
Identification of Growth-Related SNPs and Genes in the Genome of the Pearl Oyster (Pinctada fucata) Using GWAS


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. SNP Discovery and Genotyping
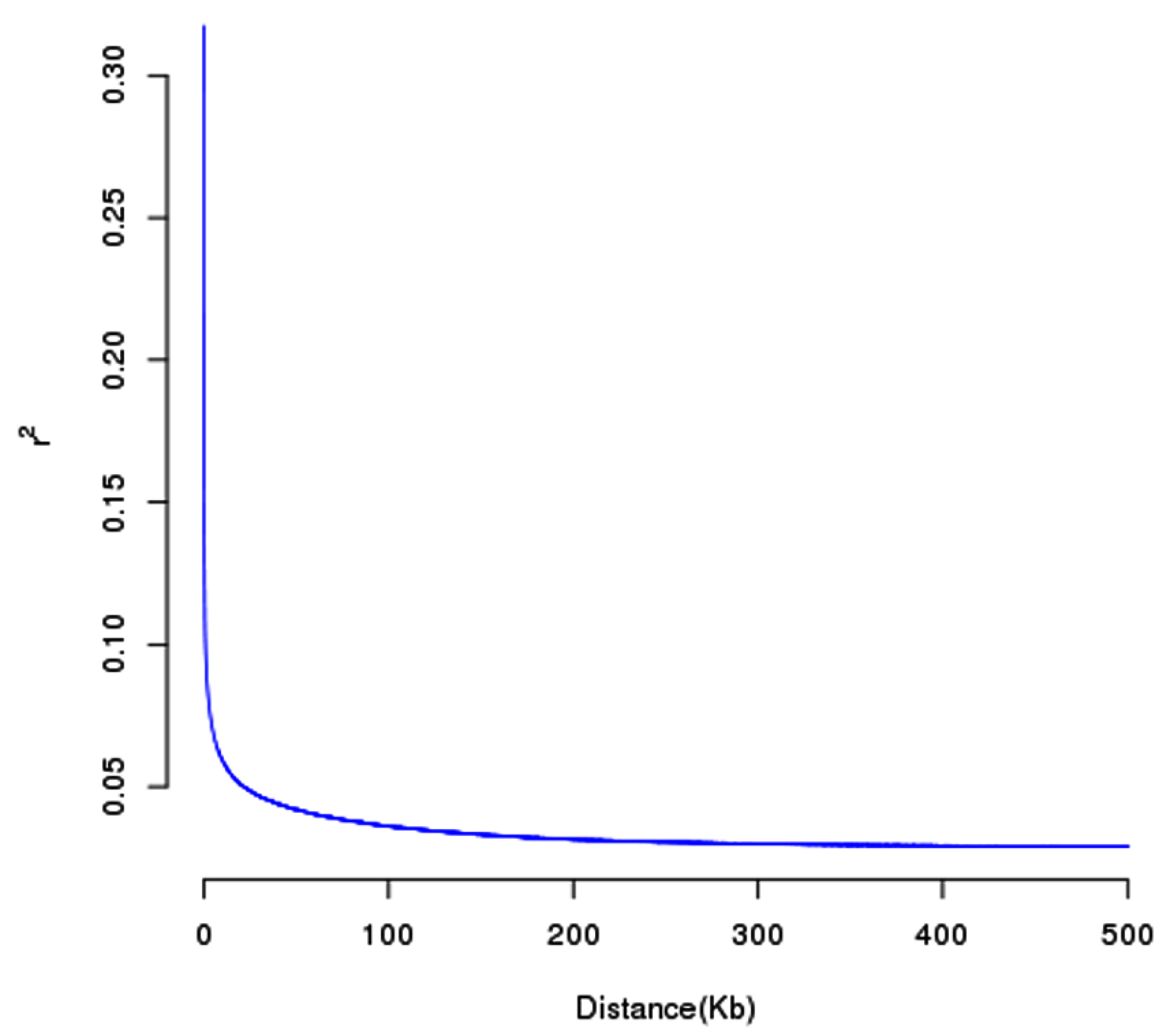
2.4. Analysis of Population Structure and Linkage Disequilibrium (LD) Decay
2.5. Statistical Analyses
2.6. Selection of Candidate Gene
2.7. KEGG Enrichment Analysis
3. Results
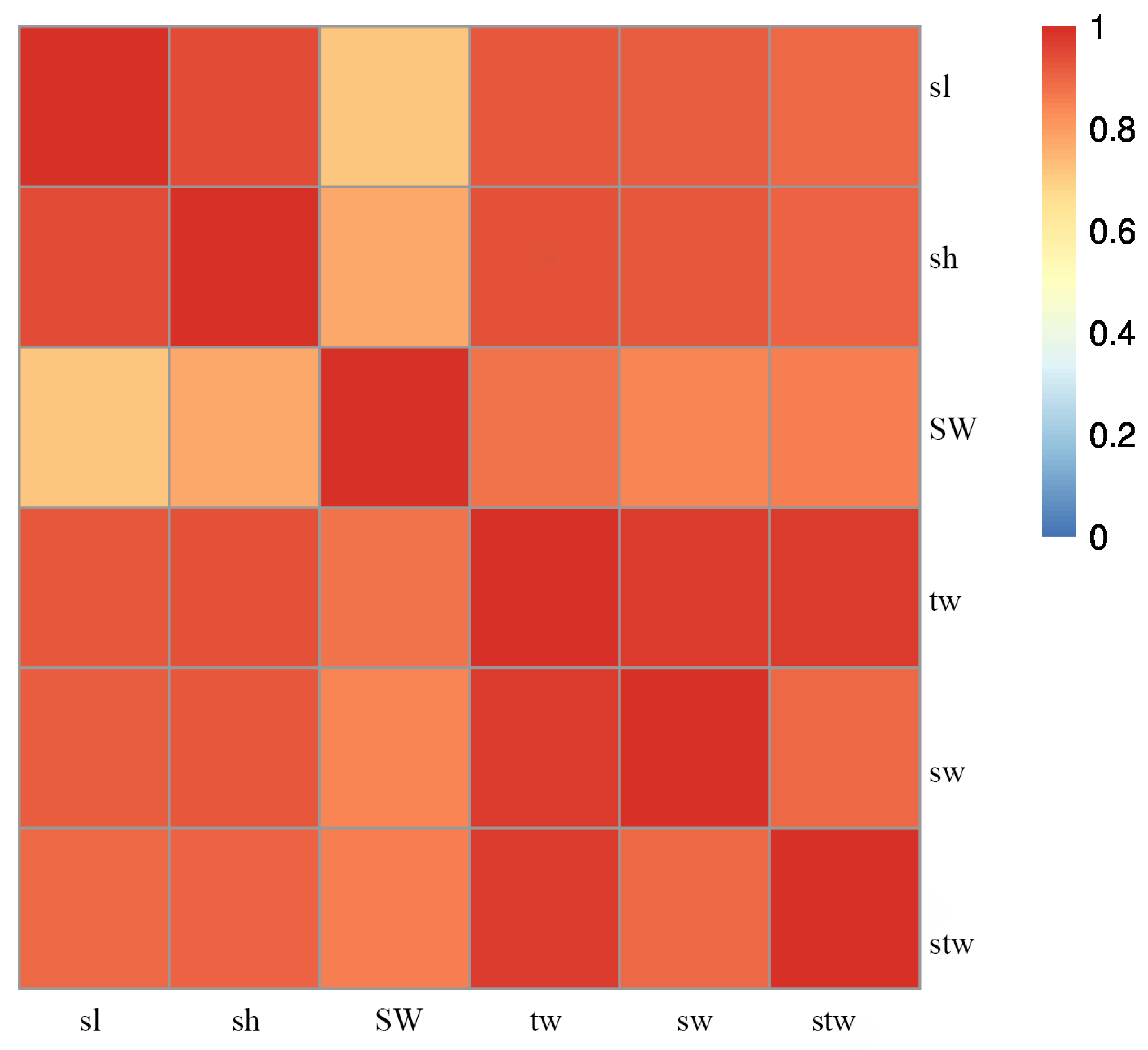
3.1. Descriptive Statistics of Growth-Related Traits
3.2. Quality Control of the Sequencing Data
3.3. Analysis of Population Structure and Genetic Relationship
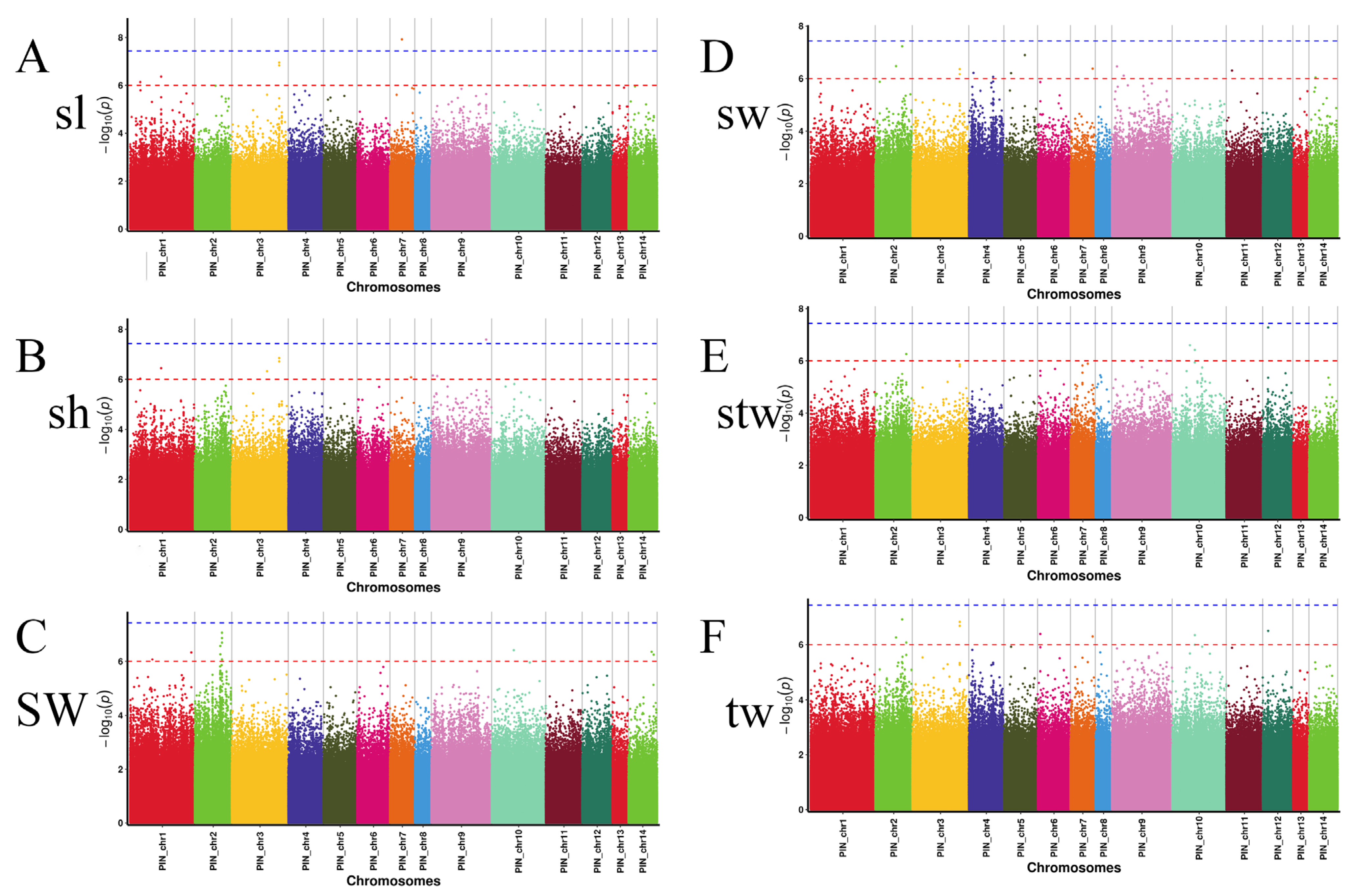
3.4. GWAS of Growth Traits
3.5. Identification of Candidate Genes and KEGG Pathway Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hao, R.J.; Du, X.D.; Yang, C.Y.; Deng, Y.W.; Zheng, Z.; Wang, Q.H. Integrated application of transcriptomics and metabolomics provides insights into unsynchronized growth in pearl oyster Pinctada fucata martensii. Sci. Total Environ. 2019, 666, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Hao, R.J.; Du, X.D.; Wang, Q.H.; Deng, Y.W.; Sun, R.J.; Zheng, Z. GC-TOF/MS-based metabolomics studies on the effect of protein sources in formulated diet for pearl oyster Pinctada fucata martensii. Aquaculture 2018, 486, 139–147. [Google Scholar] [CrossRef]
- Zhang, J.B.; Xiong, X.W.; Deng, Y.W.; Zheng, Z.; Yang, C.Y.; Du, X.D. Integrated application of transcriptomics and metabolomics provides insights into the larval metamorphosis of pearl oyster (Pinctada fucata martensii). Aquaculture 2021, 532, 736067. [Google Scholar] [CrossRef]
- Yu, D.H.; Chu, K.H. Genetic variation in wild and cultured populations of the pearl oyster Pinctada fucata from southern China. Aquaculture 2006, 258, 220–227. [Google Scholar] [CrossRef]
