




Insights into the Relationship between Intestinal Microbiota of the Aquaculture Worm Sipunculus nudus and Surrounding Sediments

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Site and Sample Collection
2.2. DNA Extraction and High-Throughput Sequencing
2.3. Statistical Analyses
3. Results
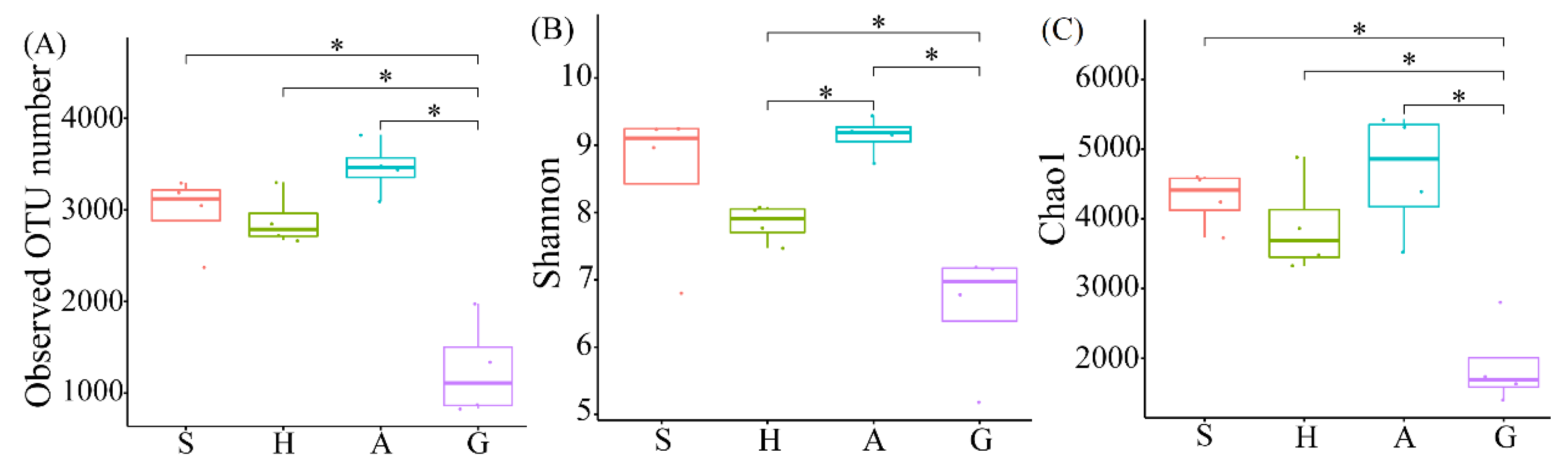
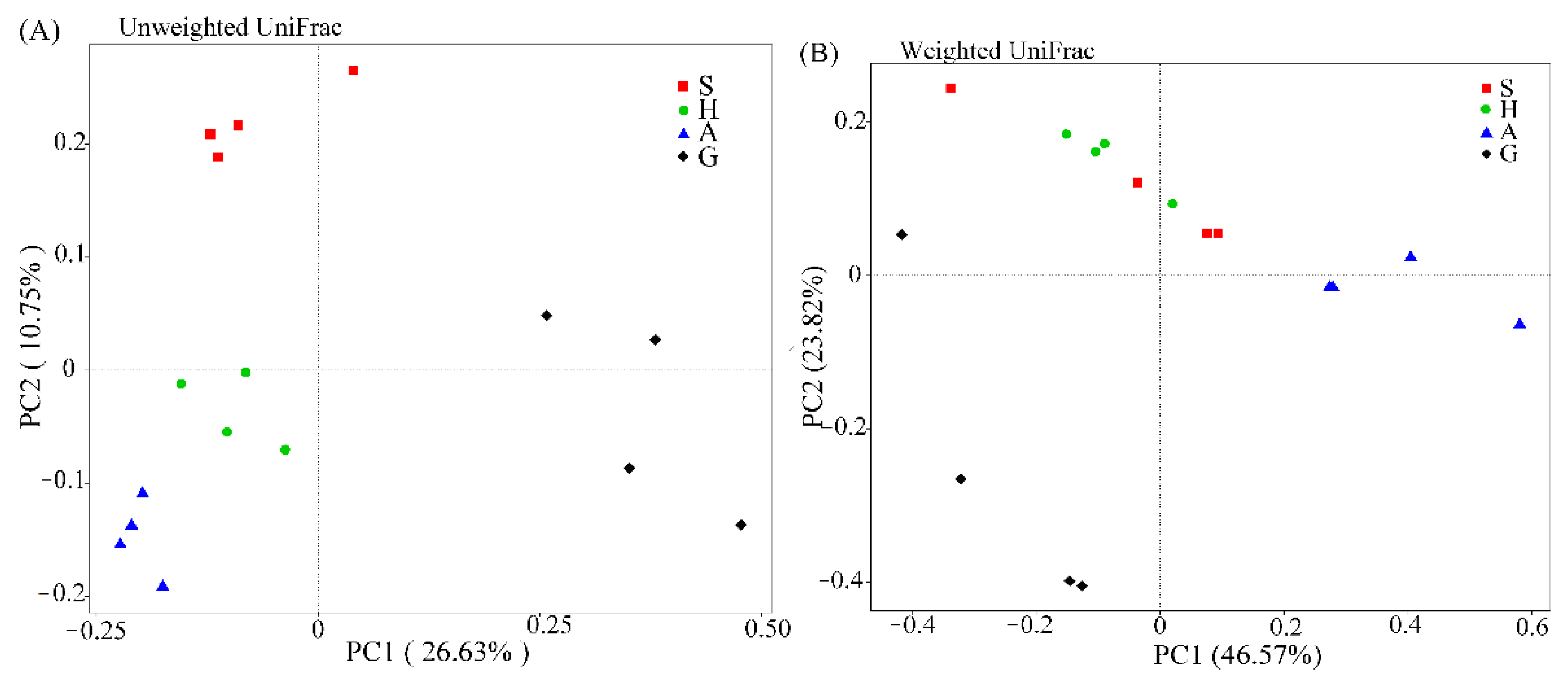
3.1. High-Throughput Sequencing Analysis and Microbial Community Diversity
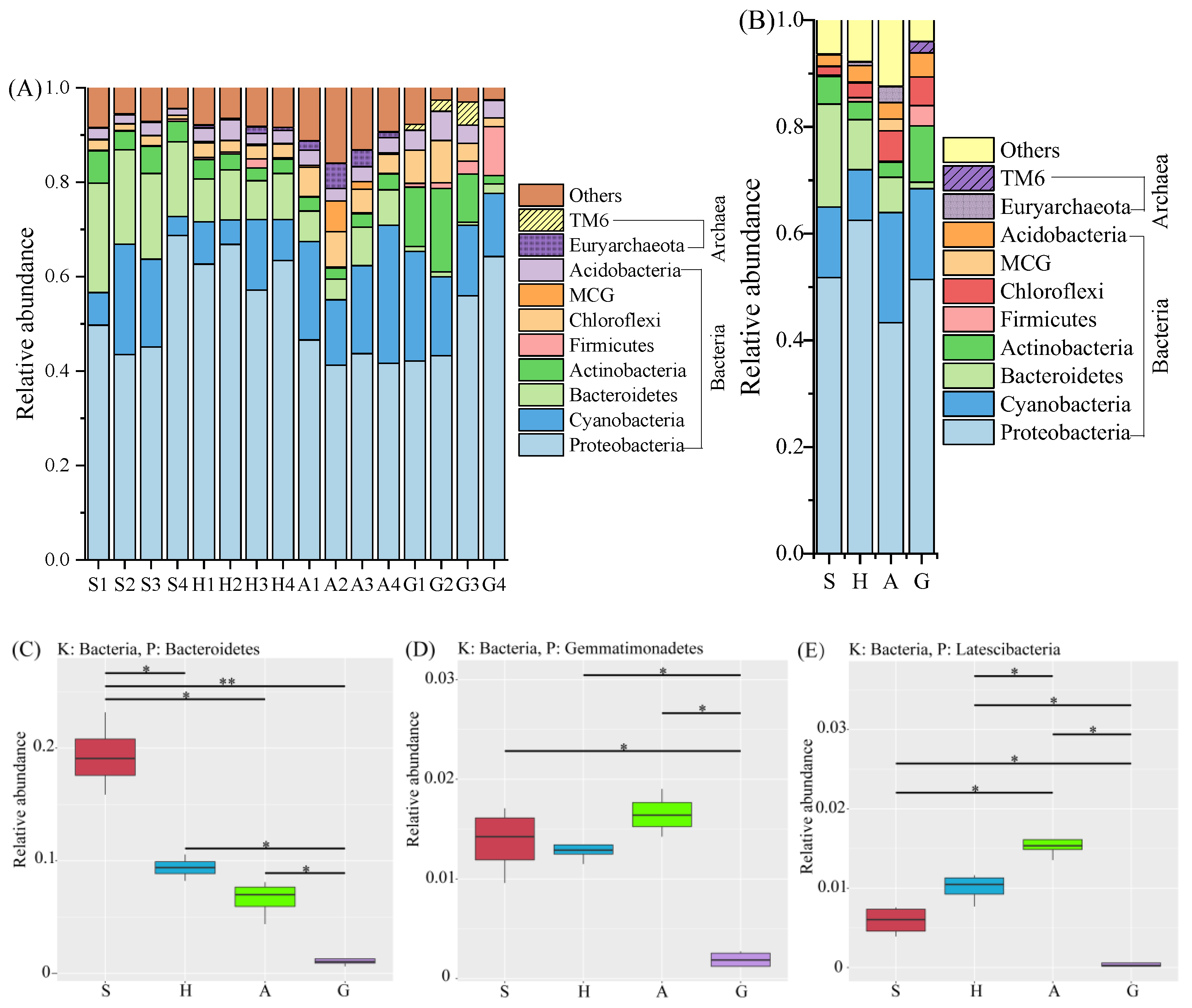
3.2. Microbial Composition and ORP Values among Intestine and Sediment Samples
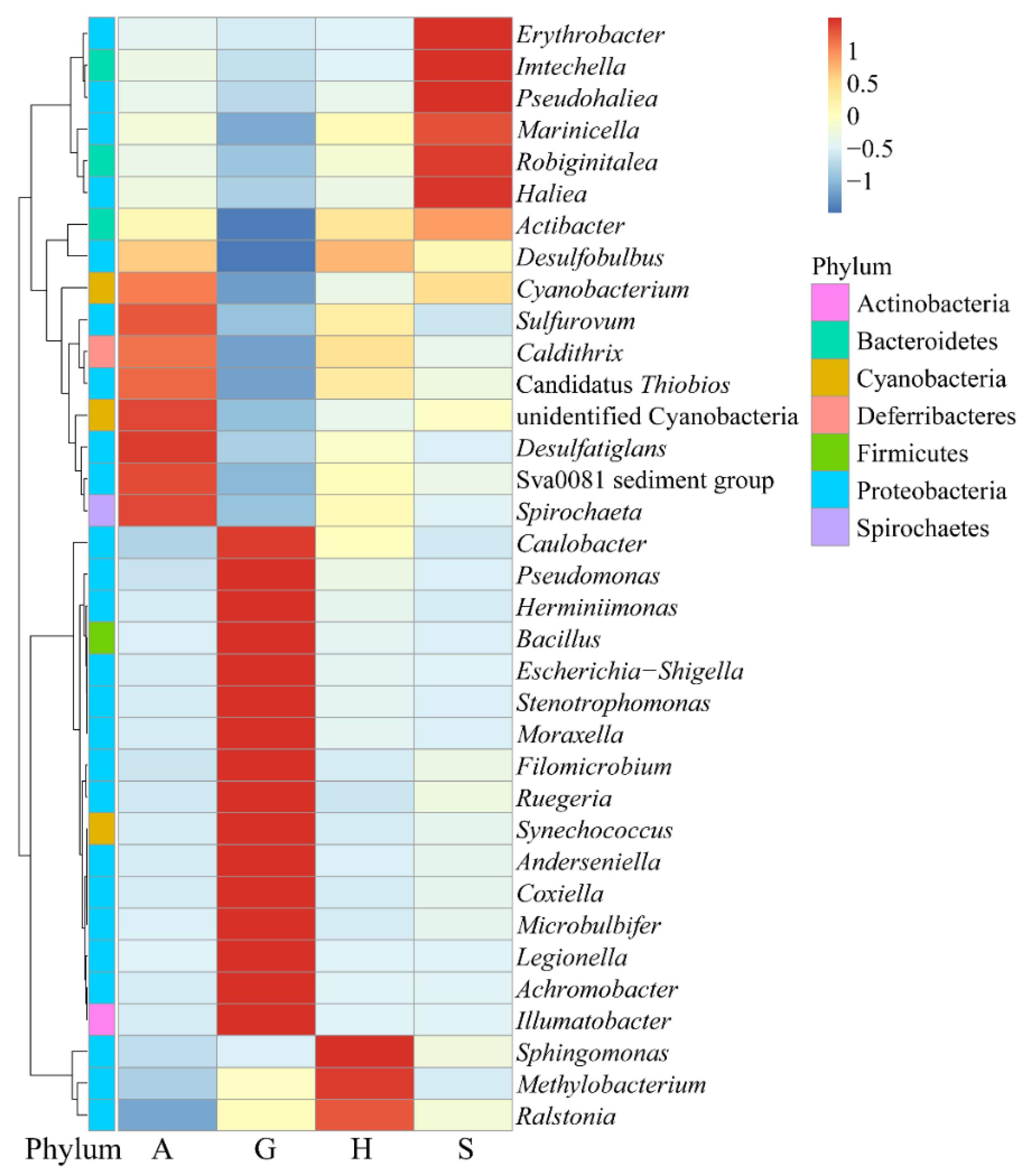
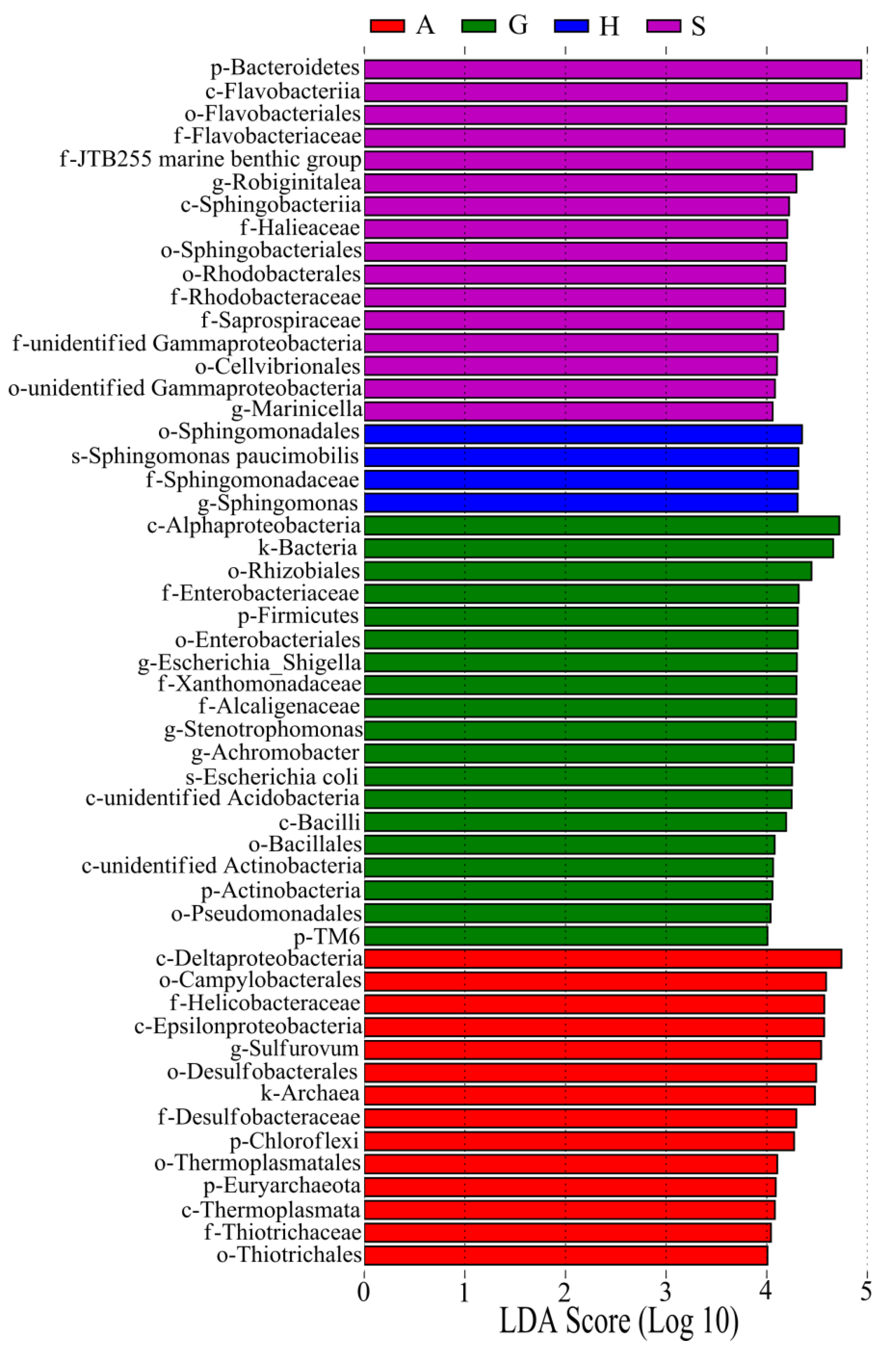
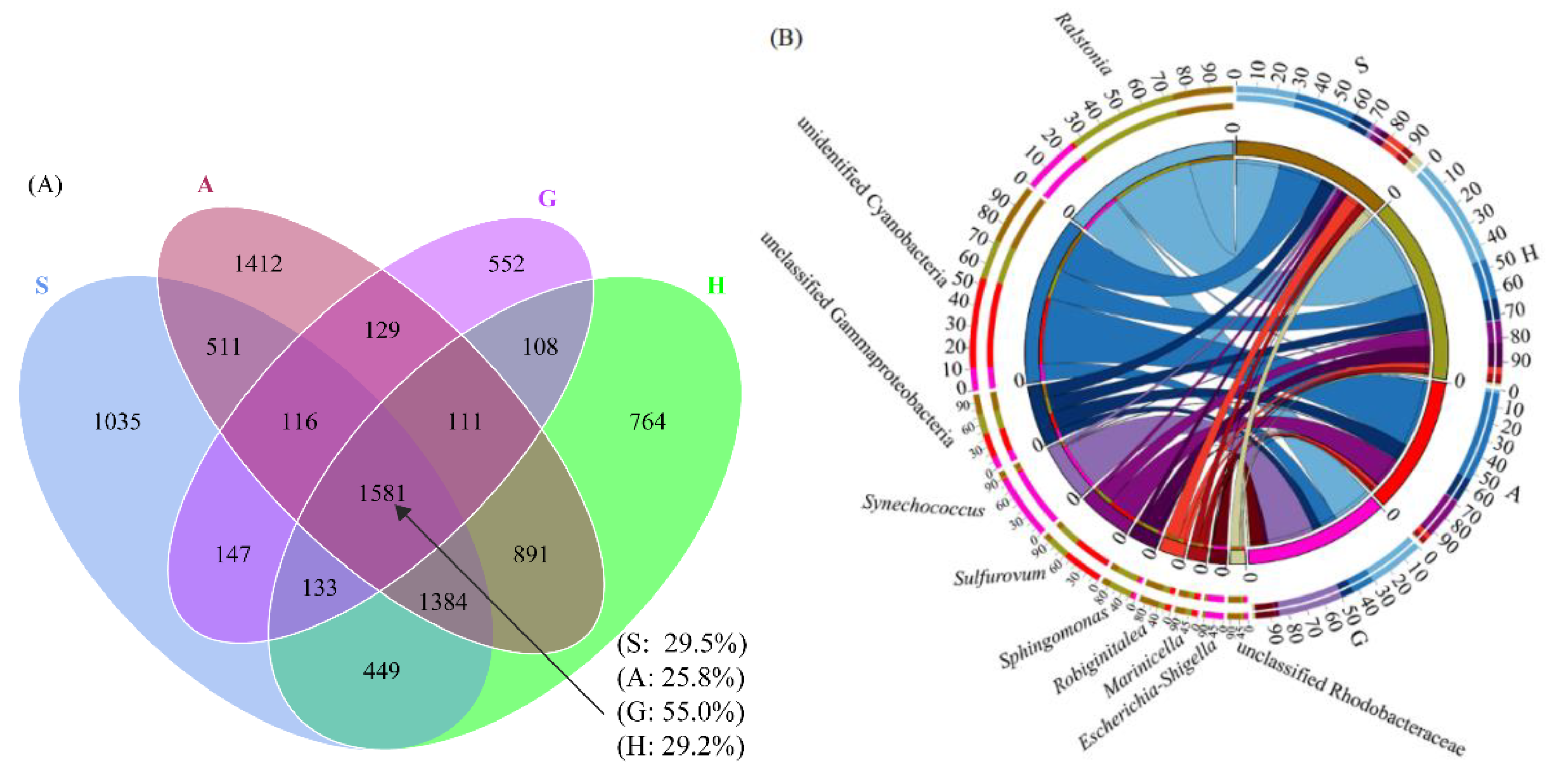
3.3. Core Microbiome of Intestine and Sediment Samples
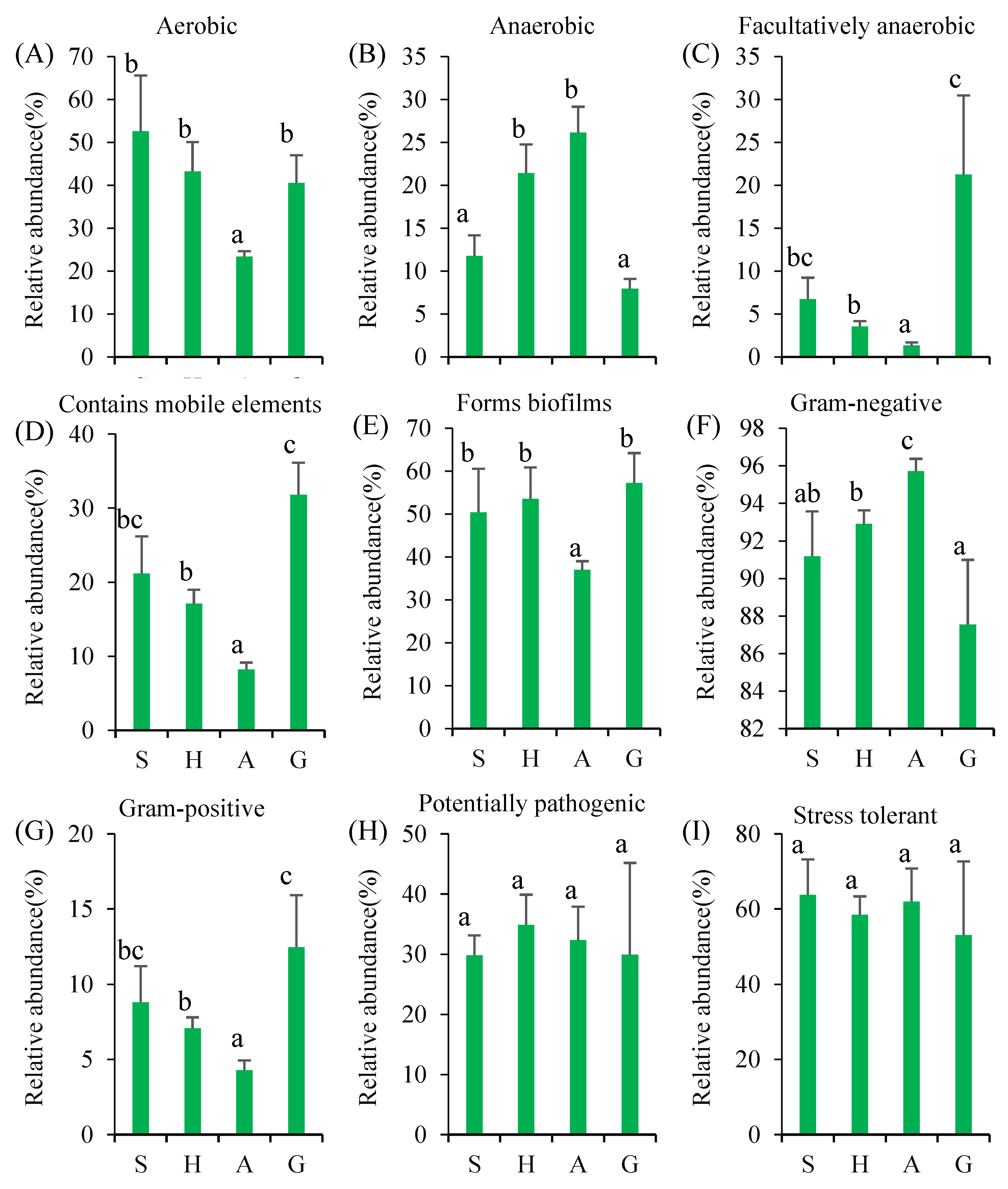
3.4. Microbial Function Changes
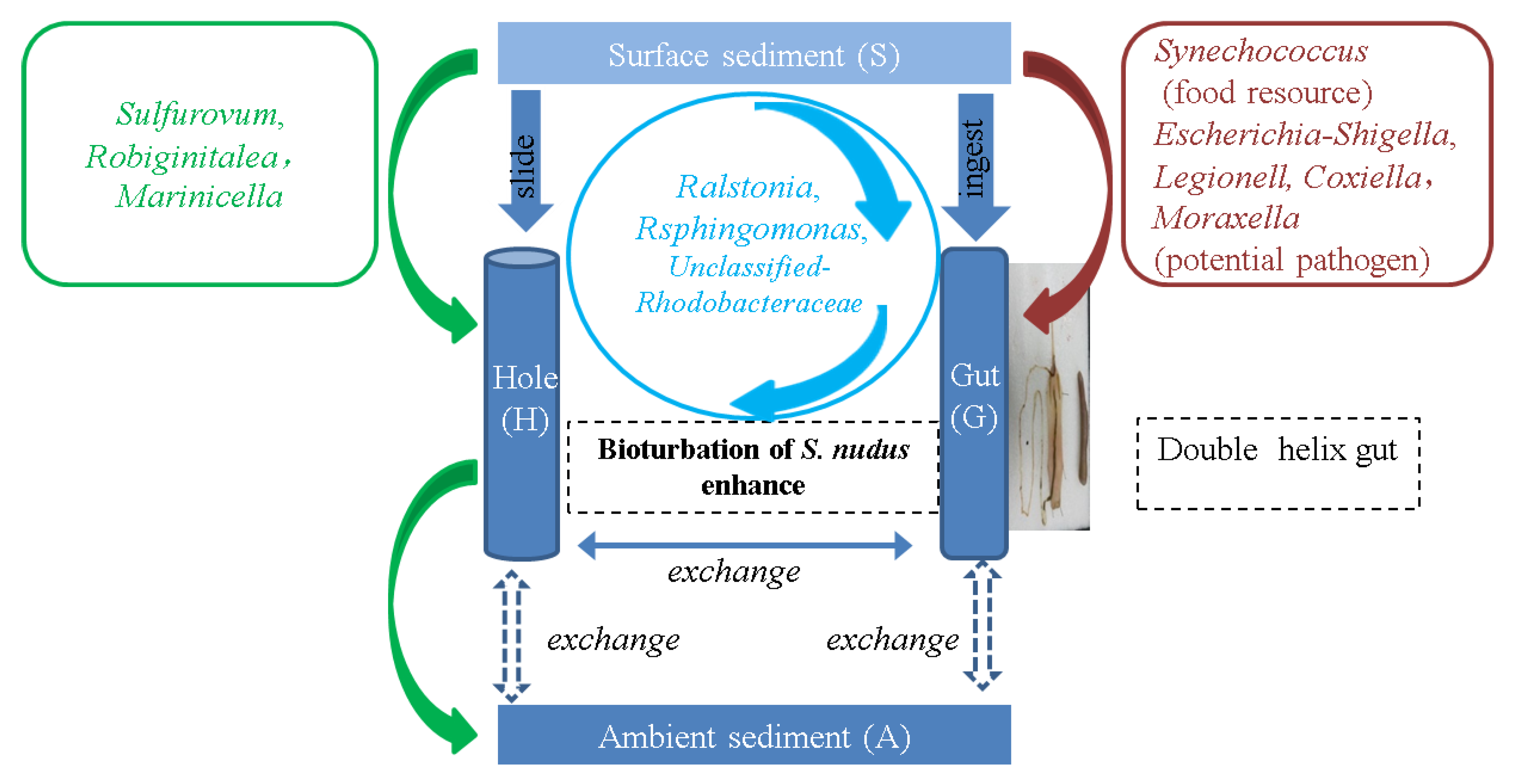
3.5. The Interactions of Microbial Composition between the Intestine of S. nudus and Surrounding Sediment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, H.B.; Chan, Y.C.; Compton, T.J.; Cheng, X.F.; Melville, D.S.; Zhang, S.D.; Zhang, Z.W.; Lei, G.C.; Ma, Z.J.; Piersma, T. Mollusc aquaculture homogenizes intertidal soft-sediment communities along the 18,400 km long coastline of China. Divers. Distrib. 2021, 27, 1553–1567. [Google Scholar] [CrossRef]
- Wang, J.; Russell, B.D.; Ding, M.W.; Dong, Y.W. Ocean acidification increases the sensitivity of and variability in physiological responses of an intertidal limpet to thermal stress. Biogeosciences 2018, 15, 2803–2817. [Google Scholar] [CrossRef]
- Fields, P.A.; Eraso, A.A. Year in the salt marsh: Seasonal changes in gill protein expression in the temperate intertidal mussel Geukensia demissa. Mar. Environ. Res. 2020, 161, 105088. [Google Scholar] [CrossRef]
