
High Abundance of Candidatus Arthromitus in Intestinal Microbiota of Seriolella violacea (Palm Ruff) under Reared Conditions

 ,
,  ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and PCR Amplicon Sequencing
2.3. Bioinformatics Analysis
2.4. Statistical Analyses
3. Results
3.1. Fish Growth
3.2. Diversity of Intestinal Microbiota
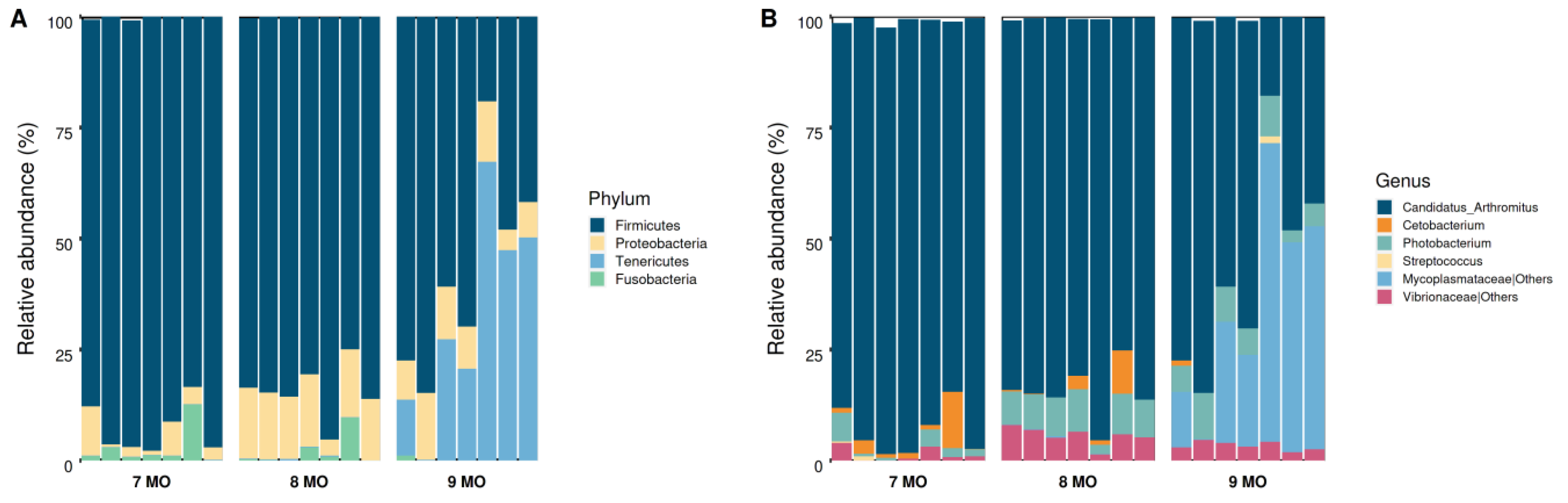
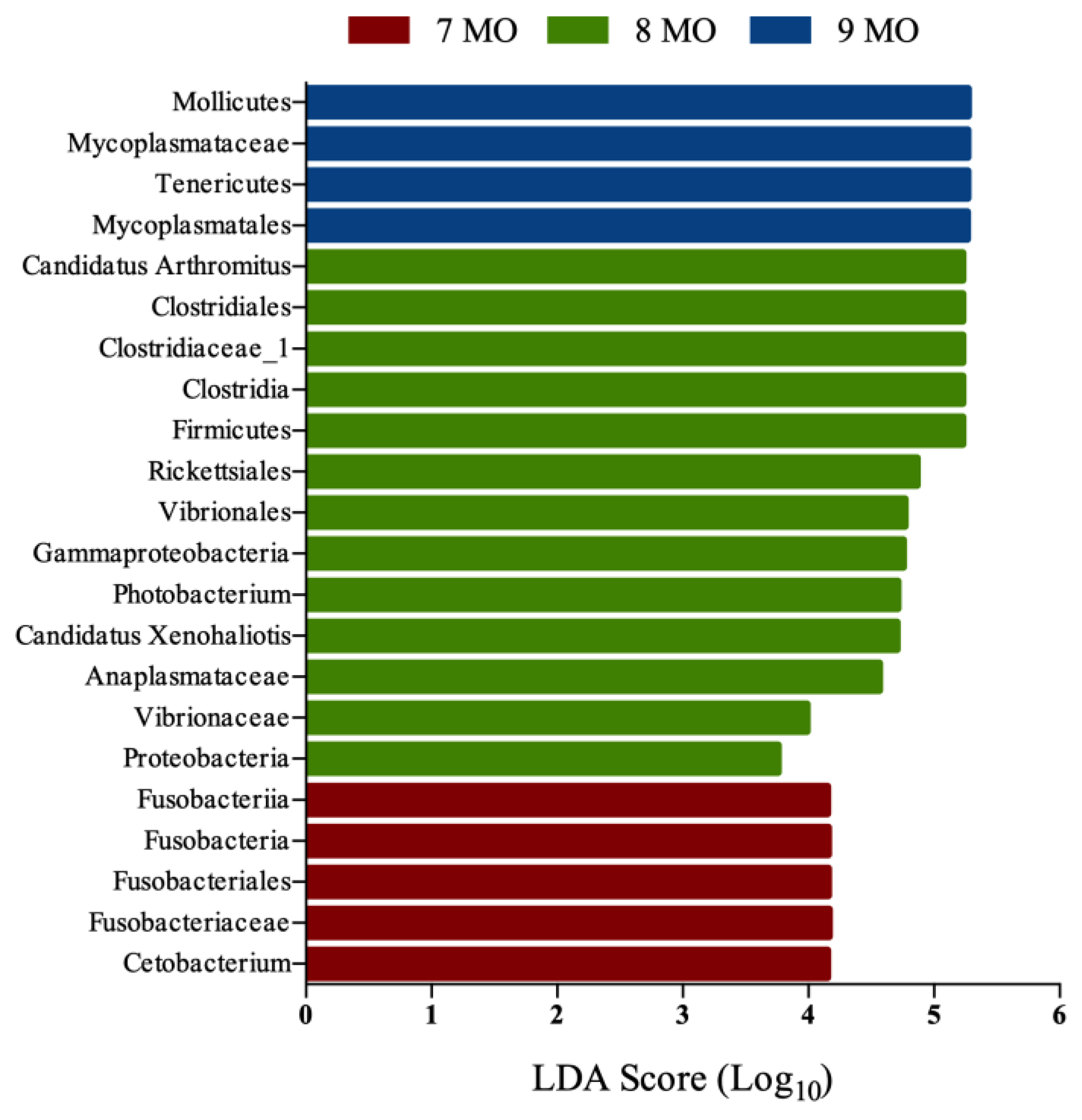
3.3. Microbial Composition of the Intestinal Microbiota
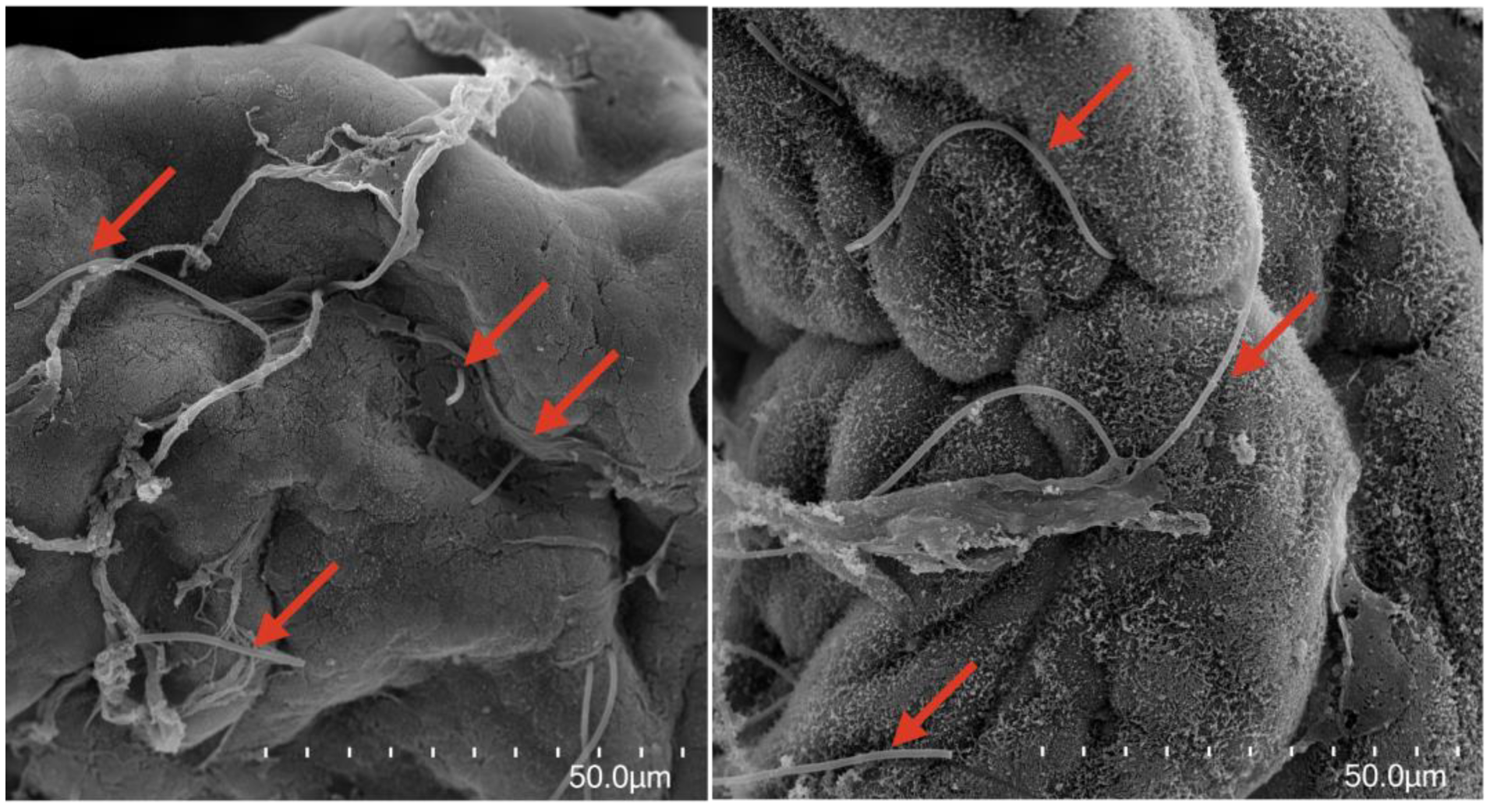
3.4. Microscopic Visualization of Filamentous Bacteria in Palm Ruff Intestine
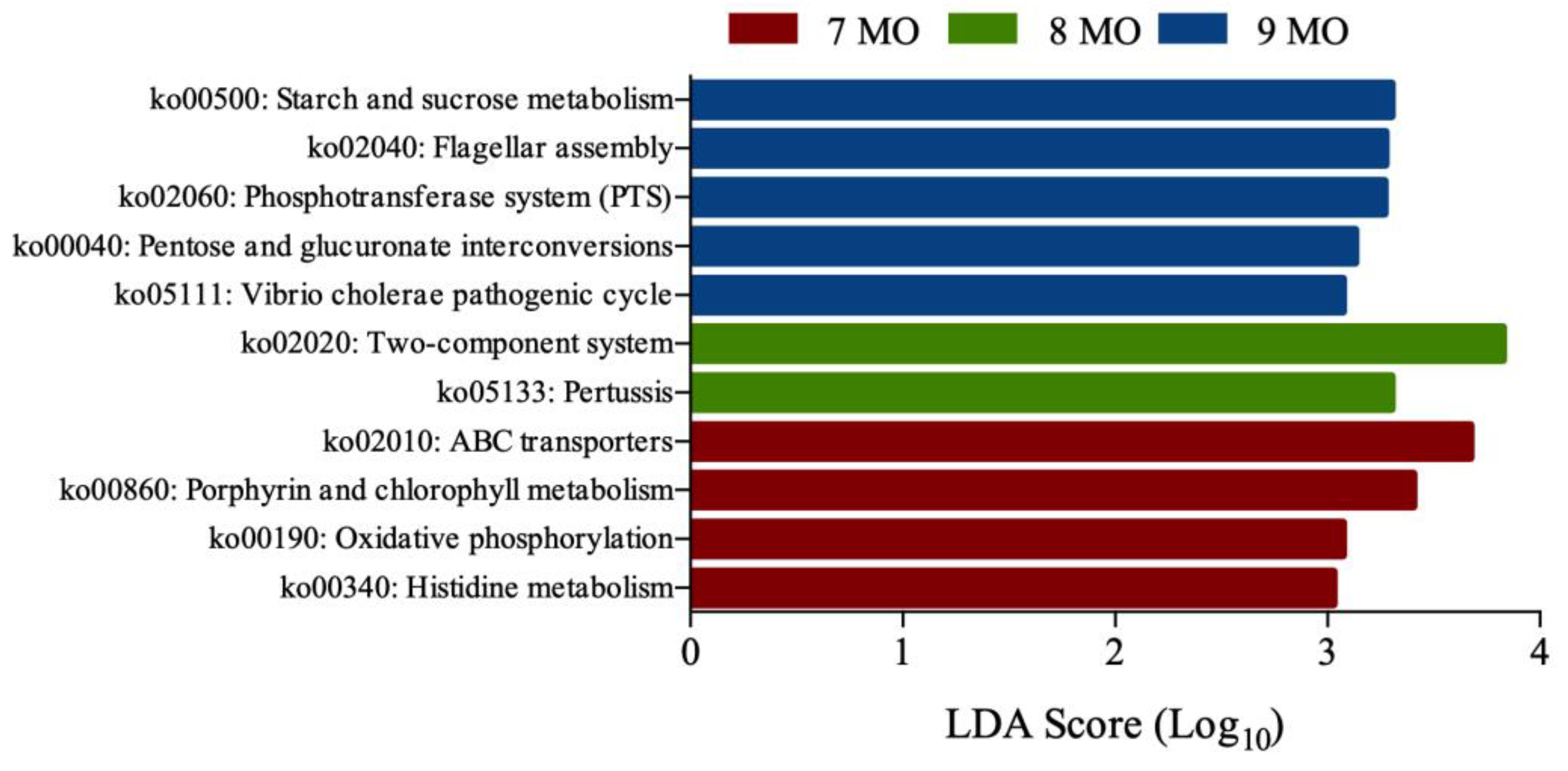
3.5. Microbial Functional Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iannacone, J. Three metazoan parasites of palm ruff Seriolella violacea Guichenot (Pisces, Centrolophidae), Callao, Peru. Rev. Bras. Zool. 2003, 20, 257–260. [Google Scholar] [CrossRef]