- Guan, Y.; He, M.; Wu, H. Differential mantle transcriptomics and characterization of growth-related genes in the diploid and triploid pearl oyster Pinctada fucata. Mar. Genom. 2017, 33, 31–38. [Google Scholar] [CrossRef]
- Krina, T.Z.; Lon, R.C. The complex interplay among factors that influence allelic association. Nat. Rev. Genet. 2004, 5, 89–100. [Google Scholar] [CrossRef]
- Jiao, W.; Fu, X.; Dou, J.; Li, H.; Su, H.; Mao, J.; Yu, Q.; Zhang, L.; Hu, X.; Huang, X.; et al. High-resolution linkage and quantitative trait locus mapping aided by genome survey sequencing: Building up an integrative genomic framework for a bivalve mollusc. DNA Res. 2014, 21, 85–101. [Google Scholar] [CrossRef]
- Jones, D.B.; Jerry, D.R.; Khatkar, M.S.; Moser, G.; Raadsma, H.W.; Taylor, J.J.; Zenger, K.R. Determining genetic contributions to host oyster shell growth: Quantitative trait loci and genetic association analysis for the silver-lipped pearl oyster, Pinctada maxima. Aquaculture 2014, 434, 367–375. [Google Scholar] [CrossRef]
- Shi, Y.H.; Wang, S.; Gu, Z.F.; Lv, J.; Zhan, X.; Yu, C.C.; Bao, Z.M.; Wang, A.M. High-density single nucleotide polymorphisms linkage and quantitative trait locus mapping of the pearl oyster, Pinctada fucata martensii Dunker. Aquaculture 2014, 434, 376–384. [Google Scholar] [CrossRef]
- Tian, M.; Li, Y.; Jing, J.; Mu, C.; Du, H.; Dou, J.; Mao, J.; Li, X.; Jiao, W.; Wang, Y.; et al. Construction of a High-Density Genetic Map and Quantitative Trait Locus Mapping in the Sea Cucumber Apostichopus japonicus. Sci. Rep. 2015, 5, 14852. [Google Scholar] [CrossRef]
- Coltell, O.; Sorli, J.V.; Asensio, E.M.; Barragan, R.; Gonzalez, J.I.; Gimenez-Alba, I.M.; Zanon-Moreno, V.; Estruch, R.; Ramirez-Sabio, J.B.; Pascual, E.C.; et al. Genome-Wide Association Study for Serum Omega-3 and Omega-6 Polyunsaturated Fatty Acids: Exploratory Analysis of the Sex-Specific Effects and Dietary Modulation in Mediterranean Subjects with Metabolic Syndrome. Nutrients 2020, 12, 310. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.X.; Zhou, T.; Wang, W.W.; Jin, Y.L.; Wang, X.Z.; Geng, X.; Luo, J.; Yuan, Z.H.; Yang, Y.J.; Shi, H.T.; et al. GWAS analysis using interspecific backcross progenies reveals superior blue catfish alleles responsible for strong resistance against enteric septicemia of catfish. Mol. Genet. Genom. 2018, 293, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Peng, W.Z.; Tang, B.; Zhang, Y.F.; Wang, Y.; Gan, Y.; Luo, X.; You, W.W.; Gwo, J.C.; Chen, N.; et al. A genome-wide association study of heat tolerance in Pacific abalone based on genome resequencing. Aquaculture 2021, 536, 736436. [Google Scholar] [CrossRef]
- Zhou, Z.X.; Han, K.H.; Wu, Y.D.; Bai, H.Q.; Ke, Q.Z.; Pu, F.; Wang, Y.L.; Xu, P. Genome-Wide Association Study of Growth and Body-Shape-Related Traits in Large Yellow Croaker (Larimichthys crocea) Using ddRAD Sequencing. Mar. Biotechnol. 2019, 21, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.P.; Matika, O.; Bean, T.P.; Houston, R.D. Genomic Selection for Growth Traits in Pacific Oyster (Crassostrea gigas): Potential of Low-Density Marker Panels for Breeding Value Prediction. Front. Genet. 2018, 9, 391. [Google Scholar] [CrossRef]
- Zhao, J.; Bai, H.; Ke, Q.; Li, B.; Zhou, Z.; Wang, H.; Chen, B.; Pu, F.; Zhou, T.; Xu, P. Genomic selection for parasitic ciliate Cryptocaryon irritans resistance in large yellow croaker. Aquaculture 2021, 531, 735786. [Google Scholar] [CrossRef]
- Brondum, R.F.; Su, G.; Janss, L.; Sahana, G.; Guldbrandtsen, B.; Boichard, D.; Lund, M.S. Quantitative trait loci markers derived from whole genome sequence data increases the reliability of genomic prediction. J. Dairy Sci. 2015, 98, 4107–4116. [Google Scholar] [CrossRef]
- Dong, L.; Xiao, S.; Wang, Q.; Wang, Z. Comparative analysis of the GBLUP, emBayesB, and GWAS algorithms to predict genetic values in large yellow croaker (Larimichthys crocea). BMC Genom. 2016, 17, 460. [Google Scholar] [CrossRef]