- Liao, M.L.; Dong, Y.W.; Somero, G.N. Thermal adaptation of mRNA secondary structure: Stability versus lability. Proc. Natl. Acad. Sci. USA 2021, 118, e2113324118. [Google Scholar] [CrossRef]
- Tang, Y.Z.; Ma, S.; Liu, Y.H.; Pi, Y.R.; Liu, Y.; Zhao, Y. Intestinal microbial diversity and functional analysis of Urechis unicinctus from two different habitats: Pond polycultured with Penaeus japonicas and coastal zone. Aquacult. EnvInterac. 2021, 13, 211–224. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Liao, M.L.; Ding, M.W.; Wang, Z.K.; Dong, Y.W. Compositional and functional features of the gut microbiota of the intertidal snail Nerita yoldii along China’s coast. J. Mar. Biol. Assoc. 2021, 101, 1103–1110. [Google Scholar] [CrossRef]
- Lemmen, C. North Sea ecosystem-scale model-based quantification of net primary productivity changes by the Benthic filter feeder Mytilus edulis. Water 2018, 10, 1527. [Google Scholar] [CrossRef]
- Li, J.W.; Hu, R.P.; Guo, Y.J.; Chen, S.W.; Xie, X.Y.; Qin, J.G.; Ma, Z.H.; Zhu, C.B.; Pei, S.R. Bioturbation of peanut worms Sipunculus nudus on the composition of prokaryotic communities in a tidal flat as revealed by 16S rRNA gene sequences. MicrobiologyOpen 2019, 8, e802. [Google Scholar] [CrossRef]
- Sun, F.L.; Wang, Y.S.; Wang, C.Z.; Zhang, L.; Tu, K.; Zheng, Z.P. Insights into the intestinal microbiota of several aquatic organisms and association with the surrounding environment. Aquaculture 2020, 507, 196–202. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef]