- Araya-Jaime, C.; Palma-Rojas, C.; Brand, E.V.; Silva, A. Cytogenetic characterization, rDNA mapping and quantification of the nuclear DNA content in Seriolella violacea Guichenot, 1848 (Perciformes, Centrolophidae). Comp. Cytogenet. 2020, 14, 319–328. [Google Scholar] [CrossRef]
- González, A.; Silva, A.; Gajardo, G.; Martinez, C. Survival and growth improvement of palm ruff, Seriolella violacea, larvae fed Artemia nauplii enriched with an experimental emulsion. J. World Aquacult. Soc. 2017, 48, 268–279. [Google Scholar] [CrossRef]
- Bustos, C.; Silva, A. Endogenous feeding and morphological changes in hatchery-reared larval palm ruff Seriolella violacea (Pisces: Centrolophidae) under starvation. Aquac. Res. 2011, 42, 892–897. [Google Scholar] [CrossRef]
- Rincón-Cervera, M.Á.; González-Barriga, V.; Romero, J.; Rojas, R.; López-Arana, S. Quantification and Distribution of Omega-3 Fatty Acids in South Pacific Fish and Shellfish Species. Foods 2020, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Nerici, C.; Silva, A.; Merino, G. Effect of two temperatures on ammonia excretion rates of Seriolella violacea (Palm fish) juveniles under rearing conditions. Aquac. Eng. 2012, 46, 47–52. [Google Scholar] [CrossRef]
- Argüello-Guevara, W.; Bohórquez-Cruz, M.; Silva, A. Effect of two temperatures on yield and increase in cranial skeletal abnormalities during early development of palm ruff, Seriolella violacea (Guichenot 1848). Aquac. Res. 2017, 48, 298–310. [Google Scholar] [CrossRef]
- Alveal, K.; Silva, A.; Lohrmann, K.B.; Viana, M.T. Morphofunctional characterization of the digestive system in the palm ruff larvae, Seriolella violacea under culture conditions. Aquaculture 2019, 501, 51–61. [Google Scholar] [CrossRef]
- Butt, R.L.; Volkoff, H. Gut microbiota and energy homeostasis in fish. Front. Endocrinol. 2019, 10, 6–8. [Google Scholar] [CrossRef]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. The Gut Microbiota of Marine Fish. Front. Microbiol. 2018, 9, 873. [Google Scholar] [CrossRef]
- Legrand, T.P.R.A.; Wynne, J.W.; Weyrich, L.S.; Oxley, A.P.A. A microbial sea of possibilities: Current knowledge and prospects for an improved understanding of the fish microbiome. Rev. Aquac. 2019, 12, 1101–1134. [Google Scholar] [CrossRef]
- Villasante, A.; Ramírez, C.; Catalán, N.; Opazo, R.; Dantagnan, P.; Romero, J. Effect of dietary carbohydrate-to-protein ratio on gut microbiota in atlantic salmon (Salmo salar). Animals 2019, 9, 89. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.; Wagner, H. Vegan: Community Ecology Package. 2015. Available online: http://CRAN.R-project.org/package=vegan (accessed on 18 December 2022).
