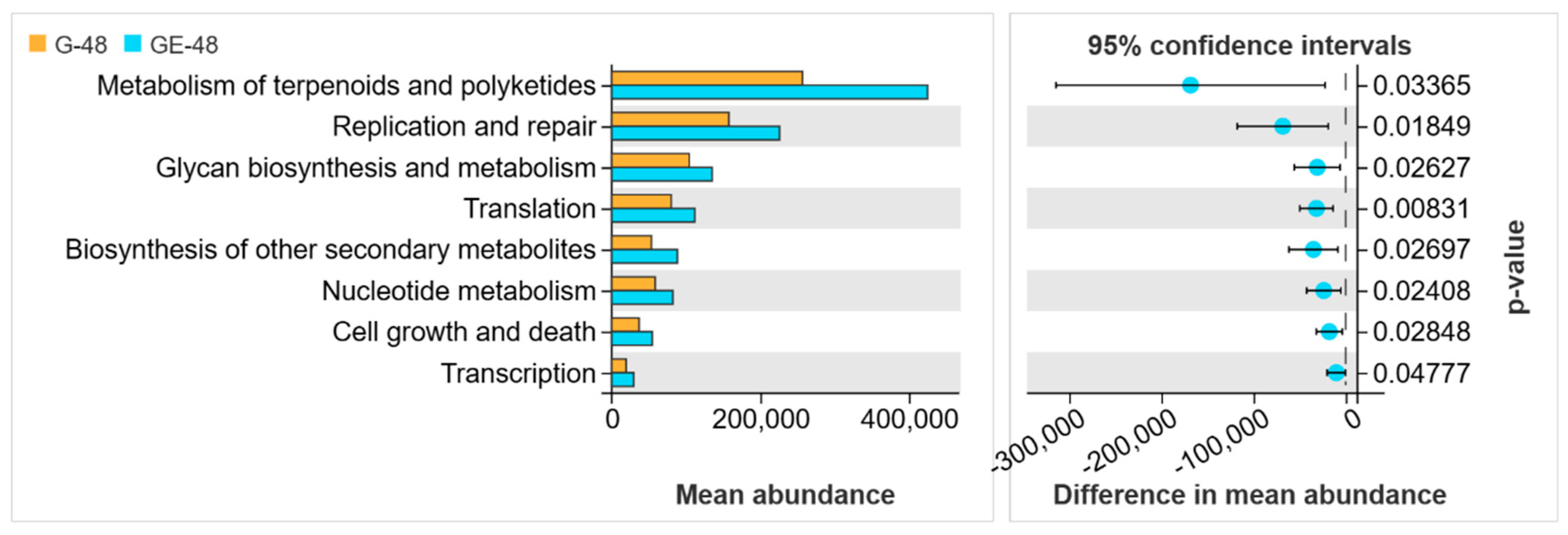
3.4. Analysis of Intestinal Microbiota
3.5. Intestinal Microbiota and DEG Association Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, S.; Wang, X.; Lu, Y.; Wang, J.; Yu, D.; Zhou, Z.; Wei, J.; Liu, L.; Liu, J.; Liu, F.; et al. Co-infections of Klebsiella pneumoniae and Elizabethkingia miricola in black-spotted frogs (Pelophylax nigromaculatus). Microb. Pathog. 2023, 180, 106150. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kim, M.K.; Lim, J.H.; Park, H.Y.; Lee, S.-T. Transfer of Chryseobacterium meningosepticum and Chryseobacterium miricola to Elizabethkingia gen. nov. as Elizabethkingia meningoseptica comb. nov. and Elizabethkingia miricola comb. nov. Int. J. Syst. Evol. Microbiol. 2005, 55, 1287–1293. [Google Scholar] [CrossRef]
- Zajmi, A.; Teo, J.; Yeo, C.C. Epidemiology and Characteristics of Elizabethkingia spp. Infections in Southeast Asia. Microorganisms 2022, 10, 882. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.X.; Yuan, J.F.; Meng, Y.; Wang, Z.; Gu, Z.M. Pathogenic Elizabethkingia miricola Infection in Cultured Black-Spotted Frogs, China, 2016. Emerg. Infect. Dis. 2017, 23, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Si, C.; Mani, R.; Keller, J.; Hoenerhoff, M.J. Septicemia caused by an emerging pathogen, Elizabethkingia miricola, in a laboratory colony of African dwarf frogs (Hymenochirus curtipes). Vet. Pathol. 2023, 60, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.D.; Cheng, Y.; Xiao, S.Y.; Liao, W.Y.; Yu, Q.; Han, S.Y.; Huang, S.S.; Shi, J.G.; Xie, Z.S.; Li, P.F. Natural occurrences and characterization of Elizabethkingia miricola infection in cultured bullfrogs (Rana catesbeiana). Front. Cell. Infect. Microbiol. 2023, 13, 1094050. [Google Scholar] [CrossRef] [PubMed]
- Trimpert, J.; Eichhorn, I.; Vladimirova, D.; Haake, A.; Schink, A.K.; Klopfleisch, R.; Lubke-Becker, A. Elizabethkingia miricolainfection in multiple anuran species. Transbound. Emerg. Dis. 2021, 68, 931–940. [Google Scholar] [CrossRef]
- Lei, X.P.; Yi, G.; Wang, K.Y.; OuYang, P.; Chen, D.F.; Huang, X.L.; Huang, C.; Lai, W.M.; Zhong, Z.J.; Huo, C.L.; et al. Elizabethkingia miricola infection in Chinese spiny frog (Quasipaa spinosa). Transbound. Emerg. Dis. 2019, 66, 1049–1053. [Google Scholar] [CrossRef]
- Hem, S.; Jarocki, V.M.; Baker, D.J.; Charles, I.G.; Drigo, B.; Aucote, S.; Donner, E.; Burnard, D.; Bauer, M.J.; Harris, P.N.A.; et al. Genomic analysis of Elizabethkingia species from aquatic environments: Evidence for potential clinical transmission. Curr. Res. Microb. Sci. 2022, 3, 100083. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef]