- Patil, Y.; Gooneratne, R.; Ju, X. Interactions between host and gut microbiota in domestic pigs: A review. Gut Microbes 2020, 11, 310–334. [Google Scholar] [CrossRef] [PubMed]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’Connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef] [PubMed]
- Dehler, C.E.; Secombes, C.J.; Martin, S.A.M. Environmental and physiological factors shape the gut microbiota of Atlantic salmon parr (Salmo salar L.). Aquaculture 2017, 467, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.A.; Drake, H.L.; Schramm, A. Nitrous oxide reductase genes (nosZ) of denitrifying microbial populationsinsoiland the earthworm gut are phylogenetically similar. Appl. Environ. Microb. 2006, 72, 1019–1026. [Google Scholar] [CrossRef]
- Mwinyi, A.; Meyer, A.; Bleidorn, C.; Lieb, B.; Bartolomaeus, T.; Podsiadlowski, L. Mitochondrial genome sequence and gene order of Sipunculus nudus give additional support for an inclusion of Sipuncula into Annelida. BMC Genom. 2009, 10, 27. [Google Scholar] [CrossRef]
- Li, J.W.; Xie, X.Y.; Zhu, C.B.; Guo, Y.J.; Chen, S.W. Edible peanut worm (Sipunculus nudus) in the Beibu Gulf: Resource, aquaculture, ecological impact and counterplan. J. Ocean. Univ. China 2017, 16, 823–830. [Google Scholar] [CrossRef]
- Adrianov, A.V.; Maiorova, A.S. Reproduction and development of common species of peanut worms (Sipuncula) from the Sea of Japan. Russ. J. Mar. Biol. 2010, 36, 1–15. [Google Scholar] [CrossRef]
- Mark, A.S.; Monika, K. A deep burrowing sipunculan of ecological and geochemical importance. Deep. Sea Res. Part I 2009, 56, 2057–2064. [Google Scholar] [CrossRef]
