
Evaluation of the Viral Diversity of Artemia Cysts from Saline Lakes in Kazakhstan Using Viral Metagenomics Analysis
 , , ,
, , ,
Abstract
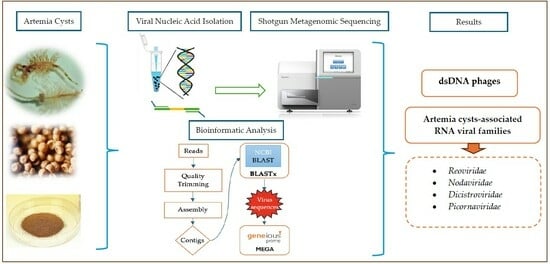
:
1. Introduction
2. Materials and Methods
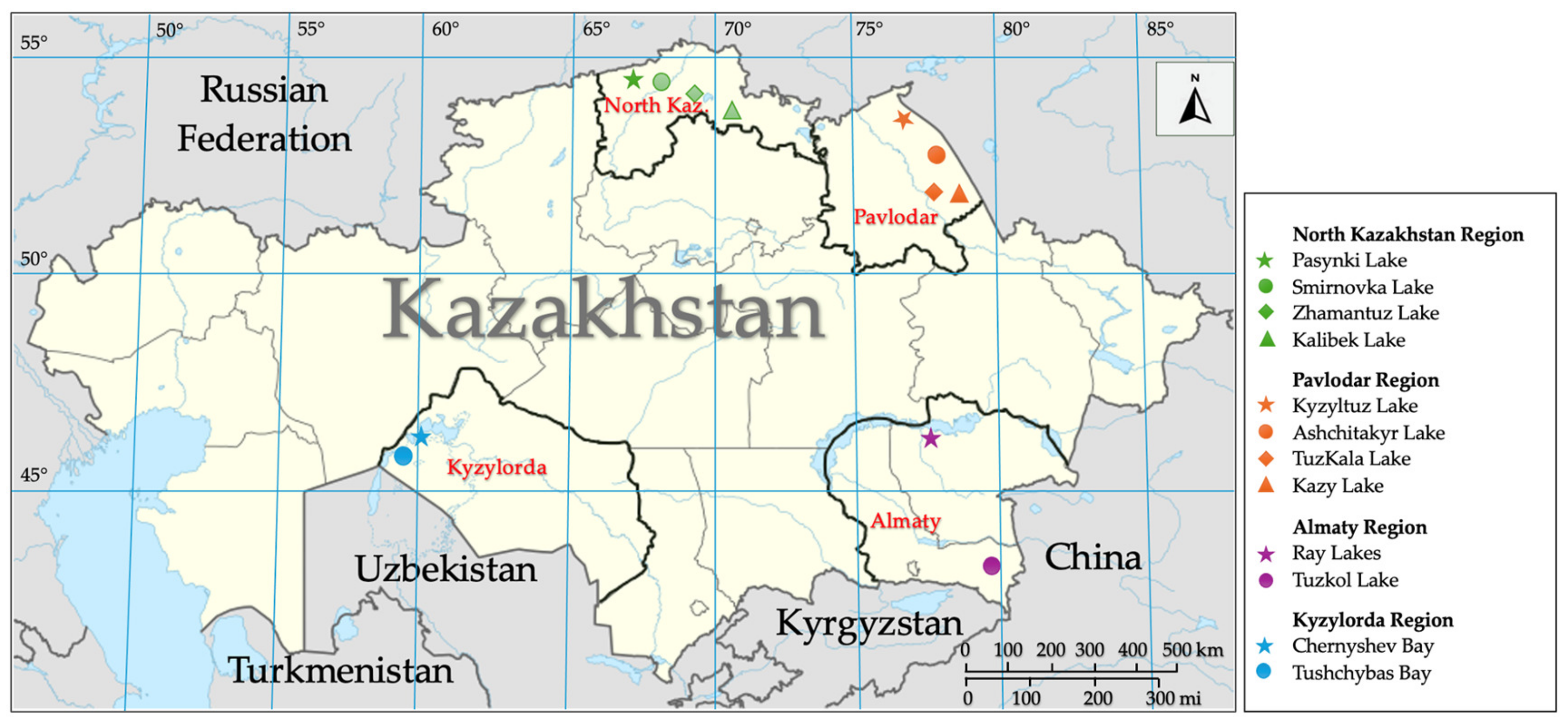
2.1. Sampling Sites and Collection
2.2. Viral Nucleic Acid Extraction and Sequencing
2.3. Metagenome Data Analysis
2.4. Phylogenetic Analysis
3. Results
3.1. Overview of Sequencing Outcomes
3.2. Contig Analysis
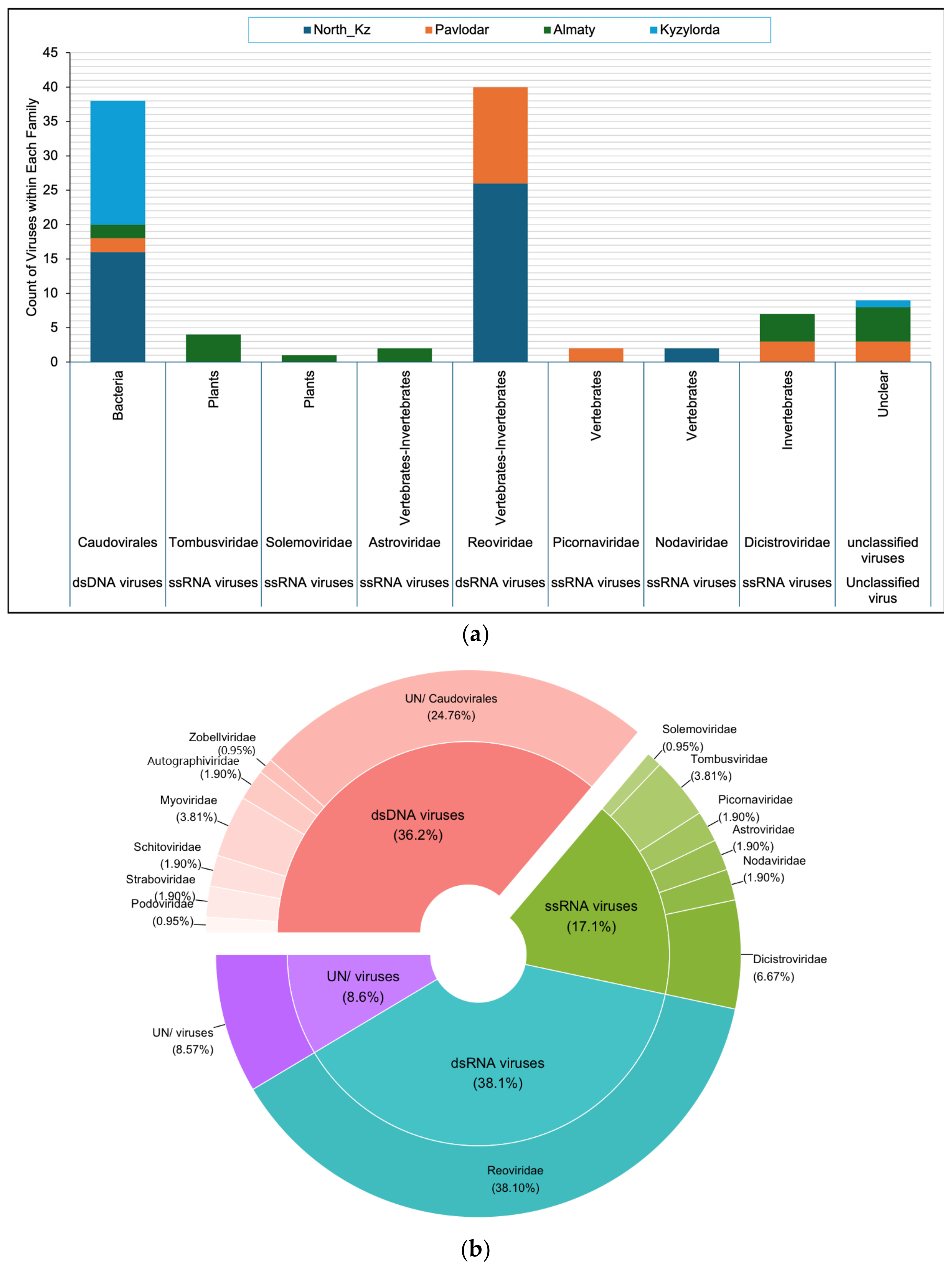
3.3. Composition of the Artemia Viral Community
3.4. Distribution of Viral Families in Lakes of Different Regions
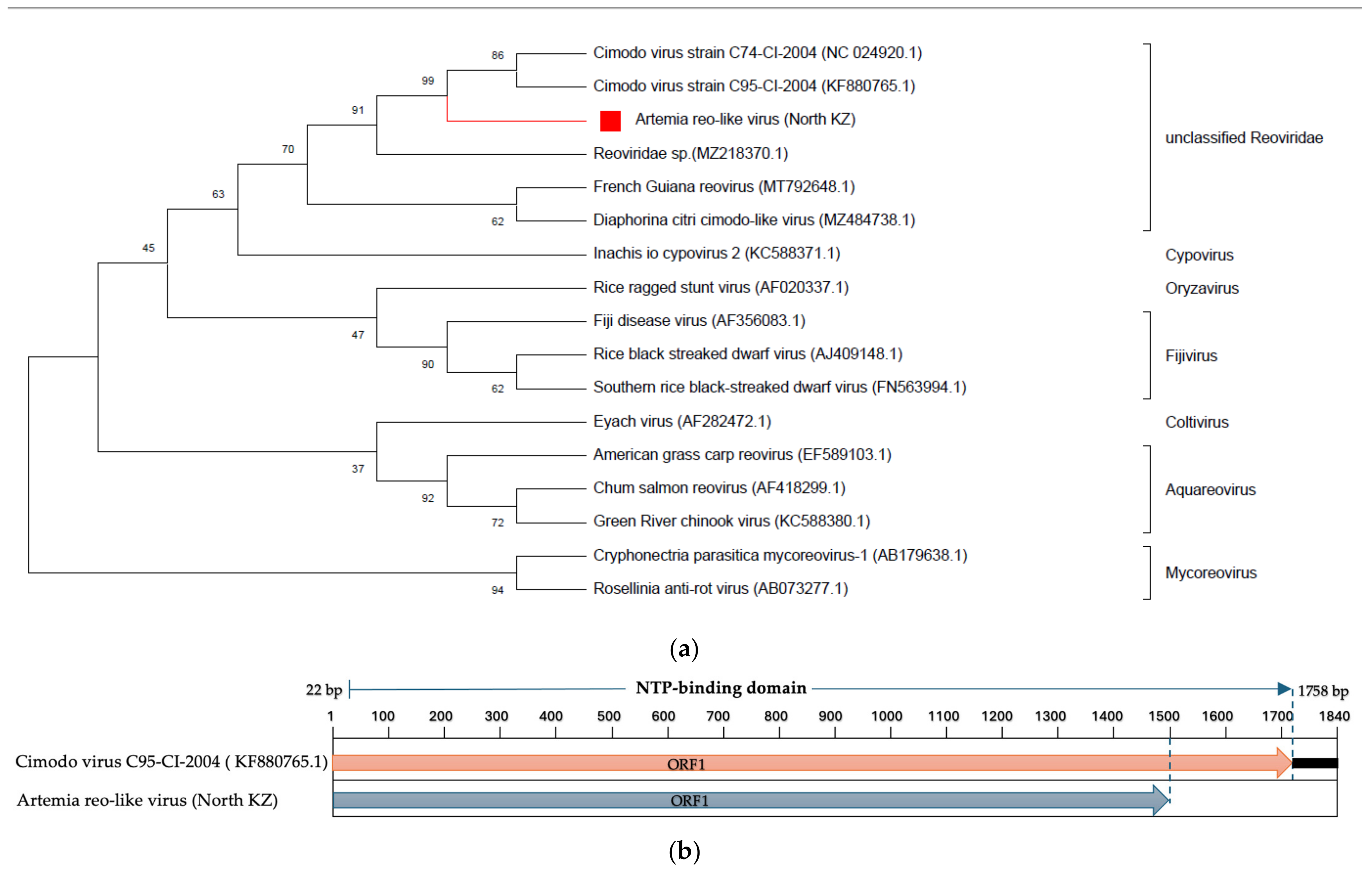
3.5. Characteristics of Selected Viruses
- Reoviridae
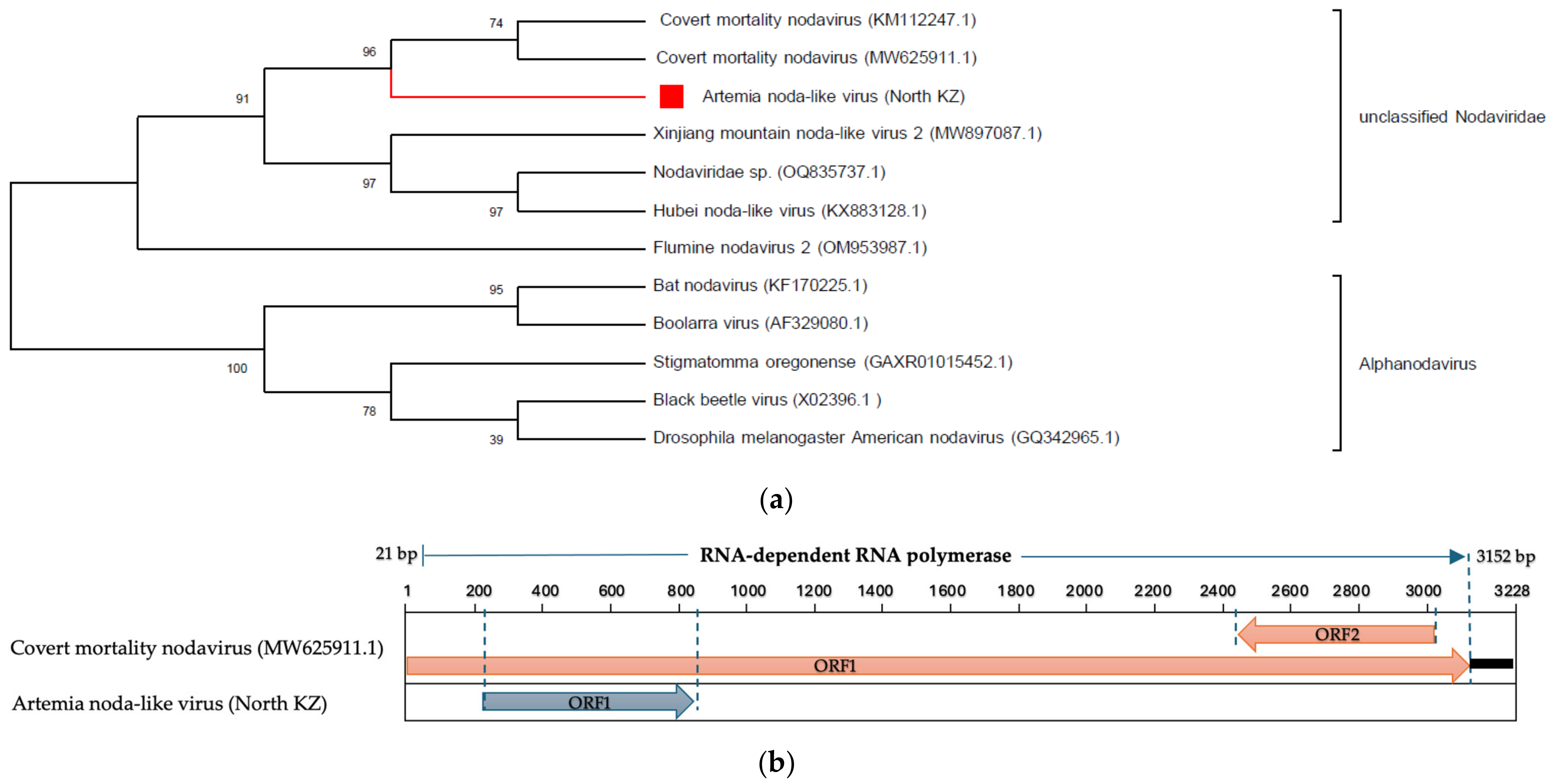
- Nodaviridae
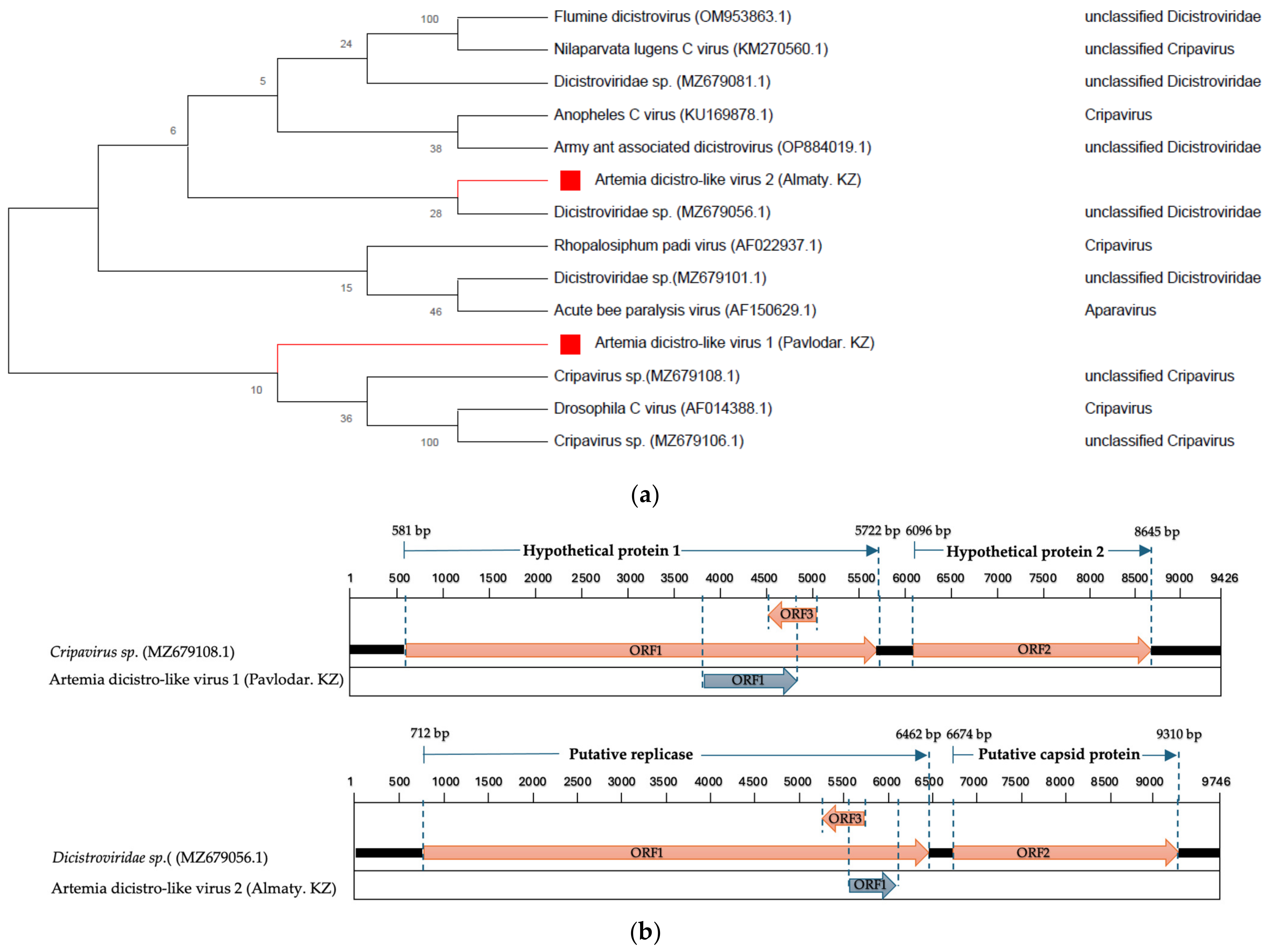
- Dicistroviridae
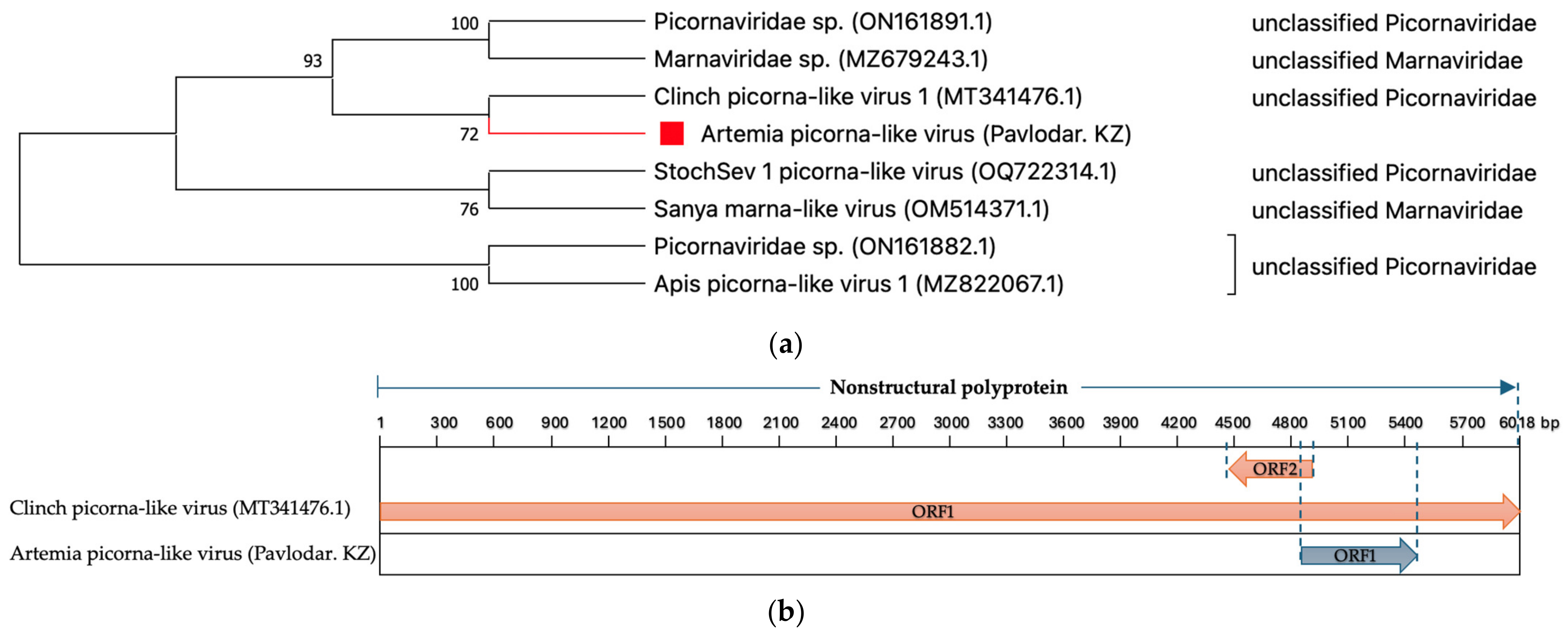
- Picornaviridae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fuhrman, J.A. Marine Viruses and Their Biogeochemical and Ecological Effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Rohwer, F. Viral Metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Elbehery, A.H.A.; Deng, L. Insights into the Global Freshwater Virome. Front. Microbiol. 2022, 13, 953500. [Google Scholar] [CrossRef] [PubMed]
- Bergh, Ø.; BØrsheim, K.Y.; Bratbak, G.; Heldal, M. High Abundance of Viruses Found in Aquatic Environments. Nature 1989, 340, 467–468. [Google Scholar] [CrossRef]
- Bassi, C.; Guerriero, P.; Pierantoni, M.; Callegari, E.; Sabbioni, S. Novel Virus Identification through Metagenomics: A Systematic Review. Life 2022, 12, 2048. [Google Scholar] [CrossRef] [PubMed]
- Varghese, F.S.; van Rij, R.P. Insect Virus Discovery by Metagenomic and Cell Culture-Based Approaches. Methods Mol. Biol. 2018, 1746, 197–213. [Google Scholar] [PubMed]
- Rosario, K.; Breitbart, M. Exploring the Viral World through Metagenomics. Curr. Opin. Virol. 2011, 1, 289–297. [Google Scholar] [CrossRef]