- Glendinning, L.; Nausch, N.; Free, A.; Taylor, D.W.; Mutapi, F. The microbiota and helminths: Sharing the same niche in the human host. Parasitology 2014, 141, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Manes, N.P.; Shulzhenko, N.; Nuccio, A.G.; Azeem, S.; Morgun, A.; Nita-Lazara, A. Multi-omics Comparative Analysis Reveals Multiple Layers of Host Signaling Pathway Regulation by the Gut Microbiota. mSystems 2017, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Bisht, V.; Nash, K.; Xu, Y.W.; Agarwal, P.; Bosch, S.; Gkoutos, G.V.; Acharjee, A. Integration of the Microbiome, Metabolome and Transcriptomics Data Identified Novel Metabolic Pathway Regulation in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 5763. [Google Scholar] [CrossRef] [PubMed]
- Salinas, I.; Magadán, S. Omics in fish mucosal immunity. Dev. Comp. Immunol. 2017, 75, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, X.; Chang, L.; Wang, Z.; Wang, T.; Zhang, Z.; Bai, J.; Liang, R.; Niu, Q.; Zhang, H. Endoplasmic reticulum rather than mitochondria plays a major role in the neuronal apoptosis induced by polybrominated diphenyl ether-153. Toxicol. Lett. 2019, 311, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Zhang, L.; Xu, C.; Huo, X.; Chang, J.; Su, J. Chitosan and anisodamine enhance the immersion immune efficacy of inactivated Elizabethkingia miricola vaccine in black spotted frogs. Fish Shellfish Immunol. 2022, 130, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Forzán, M.J.; Jones, K.M.; Ariel, E.; Whittington, R.J.; Wood, J.; Markham, R.J.F.; Daoust, P.Y. Pathogenesis of Frog Virus 3 (Ranavirus, Iridoviridae) Infection in Wood Frogs (Rana sylvatica). Vet. Pathol. 2017, 54, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wu, F.; Hao, G.; Qi, Q.; Li, R.; Li, N.; Wei, L.; Chai, T. Bacillus subtilis Improves Immunity and Disease Resistance in Rabbits. Front. Immunol. 2017, 8, 354. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Li, H. Innate immune regulatory networks in hepatic lipid metabolism. J. Mol. Med. 2019, 97, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, G.-L.; Norambuena, J.; Boyd, J.M.; Parker, D. Impact of the pentose phosphate pathway on metabolism and pathogenesis of Staphylococcus aureus. PLoS Pathog. 2023, 19, e1011531. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Li, S.; Chen, J.; Jian, C.; Hu, J.; Du, H.; Hai, H.; Wu, J.; Zeng, F.; Zhu, J.; et al. Glycerophospholipid metabolism is involved in rheumatoid arthritis pathogenesis by regulating the IL-6/JAK signaling pathway. Biochem. Biophys. Res. Commun. 2022, 600, 130–135. [Google Scholar] [CrossRef]
- Branco, L.M.; Amaral, M.P.; Boekhoff, H.; de Lima, A.B.F.; Farias, I.S.; Lage, S.L.; Pereira, G.J.S.; Franklin, B.S.; Bortoluci, K.R. Lysosomal cathepsins act in concert with Gasdermin-D during NAIP/NLRC4-dependent IL-1β secretion. Cell Death Dis. 2022, 13, 10. [Google Scholar] [CrossRef]