- Li, J.W.; Zhu, C.B.; Guo, Y.J.; Xie, X.Y.; Huang, G.Q.; Chen, S.W. Experimental study of bioturbation by Sipunculus nudus in a polyculture system. Aquaculture 2015, 437, 175–181. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.G.; Peplies, J.; Glockner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids. Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Ward, T.; Larson, J.; Meulemans, J.; Hillmann, B.; Lynch, J.; Sidiropoulos, D.; Spear, J.R.; Caporaso, G.; Blekhman, R.; Knight, R.; et al. BugBase predicts organism level microbiome phenotypes. BioRxiv 2017, BioRxiv:133462. [Google Scholar] [CrossRef]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H.J. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Wang, Y.; Han, W.; Wang, X.; Chen, H.; Zhu, F.; Wang, X.; Lei, C.L. Speciation of heavy metals and bacteria in cow dung after vermicomposting by the earthworm, Eisenia fetida. Bioresour. Technol. 2017, 245, 411–418. [Google Scholar] [CrossRef]
- Jin, R.C.; Jiang, M.; Sun, S.Y.; Dai, X.L.; Wu, H.; Zhou, J.F.; Yu, Z.L.; Zhang, F. Microbial community in Litopenaeus vannamei intestine and its aquaculture environment. J. Fish. China 2020, 44, 2037–2054, (in Chinese abstract). [Google Scholar] [CrossRef]
- Sun, M.M.; Chao, H.Z.; Zheng, X.X.; Deng, S.P.; Ye, M.; Hu, F. Ecological role of earthworm intestinal bacteria in terrestrial environments: A review. Sci. Total Environ. 2020, 740, 140008. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Hehemann, J.H.; Rebuffet, E.; Michel, G. Environmental and gut bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Choo, Y.J.; Cho, J.C. Lutimonas vermicola gen. nov., sp. nov., a member of the family Flavobacteriaceae isolated from the marine polychaete Periserrula leucophryna. Int. J. Syst. Evol. Microbiol. 2007, 57, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Ha, V.N.; Shen, Z.; Zhu, H.L.; Zhao, F.; Zhao, Z. Characteristics of bulk and rhizosphere soil microbial community in an ancient Platycladus orientalis forest. Appl. Soil. Ecol. 2018, 132, 91–98. [Google Scholar] [CrossRef]