- Hall, R.J.; Wang, J.; Todd, A.K.; Bissielo, A.B.; Yen, S.; Strydom, H.; Moore, N.E.; Ren, X.; Huang, Q.S.; Carter, P.E.; et al. Evaluation of Rapid and Simple Techniques for the Enrichment of Viruses Prior to Metagenomic Virus Discovery. J. Virol. Methods 2014, 195, 194–204. [Google Scholar] [CrossRef]
- Bhattarai, B.; Bhattacharjee, A.S.; Coutinho, F.H.; Goel, R.K. Viruses and Their Interactions with Bacteria and Archaea of Hypersaline Great Salt Lake. Front. Microbiol. 2021, 12, 701414. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Ravet, V.; Colombet, J.; Bettarel, Y.; Auguet, J.-C.; Bouvier, T.; Lucas-Staat, S.; Vellet, A.; Prangishvili, D.; et al. Analysis of Metagenomic Data Reveals Common Features of Halophilic Viral Communities across Continents. Environ. Microbiol. 2016, 18, 889–903. [Google Scholar] [CrossRef]
- Oren, A. Microbial Life at High Salt Concentrations: Phylogenetic and Metabolic Diversity. Saline Syst. 2008, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Waiser, M.J.; Robarts, R.D. Saline Inland Waters. In Encyclopedia of Inland Waters; Elsevier: Amsterdam, The Netherlands, 2009; pp. 634–644. [Google Scholar]
- Baxter, B.K. Great Salt Lake Microbiology: A Historical Perspective. Int. Microbiol. 2018, 21, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Bodaker, I.; Sharon, I.; Feingersch, R.; Rosenberg, M. Archaeal Diversity in the Dead Sea: Microbial Survival under Increasingly Harsh Conditions. Nat. Resour. Environ. Issues 2009, 15, 137–143. [Google Scholar]
- Izhitskiy, A.S.; Zavialov, P.O.; Sapozhnikov, P.V.; Kirillin, G.B.; Grossart, H.P.; Kalinina, O.Y.; Zalota, A.K.; Goncharenko, I.V.; Kurbaniyazov, A.K. Present State of the Aral Sea: Diverging Physical and Biological Characteristics of the Residual Basins. Sci. Rep. 2016, 6, 23906. [Google Scholar] [CrossRef] [PubMed]
- Aubakirova, M.; Mazhibayeva, Z.; Assylbekova, S.Z.; Isbekov, K.B.; Barbol, B.; Bolatbekova, Z.; Jussupbekova, N.; Moldrakhman, A.; Satybaldiyeva, G. The Current State of Zooplankton Diversity in the Middle Caspian Sea during Spring. Diversity 2023, 15, 798. [Google Scholar] [CrossRef]