- Hu, Y.; Li, A.; Xu, Y.; Jiang, B.; Lu, G.; Luo, X. Transcriptomic variation of locally-infected skin of Epinephelus coioides reveals the mucosal immune mechanism against Cryptocaryon irritans. Fish Shellfish Immunol. 2017, 66, 398–410. [Google Scholar] [CrossRef]
- Kubica, P.; Lara-Velazquez, M.; Bam, M.; Siraj, S.; Ong, I.; Liu, P.; Priya, R.; Salamat, S.; Brutkiewicz, R.R.; Dey, M. MR1 overexpression correlates with poor clinical prognosis in glioma patients. Neuro-Oncol. Adv. 2021, 3, vdab034. [Google Scholar] [CrossRef]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wiera, G.; Mozrzymas, J.W. Extracellular Metalloproteinases in the Plasticity of Excitatory and Inhibitory Synapses. Cells 2021, 10, 2055. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.M.Z. Investigating the Roles of HLA-DO in the Immune Response in Humans and Mice. Ph.D. Thesis, School of Graduate Studies, Rutgers University, New Brunswick, NJ, USA, 2021. [Google Scholar]
- Fan, T.-J.; Han, L.-H.; Cong, R.-S.; Liang, J. Caspase Family Proteases and Apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Yi, P.; Wang, Y.-L.; Wu, X.-J.; Yang, F.; Ma, H.-N.; Tan, M.-S. Function of NOD-like receptor protein 1 inflammasome in traumatic central nervous system injury. Zhongguo Gu Shang = China J. Orthop. Traumatol. 2021, 34, 1058–1064. [Google Scholar] [CrossRef]
- Liu, Y.; Xing, L.H.; Li, F.X.; Wang, N.; Ma, Y.Z.; Li, J.W.; Wu, Y.J.; Liang, J.; Lei, Y.X.; Wang, X.Y.; et al. Mixed lineage kinase-like protein protects against Clostridium perfringens infection by enhancing NLRP3 inflammasome-extracellular traps axis. iScience 2022, 25, 105121. [Google Scholar] [CrossRef]
- Lee, E.J.; Napier, R.J.; Vance, E.E.; Lashley, S.J.; Truax, A.D.; Ting, J.P.; Rosenzweig, H.L. The innate immune receptor Nlrp12 suppresses autoimmunity to the retina. J. Neuroinflamm. 2022, 19, 12. [Google Scholar] [CrossRef]
- Ding, J.D.; Kelly, U.; Landowski, M.; Toomey, C.B.; Groelle, M.; Miller, C.; Smith, S.G.; Klingeborn, M.; Singhapricha, T.; Jiang, H.X.; et al. Expression of Human Complement Factor H Prevents Age-Related Macular Degeneration-Like Retina Damage and Kidney Abnormalities in Aged Cfh Knockout Mice. Am. J. Pathol. 2015, 185, 29–42. [Google Scholar] [CrossRef]
- Vakhrusheva, T.V.; Grigorieva, D.V.; Gorudko, I.V.; Sokolov, A.V.; Kostevich, V.A.; Lazarev, V.N.; Vasilyev, V.B.; Cherenkevich, S.N.; Panasenko, O.M. Enzymatic and bactericidal activity of myeloperoxidase in conditions of halogenative stress. Biochem. Cell Biol. 2018, 96, 580–591. [Google Scholar] [CrossRef]
- Canney, M.; Clark, E.G.; Hiremath, S. Biomarkers in acute kidney injury: On the cusp of a new era? J. Clin. Investig. 2023, 133, 13. [Google Scholar] [CrossRef]