- Li, C.; Reimers, C.E.; Chapman, J.W.; Li, C.; Reimers, C.E.; Chapman, J.W. Microbiome analyses and presence of cable bacteria in the burrow sediment of Upogebia pugettensis. Mar. Ecol. Prog. Ser. 2020, 648, 79–94. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, W.; Wang, H.; Li, C.; Wang, L.; Li, Y.; Wang, J.H. Bacillus baekryungensis MS1 regulates the growth, non-specific immune parameters and gut microbiota of the sea cucumber Apostichopus japonicus. Fish Shellfish Immunol. 2020, 102, 133–139. [Google Scholar] [CrossRef]
- Abed, R.M.M.; Kohls, K.; Beer, D.D. Effect of salinity changes on the bacterial diversity, photosynthesis and oxygen consumption of cyanobacterial mats from an intertidal flat of the Arabian Gulf. Environ. Microbiol. 2007, 9, 1384–1392. [Google Scholar] [CrossRef]
- Zhang, M.J.; Sun, Q.Y.; Chen, P.X.; Wei, X.H.; Wang, B. How microorganisms tell the truth of potentially toxic elements pollution in environment. J. Hazard. Mater. 2002, 431, 128456. [Google Scholar] [CrossRef]
- Park, Y.J.; Ko, J.J.; Yun, S.L.; Lee, E.Y.; Kim, S.J.; Kang, S.W.; Lee, B.C.; Kim, S.K. Enhancement of bioremediation by Ralstonia sp. HM-1 in sediment polluted by Cd and Zn. Bioresour. Technol. 2008, 99, 7458–7463. [Google Scholar] [CrossRef]
- Chen, J.L.; Wong, M.H.; Wong, Y.S.; Tam, N.F.Y. Multi-factors on biodegradation kinetics of polycyclic aromatic hydrocarbons (PAHs) by Sphingomonas sp. a bacterial strain isolated from mangrove sediment. Mar. Pollut. Bull. 2008, 57, 695–702. [Google Scholar] [CrossRef]