- Motlagh, A.M.; Bhattacharjee, A.S.; Coutinho, F.H.; Dutilh, B.E.; Casjens, S.R.; Goel, R.K. Insights of Phage-Host Interaction in Hypersaline Ecosystem through Metagenomics Analyses. Front. Microbiol. 2017, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Van Stappen, G.; Sui, L.; Hoa, V.N.; Tamtin, M.; Nyonje, B.; Medeiros Rocha, R.; Sorgeloos, P.; Gajardo, G. Review on Integrated Production of the Brine Shrimp Artemia in Solar Salt Ponds. Rev. Aquac. 2020, 12, 1054–1071. [Google Scholar] [CrossRef]
- Leger, P.; Bengtson, D.A.; Simpson, K.L.; Sorgeloos, P. The Use and Nutritional Value of Artemia as a Food Source. Oceanogr. Mar. Biol. Ann. Rev. 1986, 24, 521–623. [Google Scholar]
- Conceição, L.E.C.; Yúfera, M.; Makridis, P.; Morais, S.; Dinis, M.T. Live Feeds for Early Stages of Fish Rearing. Aquac. Res. 2010, 41, 613–640. [Google Scholar] [CrossRef]
- Lavens, P.; Sorgeloos, P. The History, Present Status and Prospects of the Availability of Artemia Cysts for Aquaculture. Aquaculture 2000, 181, 397–403. [Google Scholar] [CrossRef]
- Kenzhebekov, B.K.; Nuriyeva, S.B.; Troshina, T.T.; Sharapova, L.I.; Sharipova, O.A. Artemia Populations in Modern Conditions of Salt Lakes of South-Eastern Kazakhstan. Vestn. Astrakhan State Tech. Univ. Ser. Fish. Ind. 2019, 2019, 72–82. [Google Scholar] [CrossRef]
- Sivakumar, V.K.; Sarathi, M.; Venkatesan, C.; Sivaraj, A.; Hameed, A.S.S. Experimental Exposure of Artemia to Hepatopancreatic Parvo-like Virus and Subsequent Transmission to Post-Larvae of Penaeus Monodon. J. Invertebr. Pathol. 2009, 102, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Valverde, E.J.; Labella, A.M.; Borrego, J.J.; Castro, D. Artemia Spp., a Susceptible Host and Vector for Lymphocystis Disease Virus. Viruses 2019, 11, 506. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Salgado, L.; Olveira, J.G.; Dopazo, C.P.; Bandín, I. Role of Rotifer (Brachionus plicatilis) and Artemia (Artemia salina) Nauplii in the Horizontal Transmission of a Natural Nervous Necrosis Virus (NNV) Reassortant Strain to Senegalese Sole (Solea Senegalensis) Larvae. Vet. Q. 2020, 40, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Grisez, L.; Chair, M.; Sorgeloos, P.; Ollevier, F. Mode of Infection and Spread of Vibrio Anguillarum in Turbot Scophthalmus Maximus Larvae after Oral Challenge through Live Feed. Dis. Aquat. Organ. 1996, 26, 181–187. [Google Scholar] [CrossRef]
- Borrego, J.J.; Valverde, E.J.; Labella, A.M.; Castro, D. Lymphocystis Disease Virus: Its Importance in Aquaculture. Rev. Aquac. 2017, 9, 179–193. [Google Scholar] [CrossRef]
- Padrós, F.; Caggiano, M.; Toffan, A.; Constenla, M.; Zarza, C.; Ciulli, S. Integrated Management Strategies for Viral Nervous Necrosis (VNN) Disease Control in Marine Fish Farming in the Mediterranean. Pathogens 2022, 11, 330. [Google Scholar] [CrossRef] [PubMed]
- da Silva, S.M.B.C.; Lavander, H.D.; de Santana Luna, M.M.; de Melo Eloi da Silva, A.O.; Gálvez, A.O.; Coimbra, M.R.M. Artemia Franciscana as a Vector for Infectious Myonecrosis Virus (IMNV) to Litopenaeus Vannamei Juvenile. J. Invertebr. Pathol. 2015, 126, 1–5. [Google Scholar] [CrossRef]
- Zhang, J.-S.; Dong, S.-L.; Dong, Y.-W.; Tian, X.-L.; Cao, Y.-C.; Li, Z.-J.; Yan, D.-C. Assessment of the Role of Brine Shrimp Artemia in White Spot Syndrome Virus (WSSV) Transmission. Vet. Res. Commun. 2010, 34, 25–32. [Google Scholar] [CrossRef]
- Sudhakaran, R.; Yoganandhan, K.; Ishaq Ahmed, V.; Sahul Hameed, A. Artemia as a Possible Vector for Macrobrachium Rosenbergii Nodavirus (MrNV) and Extra Small Virus Transmission (XSV) to Macrobrachium Rosenbergii Post-Larvae. Dis. Aquat. Organ. 2006, 70, 161–166. [Google Scholar] [CrossRef]