- Iwai, S.; Weinmaier, T.; Schmidt, B.L.; Albertson, D.G.; Poloso, N.J.; Dabbagh, K.; DeSantis, T.Z. Piphillin: Improved prediction of metagenomic content by direct inference from human microbiomes. PLoS ONE 2016, 11, e0166104. [Google Scholar] [CrossRef] [PubMed]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ramírez, C.; Romero, J. Fine flounder (Paralichthys adspersus) microbiome showed important differences between wild and reared specimens. Front. Microbiol. 2017, 8, 271. [Google Scholar] [CrossRef]
- Rimoldi, S.; Terova, G.; Ascione, C.; Giannico, R.; Brambilla, F. Next generation sequencing for gut microbiome characterization in rainbow trout (Oncorhynchus mykiss) fed animal by-product meals as an alternative to fishmeal protein sources. PLoS ONE 2018, 13, e0193652. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.; Díaz, O.; Miranda, C.D.; Rojas, R. Red Cusk-Eel (Genypterus chilensis) Gut Microbiota Description of Wild and Aquaculture Specimens. Microorganisms 2022, 10, 105. [Google Scholar] [CrossRef]
- Ramírez, C.; Romero, J. The microbiome of Seriola lalandi of wild and aquaculture origin reveals differences in composition and potential function. Front. Microbiol. 2017, 8, 1844. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, D.Y. Microbial diversity in the intestine of olive flounder (Paralichthys olivaceus). Aquaculture. 2014, 41, 103–108. [Google Scholar] [CrossRef]
- Romero, J.; Ringø, E.; Merrifield, D.L. The gut microbiota of fish. In Aquaculture Nutrition: Gut Health, Probiotics and Prebiotics; Merrifield, D., Ringø, E., Eds.; Wiley-Blackwell Publishing: Oxford, UK, 2014; pp. 75–100. [Google Scholar]
- Del-Pozo, J.; Crumlish, M.; Turnbull, J.F.; Ferguson, H.W. Histopathology and Ultrastructure of segmented filamentous bacteria– associated rainbow trout gastroenteritis. Vet. Pathol. 2010, 47, 220–230. [Google Scholar] [CrossRef]
- Geraylou, Z.; Souffreau, C.; Rurangwa, E.; Maes, G.E.; Spanier, K.I.; Courtin, C.M.; Delcour, J.A.; Buyse, J.; Ollevier, F. Prebiotic effects of arabinoxylan oligosaccharides on juvenile Siberian sturgeon (Acipenser baerii) with emphasis on the modulation of the gut microbiota using 454 pyrosequencing. FEMS Microbiol. Ecol. 2013, 86, 357–371. [Google Scholar] [CrossRef]
- Rossi, L.T.; Sharpen, A.R.; Zimmermann, J.A.; Olivero, C.R.; Zbrun, M.V.; Frizzo, L.S.; Signorini, M.L.; Bacchetta, C.; Cian, R.E.; Cazenave, J.; et al. Intestinal microbiota modulation in juvenile Pacú (Piaractus mesopotamicus) by supplementation with Pyropia columbina and β-carotene. Aquac. Int. 2020, 28, 1001–1016. [Google Scholar] [CrossRef]
- Kokou, F.; Sasson, G.; Nitzan, T.; Doron-Faigenboim, A.; Harpaz, S.; Cnaani, A.; Mizrahi, I. Host genetic selection for cold tolerance shapes microbiome composition and modulates its response to temperature. Elife 2018, 7, e36398. [Google Scholar] [CrossRef]
- Hedblom, G.A.; Reiland, H.A.; Sylte, M.J.; Johnson, T.J.; Baumler, D.J. Segmented filamentous bacteria—Metabolism meets immunity. Front. Microbiol. 2018, 9, 1991. [Google Scholar] [CrossRef]