- Antonia, A.L.; Gibbs, K.D.; Trahair, E.D.; Pittman, K.J.; Martin, A.T.; Schott, B.H.; Smith, J.S.; Rajagopal, S.; Thompson, J.W.; Reinhardt, R.L.; et al. Pathogen Evasion of Chemokine Response Through Suppression of CXCL10. Front. Cell. Infect. Microbiol. 2019, 9, 280. [Google Scholar] [CrossRef]
- Peachey, L.E.; Jenkins, T.P.; Cantacessi, C. This Gut Ain’t Big Enough for Both of Us. Or Is It? Helminth–Microbiota Interactions in Veterinary Species. Trends Parasitol. 2017, 33, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Xiao, S.; Liao, W.; Yu, Q.; Han, S.; Shi, J.; He, J.; Li, P. The occurrence of Morganella morganii caused large death in cultured American bullfrog (Rana catebeiana). Aquaculture 2023, 568, 739343. [Google Scholar] [CrossRef]
- Ramkumar, R.; Ravi, M.; Jayaseelan, C.; Rahuman, A.A.; Anandhi, M.; Rajthilak, C.; Perumal, P. Description of Providencia vermicola isolated from diseased Indian major carp, Labeo rohita (Hamilton, 1822). Aquaculture 2014, 420, 193–197. [Google Scholar] [CrossRef]
- Camus, A.C.; Hawke, J.P. Providencia rettgeri-associated Septicemia and Meningoencephalitis in Juvenile Farmed American Alligators Alligator mississippiensis. J. Aquat. Anim. Health 2002, 14, 149–153. [Google Scholar] [CrossRef]
- Yang, P.K.; Zheng, Y.Z.; Aweya, J.J.; Zou, X.H.; Lin, M.; Liu, Y.Q.; Zhang, Z.X.; Sun, Y.J.; Wang, H.J. iTRAQ-based comparative proteomic analysis of the Lithobates catesbeianus bullfrog spleen following challenge with Citrobacter freundii. Aquac. Rep. 2022, 23, 101037. [Google Scholar] [CrossRef]
- Ramakrishna, C.; Kujawski, M.; Chug, H.; Li, L.; Mazmanian, S.K.; Cantin, E.M. Bacteroides fragilis polysaccharide A induces IL-10 secreting B and T cells that prevent viral encephalitis. Nat. Commun. 2019, 10, 2153. [Google Scholar] [CrossRef] [PubMed]
- Arnolds, K.L.; Yamada, E.; Neff, C.P.; Schneider, J.M.; Palmer, B.E.; Lozupone, C.A. Disruption of Genes Encoding Putative Zwitterionic Capsular Polysaccharides of Diverse Intestinal Bacteroides Reduces the Induction of Host Anti-Inflammatory Factors. Microb. Ecol. 2023, 85, 1620–1629. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers for RT-qPCR | Primer Sequence (5′ to 3′) |
|---|---|
| Actb-F | CGTCCAGAGGCATACAGAGA |
| Actb-R | ACCCCAAAGCAAACCGAGAA |
| Taldo1-F | AAGCCACAGGATGCCACTAC |
| Taldo1-R | TAGAAACCCGACCAGGAACC |
| LAPTM4A-F | GAGGTTAGGAGGATTCGCTG |
| LAPTM4A-R | GCCTGCGTGCTCTTTGC |
| CD63-F | AACACTTCACAGCCAGCCAG |
| CD63-R | CAAGCAGCAAGCAAAGACGA |
| CATHL2-F | TGTGACTACAAGGAGGACGG |
| CATHL2-R | GATTATTTCTGGTGAAGCGGAG |
| rpl4-b-F | CATCTGGACGGAGAGTGCC |
| rpl4-b-R | TGCCTTGGGTTTCTTTGCCT |
| Bhmt-F | CCCAACAACAGAGGCACCA |
| Bhmt-R | ATGTGGAAGAGGCAGTGTGG |