- Victoria, J.G.; Kapoor, A.; Li, L.; Blinkova, O.; Slikas, B.; Wang, C.; Naeem, A.; Zaidi, S.; Delwart, E. Metagenomic Analyses of Viruses in Stool Samples from Children with Acute Flaccid Paralysis. J. Virol. 2009, 83, 4642–4651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, L.; Deng, X.; Kapusinszky, B.; Pesavento, P.A.; Delwart, E. Faecal Virome of Cats in an Animal Shelter. J. Gen. Virol. 2014, 95, 2553–2564. [Google Scholar] [CrossRef] [PubMed]
- Bağcı, C.; Patz, S.; Huson, D.H. DIAMOND + MEGAN: Fast and Easy Taxonomic and Functional Analysis of Short and Long Microbiome Sequences. Curr. Protoc. 2021, 1, e59. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Compares BWA to Other Long Read Aligners like CUSHAW2 Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Felderhoff, H.; Bağci, C.; Huson, D.H. Using AnnoTree to Get More Assignments, Faster, in DIAMOND+MEGAN Microbiome Analysis. mSystems 2022, 7, e0140821. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus Taxonomy: The Database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef]
- Hulo, C.; de Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A Knowledge Resource to Understand Virus Diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Attoui, H.; Mertens, P.P.C.; Becnel, J.; Belaganahalli, S.; Bergoin, M.; Brussaard, C.P.; Chappell, J.D.; Ciarlet, M.; del Vas, M.; Dermody, T.S.; et al. Family: Reoviridae. Ninth Report of the International Committee on Taxonomy of Viruses. 2011. Available online: https://ictv.global/report_9th/dsRNA/Reoviridae (accessed on 10 July 2023).
- Hermanns, K.; Zirkel, F.; Kurth, A.; Drosten, C.; Junglen, S. Cimodo Virus Belongs to a Novel Lineage of Reoviruses Isolated from African Mosquitoes. J. Gen. Virol. 2014, 95, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Sahul Hameed, A.S.; Ninawe, A.S.; Nakai, T.; Chi, S.C.; Johnson, K.L. ICTV Virus Taxonomy Profile: Nodaviridae. J. Gen. Virol. 2019, 100, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Warsaba, R.; Sadasivan, J.; Jan, E. Dicistrovirus-Host Molecular Interactions. Curr. Issues Mol. Biol. 2020, 34, 83–112. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E. ICTV Virus Taxonomy Profile: Dicistroviridae. J. Gen. Virol. 2017, 98, 355–356. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Ryu, W.-S. Picornavirus. In Molecular Virology of Human Pathogenic Viruses; Elsevier: Amsterdam, The Netherlands, 2017; pp. 153–164. [Google Scholar]
- Ribeiro, G.d.O.; Monteiro, F.J.C.; Rego, M.O.d.S.; Ribeiro, E.S.D.; Castro, D.F.d.; Caseiro, M.M.; Souza Marinho, R.d.S.; Komninakis, S.V.; Witkin, S.S.; Deng, X.; et al. Detection of RNA-Dependent RNA Polymerase of Hubei Reo-Like Virus 7 by Next-Generation Sequencing in Aedes Aegypti and Culex Quinquefasciatus Mosquitoes from Brazil. Viruses 2019, 11, 147. [Google Scholar] [CrossRef]
- Truchado, D.A.; Llanos-Garrido, A.; Oropesa-Olmedo, D.A.; Cerrada, B.; Cea, P.; Moens, M.A.J.; Gomez-Lucia, E.; Doménech, A.; Milá, B.; Pérez-Tris, J.; et al. Comparative Metagenomics of Palearctic and Neotropical Avian Cloacal Viromes Reveal Geographic Bias in Virus Discovery. Microorganisms 2020, 8, 1869. [Google Scholar] [CrossRef]
- Guo, L.; Lu, X.; Liu, X.; Li, P.; Wu, J.; Xing, F.; Peng, H.; Xiao, X.; Shi, M.; Liu, Z.; et al. Metatranscriptomic Analysis Reveals the Virome and Viral Genomic Evolution of Medically Important Mites. J. Virol. 2021, 95, e01686-20. [Google Scholar] [CrossRef] [PubMed]
- Britt, K.; Stevens, K.; Gebben, S.; Levy, A.; Al Rwahnih, M.; Batuman, O. Partial Genome Sequence of a Novel Reo-Like Virus Detected in Asian Citrus Psyllid (Diaphorina Citri) Populations from Florida Citrus Groves. Microbiol. Resour. Announc. 2021, 10, e0056321. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Li, C.; Wang, Y.; Hu, T.; Zhang, F.; Meng, F.; Han, X.; Wang, G.; Qin, J.; Nauwynck, H.; et al. Diversity and Connectedness of Brine Shrimp Viruses in Global Hypersaline Ecosystems. Sci. China Life Sci. 2023. [Google Scholar]