- Snel, J.; Heinen, P.P.; Blok, H.J.; Carman, R.J.; Duncan, A.J.; Allen, P.C.; Collins, M.D. Comparison of 16s rRNA Sequences of Segmented Filamentous Bacteria Isolated from Mice, Rats, and Chickens and Proposal of “Candidatus Arthromitus.”. Int. J. Syst. Bacteriol. 1995, 45, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Savage, D. Habitat, succession, attachment, and morphology of segmented, filamentous microbes indigenous to the murine gastrointestinal tract. Infect Immun. 1974, 10, 948–956. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Yang, Y.; Torchinsky, M.; Gobert, M.; Xiong, H.; Xu, M.; Linehan, J.; Alonzo, F.; Ng, C.; Chen, A.; Lin, X.; et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 2014, 510, 152–156. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, Y.; Chen, X.; Zhao, Y.; Wu, Y.; Li, Y.; Wang, X.; Chen, H.; Xiang, C. Induction of Intestinal Th17 Cells by Flagellins From Segmented Filamentous Bacteria. Front. Immunol. 2019, 10, 2750. [Google Scholar] [CrossRef]
- Goto, Y.; Panea, C.; Nakato, G.; Cebula, A.; Lee, C.; Diez, M.; Laufer, T.M.; Ignatowicz, L.; Ivanov, I.I. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 2014, 40, 594–607. [Google Scholar] [CrossRef]
- Sczesnak, A.; Segata, N.; Qin, X.; Gevers, D.; Petrosino, J.; Huttenhower, C.; Littman, D.R.; Ivanov, I.I. The genome of Th17 cell-inducing segmented filamentous bacteria reveals extensive auxotrophy and adaptations to the intestinal environment. Cell Host Microbe. 2011, 10, 260–272. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, Y.; Zhu, L.; Liu, W.; Liao, N.; Jiang, M.; Zhu, B.; Yu, H.; Xiang, C.; Wang, X. Comparative analysis of the distribution of segmented filamentous bacteria in humans, mice and chickens. ISME J. 2013, 7, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Liao, N.; Yin, Y.; Sun, G.; Xiang, C.; Liu, D.; Yu, H.; Wang, X. Colonization and distribution of segmented filamentous bacteria (SFB) in chicken gastrointestinal tract and their relationship with host immunity. FEMS Microb. Ecol. 2012, 81, 395–406. [Google Scholar] [CrossRef]
- Catalán, N.; Villasante, A.; Wacyk, J.; Ramírez, C.; Romero, J. Fermented Soybean Meal Increases Lactic Acid Bacteria in Gut Microbiota of Atlantic Salmon (Salmo salar). Probiotics Antimicro Prot. 2018, 10, 566–576. [Google Scholar] [CrossRef]
- Desai, A.R.; Links, M.G.; Collins, S.A.; Mansfield, G.S.; Drew, M.D.; Van Kessel, A.G.; Hill, J.E. Effects of plant-based diets on the distal gut microbiome of rainbow trout (Oncorhynchus mykiss). Aquaculture 2012, 350, 134–142. [Google Scholar] [CrossRef]
- Zhao, R.; Symonds, J.E.; Walker, S.P.; Steiner, K.; Carter, C.G.; Bowman, J.P.; Nowak, B.F. Salinity and fish age affect the gut microbiota of farmed Chinook salmon (Oncorhynchus tshawytscha). Aquaculture 2020, 528, 735539. [Google Scholar] [CrossRef]
- Llewellyn, M.S.; McGinnity, P.; Dionne, M.; Letourneau, J.; Thonier, F.; Carvalho, G.R.; Creer, S.; Derome, N. The biogeography of the atlantic salmon (Salmo salar) gut microbiome. ISME J. 2016, 10, 1280–1284. [Google Scholar] [CrossRef]
- Lokesh, J.; Kiron, V.; Sipkema, D.; Fernandes, J.M.O.; Moum, T. Succession of embryonic and the intestinal bacterial communities of Atlantic salmon (Salmo salar) reveals stage-specific microbial signatures. MicrobiologyOpen 2018, 8, e00672. [Google Scholar] [CrossRef]
- Dehler, C.E.; Secombes, C.J.; Martin, S.A.M. Seawater transfer alters the intestinal microbiota profiles of Atlantic salmon (Salmo salar L.). Sci. Rep. 2017, 7, 13877. [Google Scholar] [CrossRef] [PubMed]