| RAC2-F | GAAAGGGTCGGTGGCAAAGG |
| RAC2-F | ACCAAAAGCCCCAAGTGTGT |
| MYL9-F | GAATGGTGCTGCCGAGGTAA |
| MYL9-F | GGAGCCAAAGACAAGGATGA |
| hba2-F | GTTTCGCCGTCCAGTTTGT |
| hba2-F | TGCCCTATTTCACCTCTTGCT |
| CTRB1-F | TCCAGAATCGTGAATGGTGAGG |
| CTRB1-F | GTGGTTGAGCAGTGACAGGAG |
| Atp5me-F | AAGTGTCCCCGCTCATCAA |
| Atp5me-F | CCTCTGCCTCAATCCTTCTGT |
| Gene ID | Annotation | Log2fc | p-Value |
|---|---|---|---|
| CASP3 | caspase 3 | 12.95595 | 4.16 × 10−9 |
| Mr1 | major histocompatibility complex, class I-related | 4.073231 | 0.011 |
| MMP3 | matrix metallopeptidase 3 | 3.588169 | 0.000742 |
| CXCR1 | C-X-C motif chemokine receptor 1 | −2.96424 | 0.018407 |
| CYBB | cytochrome b-245 beta chain | −3.30264 | 0.000655 |
| C3 | complement C3 | −3.71698 | 0.000203 |
| CLEC4M | C-type lectin domain family 4 member M | −4.19152 | 0.021062 |
| NAIP | NLR family apoptosis inhibitory protein | −4.41834871 | 0.005662 |
| GBP4 | guanylate binding protein 4 | −9.52290841 | 0.03983 |
| Nlrp3 | NLR family pyrin domain containing 3 | −10.5209 | 0.014988 |
| Gene ID | Annotation | Log2fc | p-Value |
|---|---|---|---|
| NLRP12 | NLR family pyrin domain containing 12 | 10.37032 | 0.037796 |
| Mr1 | major histocompatibility complexes, class I-related | 8.930455 | 0.000588 |
| CASP3 | caspase 3 | 6.589579 | 1.05 × 10−24 |
| Prkcd | protein kinase C delta | 3.943372 | 0.038313 |
| Cxcl9 | C-X-C motif chemokine ligand 9 | 1.894896 | 7.80 × 10−8 |
| MMP3 | matrix metallopeptidase 3 | 1.879163 | 0.003148 |
| MPO | Myeloperoxidase | −3.4607 | 2.49 × 10−12 |
| IGHV2-26 | immunoglobulin heavy variable 2-26 | −6.63979 | 0.013474 |
| HLA-DRA | major histocompatibility complex, class II, DR alpha | −10.2669 | 1.83 × 10−74 |
| Cfh | complement factor H | −14.7934 | 0.009374 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Q.; Wang, D.; Wei, K.; Xu, B.; Xu, J. The Mechanism of Elizabethkingia miricola Infection of the Black Spotted Frog as Revealed by Multi-Omics Analysis. Fishes 2024, 9, 91. https://doi.org/10.3390/fishes9030091
Wei Q, Wang D, Wei K, Xu B, Xu J. The Mechanism of Elizabethkingia miricola Infection of the Black Spotted Frog as Revealed by Multi-Omics Analysis. Fishes. 2024; 9(3):91. https://doi.org/10.3390/fishes9030091
Chicago/Turabian StyleWei, Qingcong, Dan Wang, Kaijin Wei, Bin Xu, and Jin Xu. 2024. "The Mechanism of Elizabethkingia miricola Infection of the Black Spotted Frog as Revealed by Multi-Omics Analysis" Fishes 9, no. 3: 91. https://doi.org/10.3390/fishes9030091
APA StyleWei, Q., Wang, D., Wei, K., Xu, B., & Xu, J. (2024). The Mechanism of Elizabethkingia miricola Infection of the Black Spotted Frog as Revealed by Multi-Omics Analysis. Fishes, 9(3), 91. https://doi.org/10.3390/fishes9030091