- Zhang, Q.; Liu, Q.; Liu, S.; Yang, H.; Liu, S.; Zhu, L.; Yang, B.; Jin, J.; Ding, L.; Wang, X.; et al. A New Nodavirus Is Associated with Covert Mortality Disease of Shrimp. J. Gen. Virol. 2014, 95, 2700–2709. [Google Scholar] [CrossRef]
- Liu, S.; Wang, X.; Xu, T.; Li, X.; Du, L.; Zhang, Q. Vectors and Reservoir Hosts of Covert Mortality Nodavirus (CMNV) in Shrimp Ponds. J. Invertebr. Pathol. 2018, 154, 29–36. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, T.; Wan, X.; Liu, S.; Wang, X.; Li, X.; Dong, X.; Yang, B.; Huang, J. Prevalence and Distribution of Covert Mortality Nodavirus (CMNV) in Cultured Crustacean. Virus Res. 2017, 233, 113–119. [Google Scholar] [CrossRef]
- Liu, S.; Xia, J.; Tian, Y.; Yao, L.; Xu, T.; Li, X.; Li, X.; Wang, W.; Kong, J.; Zhang, Q. Investigation of Pathogenic Mechanism of Covert Mortality Nodavirus Infection in Penaeus Vannamei. Front. Microbiol. 2022, 13, 904358. [Google Scholar] [CrossRef]
- Sanfaçon, H.; Wellink, J.; Le Gall, O.; Karasev, A.; van der Vlugt, R.; Wetzel, T. Secoviridae: A Proposed Family of Plant Viruses within the Order Picornavirales That Combines the Families Sequiviridae and Comoviridae, the Unassigned Genera Cheravirus and Sadwavirus, and the Proposed Genus Torradovirus. Arch. Virol. 2009, 154, 899–907. [Google Scholar] [CrossRef]
- Yang, S.; He, Y.; Chen, X.; Kalim, U.; Wang, Y.; Yang, S.; Qi, H.; Cheng, H.; Lu, X.; Wang, X.; et al. Viral Metagenomics Reveals Diverse Viruses in the Feces Samples of Raccoon Dogs. Front. Vet. Sci. 2021, 8, 693564. [Google Scholar] [CrossRef]
- Saccò, M.; White, N.E.; Harrod, C.; Salazar, G.; Aguilar, P.; Cubillos, C.F.; Meredith, K.; Baxter, B.K.; Oren, A.; Anufriieva, E.; et al. Salt to Conserve: A Review on the Ecology and Preservation of Hypersaline Ecosystems. Biol. Rev. 2021, 96, 2828–2850. [Google Scholar] [CrossRef]
- WOAH. Chapter 1.3. Diseases Listed by WOAH. In Aquatic Animal Health Code; WOAH: Paris, France, 2023. [Google Scholar]
- Pica, N.; Bouvier, N.M. Environmental Factors Affecting the Transmission of Respiratory Viruses. Curr. Opin. Virol. 2012, 2, 90–95. [Google Scholar] [CrossRef]