- Chakraborty, S.; Saha, A.; Neelavar Ananthram, A. Comparison of DNA extraction methods for non-marine molluscs: Is modified CTAB DNA extraction method more efficient than DNA extraction kits? 3 Biotech 2020, 10, 69. [Google Scholar] [CrossRef]
- Du, X.; Fan, G.; Jiao, Y.; Zhang, H.; Guo, X.; Huang, R.; Zheng, Z.; Bian, C.; Deng, Y.; Wang, Q.; et al. The pearl oyster Pinctada fucata martensii genome and multi-omic analyses provide insights into biomineralization. GigaScience 2017, 6, gix059. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Yang, J.A.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Team, R. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Computing 2009, 14, 12–21. [Google Scholar]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Hardy, O.J.; Vekemans, X. spagedi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Loiselle, B.; Sork, V.; Nason, J.; Graham, C.; Loiselle, B.A.; Sork, V.L.; Nason, J.; Graham, C. Spatial genetic-structure of a tropical understory shrub, Psychotria Officinalis (Rubiaceae). Am. J. Bot. 1995, 82, 1420–1425. [Google Scholar] [CrossRef]
- Fan, S.; Wang, J.; Huang, G.; Liu, B.; Yu, D. Identification of twenty novel polymorphic microsatellite DNA markers from transcripts of the pearl oyster Pinctada fucata using next-generation sequencing approach. J. Genet. 2014, 93, e82–e85. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.; Li, X.; Fu, Q.; Jiao, W.; Li, Y.; Li, T.; Wang, Y.; Hu, X.; Wang, S.; Bao, Z. Evaluation of the 2b-RAD method for genomic selection in scallop breeding. Sci. Rep. 2016, 6, srep19244. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Song, K.; Meng, J.; Xu, F.; Li, L.; Zhang, G. Construction of a high-density genetic map and fine QTL mapping for growth and nutritional traits of Crassostrea gigas. BMC Genom. 2018, 19, 626. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, X.; Guo, X.; Gao, Q.; Zhao, H.; Zhang, G. A preliminary genetic linkage map of the Pacific abalone Haliotis discus hannai Ino. Mar. Biotechnol. 2006, 8, 386–397. [Google Scholar] [CrossRef]