- Panserat, S.; Kaushik, S.; Médale, F. Rainbow trout as a model for nutrition and nutrient metabolism studies. In Trout: From Physiology to Conservation; Polakof, S., Moon, T.W., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2013; pp. 131–153. [Google Scholar]
- Liu, H.; Guo, X.; Gooneratne, R.; Lai, R.; Zeng, C.; Zhan, F.; Wang, W. The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 2016, 6, 24340. [Google Scholar] [CrossRef]
- Prakash, T.; Oshima, K.; Morita, H.; Fukuda, S.; Imaoka, A.; Kumar, N.; Sharma, V.K.; Kim, S.-W.; Takahashi, M.; Saitou, N.; et al. Complete genome sequences of rat and mouse segmented filamentous bacteria, a potent inducer of th17 cell differentiation. Cell Host Microbe 2011, 10, 273–284. [Google Scholar] [CrossRef]
- Hedblom, G.A.; Dev, K.; Bowden, S.D.; Baumler, D. Comparative genome analysis of commensal segmented filamentous bacteria (SFB) from turkey and murine hosts reveals distinct metabolic features. BMC Genom. 2022, 23, 659. [Google Scholar] [CrossRef] [PubMed]
- Nearing, J.T.; Comeau, A.M.; Langille, M.G.I. Identifying biases and their potential solutions in human microbiome studies. Microbiome 2021, 9, 113. [Google Scholar] [CrossRef]
- Brooks, J.P.; Edwards, D.J.; Harwich, M.D.; Rivera, M.C.; Fettweis, J.M.; Serrano, M.G.; Reris, R.A.; Sheth, N.U.; Huang, B.; Girerd, P.; et al. The truth about metagenomics: Quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 2015, 15, 66. [Google Scholar] [CrossRef]
- Bergsten, E.; Mestivier, D.; Sobhani, I. The Limits and Avoidance of Biases in Metagenomic Analyses of Human Fecal Microbiota. Microorganisms 2020, 8, 1954. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indexes | 7MO | 8MO | 9MO |
|---|---|---|---|
| Chao1 | 52.80 ± 24.94 a | 71.10 ± 23.64 a | 59.40 ± 21.50 a |
| Shannon | 3.34 ± 0.01 a | 3.41 ± 0.10 a | 3.67 ± 0.18 b |
| Simpson | 0.96 ± 0.00 a | 0.96 ± 0.00 a | 0.97 ± 0.01 b |
| Group | FTU Scores Range (Average ± SD) |
|---|---|
| 7MO | 0.78–0.97 (0.87 ± 0.08) |
| 8MO | 0.57–0.91 (0.70 ± 0.10) |
| 9MO | 0.68–0.89 (0.77 ± 0.07) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero, J.; Catalán, N.; Ramírez, C.; Miranda, C.D.; Oliva, M.; Flores, H.; Romero, M.S.; Rojas, R. High Abundance of Candidatus Arthromitus in Intestinal Microbiota of Seriolella violacea (Palm Ruff) under Reared Conditions. Fishes 2023, 8, 109. https://doi.org/10.3390/fishes8020109
Romero J, Catalán N, Ramírez C, Miranda CD, Oliva M, Flores H, Romero MS, Rojas R. High Abundance of Candidatus Arthromitus in Intestinal Microbiota of Seriolella violacea (Palm Ruff) under Reared Conditions. Fishes. 2023; 8(2):109. https://doi.org/10.3390/fishes8020109
Chicago/Turabian StyleRomero, Jaime, Natalia Catalán, Carolina Ramírez, Claudio D. Miranda, Marcia Oliva, Héctor Flores, María Soledad Romero, and Rodrigo Rojas. 2023. "High Abundance of Candidatus Arthromitus in Intestinal Microbiota of Seriolella violacea (Palm Ruff) under Reared Conditions" Fishes 8, no. 2: 109. https://doi.org/10.3390/fishes8020109
APA StyleRomero, J., Catalán, N., Ramírez, C., Miranda, C. D., Oliva, M., Flores, H., Romero, M. S., & Rojas, R. (2023). High Abundance of Candidatus Arthromitus in Intestinal Microbiota of Seriolella violacea (Palm Ruff) under Reared Conditions. Fishes, 8(2), 109. https://doi.org/10.3390/fishes8020109