- Li, J.X.; Gu, J.D. Complete degradation of dimethyl isophthalate requires the biochemical cooperation between Klebsiella oxytoca Sc and Methylobacterium mesophilicum Sr Isolated from Wetland sediment. Sci. Total Environ. 2007, 380, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, J.; Wheeler, P.E.; Obafemi, S.; Valdez, J.; Forstner, M.R.J.; Bonner, T.H.; Dittmar, H. Detection of salmonellae from fish in a natural river system. J. Aquat. Anim. Health 2008, 20, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Gainza, O.; Ramírez, C.; Ramos, A.S.; Romero, J. Intestinal microbiota of white shrimp Penaeus vannamei under intensive cultivation conditions in Ecuador. Microb. Ecol. 2018, 75, 562–568. [Google Scholar] [CrossRef]
- Navarrete, P.; Magne, F.; Araneda, C.; Fuentes, P.; Barros, L.; Opazo, R.; Espejo, R.; Romero, J. PCR-TTGE analysis 16S rRNA from rainbow trout (Oncorhynchus mykiss) gut microbiota reveals host-specific communities of active bacteria. PLoS ONE 2012, 7, e31335. [Google Scholar] [CrossRef] [PubMed]
- Givens, C.E.; Ransom, B.; Bano, N.; Hollibaugh, J.T. Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar. Ecol. Prog. Ser. 2015, 518, 209–223. [Google Scholar] [CrossRef]
- Zarkasi, K.Z.; Taylor, R.S.; Abell, G.C.J.; Tamplin, M.L.; Glencross, B.D.; Bowman, J.P. Atlantic salmon (Salmo salar L.) gastrointestinal microbial community dynamics in relation to digesta properties and diet. Microb. Ecol. 2016, 71, 589–603. [Google Scholar] [CrossRef]
- Duan, Y.F.; Huang, J.H.; Wang, Y.; Zhang, J.S. Characterization of bacterial community in intestinal and rearing water of Penaeus monodon differing growth performances in outdoor and indoor ponds. Aquac. Res. 2020, 51, 4279–4289. [Google Scholar] [CrossRef]