- Shi, Y.H.; Kui, H.; Guo, X.M.; Gu, Z.F.; Wang, Y.; Wang, A.M. Genetic linkage map of the pearl oyster, Pinctada martensii (Dunker). Aquac. Res. 2009, 41, 35–44. [Google Scholar] [CrossRef]
- Li, Y.G.; He, M.X. Genetic Mapping and QTL Analysis of Growth-Related Traits in Pinctada fucata Using Restriction-Site Associated DNA Sequencing. PLoS ONE 2014, 9, 0111707. [Google Scholar] [CrossRef]
- Bentley, D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006, 16, 545–552. [Google Scholar] [CrossRef]
- Balding, D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006, 7, 781–791. [Google Scholar] [CrossRef]
- Yang, J.; Luo, S.; Li, J.; Zheng, Z.; Du, X.; Deng, Y. Transcriptome analysis of growth heterosis in pearl oyster Pinctada fucata martensii. FEBS Open Bio 2018, 8, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Guo, X. Identification and mapping of disease-resistance QTLs in the eastern oyster, Crassostrea virginica Gmelin. Aquaculture 2006, 254, 160–170. [Google Scholar] [CrossRef]
- Myllyharju, J.; Kivirikko, K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004, 20, 33–43. [Google Scholar] [CrossRef]
- Liang, J.; Zheng, Z.; Huang, R.; Wang, Q.; Deng, Y.; Jiao, Y.; Du, X. Cloning and expression analysis of PmCOLVIA6 gene of Pinctada martensii. Genom. Appl. Biol. 2016, 35, 811–819. [Google Scholar]
- Bonaldo, P.; Russo, V.; Bucciotti, F.; Doliana, R.; Colombatti, A. Structural and functional features of the alpha 3 chain indicate a bridging role for chicken collagen VI in connective tissues. Biochemistry 1990, 29, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.A.; Tillet, E.; Timpl, R.; Stallcup, W.B. Binding of the NG2 proteoglycan to type VI collagen and other extracellular matrix molecules. J. Biol. Chem. 1996, 271, 26110–26116. [Google Scholar] [CrossRef] [PubMed]
- Doane, K.J.; Yang, G.; Birk, D.E. Corneal cell-matrix interactions: Type VI collagen promotes adhesion and spreading of corneal fibroblasts. Exp. Cell Res. 1992, 200, 490–499. [Google Scholar] [CrossRef]
- Heino, J. The collagen family members as cell adhesion proteins. Bioessays 2007, 29, 1001–1010. [Google Scholar] [CrossRef]
- Keene, D.R.; Engvall, E.; Glanville, R.W. Ultrastructure of type VI collagen in human skin and cartilage suggests an anchoring function for this filamentous network. J. Cell Biol. 1988, 107, 1995–2006. [Google Scholar] [CrossRef]
- Kuo, H.J.; Maslen, C.L.; Keene, D.R.; Glanville, R.W. Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. J. Biol. Chem. 1997, 272, 26522–26529. [Google Scholar] [CrossRef]
- McDevitt, C.A.; Marcelino, J.; Tucker, L. Interaction of intact type VI collagen with hyaluronan. FEBS Lett. 1991, 294, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, M.; Aumailley, M.; Specks, U.; Knolle, J.; Zerwes, H.G.; Timpl, R. Integrin and Arg-Gly-Asp dependence of cell adhesion to the native and unfolded triple helix of collagen type VI. Exp. Cell Res. 1993, 206, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, C.; Klatt, A.R.; Wagener, R.; Paulsson, M.; Bateman, J.F.; Heinegard, D.; Morgelin, M. Complexes of matrilin-1 and biglycan or decorin connect collagen VI microfibrils to both collagen II and aggrecan. J. Biol. Chem. 2003, 278, 37698–37704. [Google Scholar] [CrossRef]
- Zheng, X.C.; Zhang, L.; Jiang, W.B.; Abasubong, K.P.; Zhang, C.Y.; Zhang, D.D.; Li, X.F.; Jiang, G.Z.; Chi, C.; Liu, W.B. Effects of dietary icariin supplementation on the ovary development-related transcriptome of Chinese mitten crab (Eriocheir sinensis). Comp. Biochem. Phys. D 2021, 37, 100756. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | N | Mean | SD | Max | Min | CV |
|---|---|---|---|---|---|---|