- Karsten, G.R.; Drake, H.L. Comparative assessment of the aerobic and anaerobic microfloras of earthworm guts and forest soils. Appl. Environ. Microb. 1995, 67, 1039–1044. [Google Scholar] [CrossRef]
- Gajardo, K.; Rodiles, A.; Kortner, T.M.; Krogdahl, A.; Bakke, A.M.; Merrifield, D.L. A high resolution map of the gut microbiota in Atlantic salmon (Salmo salar): A basis for comparative gut microbial research. Sci. Rep. 2016, 6, 30893. [Google Scholar] [CrossRef]
- Zheng, Y.; Wu, W.; Hu, G.; Qiu, L.; Meng, S.L.; Song, C. Gut microbiota analysis of juvenile genetically improved farmed tilapia (Oreochromis niloticus) by dietary supplementation of different resveratrol concentrations. Fish Shellfish Immunol. 2018, 77, 200–207. [Google Scholar] [CrossRef]
- Stevens, H.; Brinkhoff, T.; Rink, B.; Vollmers, J.; Simon, M. Diversity and abundance of Gram positive bacteria in a tidal flat ecosystem. Environ. Microbiol. 2007, 9, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Clements, K.D.; Angert, E.R.; Montgomery, W.L. Intestinal microbiota in fishes: What’s known and what’s not. Mol. Ecol. 2014, 23, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Burja, A.M.; Banaigs, B.; Abou-Mansour, E.; Burgess, J.G.; Wright, P.C. Marine cyanobacteria—A prolific source of natural products. Tetrahedron 2001, 57, 9347–9377. [Google Scholar] [CrossRef]
- Singh, A.; Singh, D.P.; Tiwari, R.; Kumar, K.; Singh, R.; Singh, S.; Prasanna, R.; Saxena, A.K.; Nain, L. Taxonomic and functional annotation of gut bacterial communities of Eisenia foetida and Perionyx excavates. Microbiol. Res. 2015, 175, 48–56. [Google Scholar] [CrossRef]