| Shell length (sl, mm) | 60 | 57.88 | 8.48 | 70.68 | 46.64 | 14.65 |
| Shell width (SW, mm) | 60 | 20.39 | 2.00 | 25.02 | 16.69 | 9.80 |
| Shell height (sh, mm) | 60 | 60.10 | 8.12 | 75.06 | 48.22 | 13.52 |
| Total weight (tw, g) | 60 | 26.44 | 8.24 | 39.75 | 15.12 | 31.20 |
| Shell weight (sw, g) | 60 | 13.38 | 4.04 | 20.53 | 7.47 | 30.20 |
| Soft tissue weight (stw, g) | 60 | 13.05 | 4.41 | 22.69 | 5.92 | 33.80 |
| Groups | sl | SW | sh | tw | sw | stw |
|---|---|---|---|---|---|---|
| L | 65.85 ± 3.44 a | 21.97 ± 1.35 | 67.81 ± 2.89 a | 34.15 ± 3.40 a | 17.14 ± 1.84 a | 17.01 ± 2.26 |
| S | 49.92 ± 1.79 b | 18.81 ± 1.07 | 52.39 ± 1.73 b | 18.73 ± 1.8 b | 9.63 ± 0.80 b | 9.10 ± 1.44 |
| p-value | 0.004 | 0.166 | 0.03 | 0.0001 | 0.001 | 0.075 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Thaimuangphol, W.; Hong, Y.; Yan, Z.; Chen, Z.; Jin, M.; Zheng, A.; Wang, B.; Wang, Z. Identification of Growth-Related SNPs and Genes in the Genome of the Pearl Oyster (Pinctada fucata) Using GWAS. Fishes 2023, 8, 296. https://doi.org/10.3390/fishes8060296
Zhao M, Thaimuangphol W, Hong Y, Yan Z, Chen Z, Jin M, Zheng A, Wang B, Wang Z. Identification of Growth-Related SNPs and Genes in the Genome of the Pearl Oyster (Pinctada fucata) Using GWAS. Fishes. 2023; 8(6):296. https://doi.org/10.3390/fishes8060296
Chicago/Turabian StyleZhao, Mingming, Wipavee Thaimuangphol, Yujie Hong, Ziqi Yan, Zongfa Chen, Minxuan Jin, Anna Zheng, Bei Wang, and Zhongliang Wang. 2023. "Identification of Growth-Related SNPs and Genes in the Genome of the Pearl Oyster (Pinctada fucata) Using GWAS" Fishes 8, no. 6: 296. https://doi.org/10.3390/fishes8060296
APA StyleZhao, M., Thaimuangphol, W., Hong, Y., Yan, Z., Chen, Z., Jin, M., Zheng, A., Wang, B., & Wang, Z. (2023). Identification of Growth-Related SNPs and Genes in the Genome of the Pearl Oyster (Pinctada fucata) Using GWAS. Fishes, 8(6), 296. https://doi.org/10.3390/fishes8060296