- Montagna, M.; Berruti, A.; Bianciotto, V.; Cremonesi, P.; Giannico, R.; Gusmeroli, F. Differential biodiversity responses between kingdoms (plants, fungi, bacteria and metazoa) along an Alpine succession gradient. Mol. Ecol. 2018, 27, 3671–3685. [Google Scholar] [CrossRef]
- Pass, D.A.; Morgan, A.J.; Read, D.; Field, D.; Weightman, A.; Kille, P. The effect of anthropogenic arsenic contamination on the earthworm microbiome. Environ. Microbiol. 2015, 17, 1884–1896. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Groups | Similarity | ANOSIM | Adonis | ||
|---|---|---|---|---|---|
| R | p | R2 | p | ||
| G/S | 34.8% | 0.896 | 0.034 | 0.561 | 0.048 |
| G/A | 29.9% | 1.0 | 0.028 | 0.631 | 0.001 |
| G/H | 36.3% | 0.74 | 0.036 | 0.539 | 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Chen, S.; Wu, P.; Zhu, C.; Hu, R.; Li, T.; Guo, Y. Insights into the Relationship between Intestinal Microbiota of the Aquaculture Worm Sipunculus nudus and Surrounding Sediments. Fishes 2023, 8, 32. https://doi.org/10.3390/fishes8010032
Li J, Chen S, Wu P, Zhu C, Hu R, Li T, Guo Y. Insights into the Relationship between Intestinal Microbiota of the Aquaculture Worm Sipunculus nudus and Surrounding Sediments. Fishes. 2023; 8(1):32. https://doi.org/10.3390/fishes8010032
Chicago/Turabian StyleLi, Junwei, Suwen Chen, Peng Wu, Changbo Zhu, Ruiping Hu, Ting Li, and Yongjian Guo. 2023. "Insights into the Relationship between Intestinal Microbiota of the Aquaculture Worm Sipunculus nudus and Surrounding Sediments" Fishes 8, no. 1: 32. https://doi.org/10.3390/fishes8010032
APA StyleLi, J., Chen, S., Wu, P., Zhu, C., Hu, R., Li, T., & Guo, Y. (2023). Insights into the Relationship between Intestinal Microbiota of the Aquaculture Worm Sipunculus nudus and Surrounding Sediments. Fishes, 8(1), 32. https://doi.org/10.3390/fishes8010032