- Barata, C.; Hontoria, F.; Amat, F. Life History, Resting Egg Formation, and Hatching May Explain the Temporal-Geographical Distribution of Artemia Strains in the Mediterranean Basin. Hydrobiologia 1995, 298, 295–305. [Google Scholar] [CrossRef]
- Gong, Z.; Liang, Y.; Wang, M.; Jiang, Y.; Yang, Q.; Xia, J.; Zhou, X.; You, S.; Gao, C.; Wang, J.; et al. Viral Diversity and Its Relationship with Environmental Factors at the Surface and Deep Sea of Prydz Bay, Antarctica. Front. Microbiol. 2018, 9, 2981. [Google Scholar] [CrossRef]
- Umiliana, M.; Sarjito, S.; Desrina, D. Pengaruh Salinitas Terhadap Infeksi Infectious Myonecrosis Virus (Imnv) Pada Udang Vaname Litopenaeus Vannamei (Boone, 1931). J. Aquac. Manag. Technol. 2016, 5, 73–81. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Region | Raw Paired End Reads | Filtered Paired End Reads | Host-Genome Removed Paired End Reads | GC% | Contigs Post-Assembly | Average Contig Length (bp) | Min Contig Length (bp) | Max Contig Length (bp) | No. of Viral Contigs | Percent of Virus Contigs | SRA Accession No. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| North_KZ | 1,968,713 | 1,836,619 (93.3 %) | 1,788,961 (90.9%) | 44.3% | 18,182 | 487 | 206 | 9836 | 44 | 0.24 % | SRR25455641 |
| Pavlodar | 2,121,769 | 1,948,430 (91.8%) | 1,941,957 (91.5%) | 53.0% | 5526 | 468 | 200 | 9067 | 24 | 0.43 % | SRR25455640 |
| Almaty | 2,533,969 | 2,330,154 (91.9%) | 2,313,732 (91.3%) | 48.1% | 5205 | 507 | 200 | 9717 | 18 | 0.34 % | SRR25455639 |
| Kyzylorda | 1,484,062 | 1,371,449 (92.4%) | 1,357,628 (91.5%) | 42.1% | 10,950 | 473 | 200 | 11,780 | 19 | 0.17% | SRR25455638 |
| Mean/(95% CI) | 2,027,128 (1.33–2.72 M) | 1,871,663 | 1,850,570 (1.22–2.48 M) | 46.88 % | 9966 | 484 | 201 | 10,100 | 26 | 0.26 % | - |
| Total | 8,108,513 | 7,486,652 | 7,402,278 | - | 39,863 | - | - | - | 105 | - | - |
| Region | Order | Family | Species | Alignment Length a, (nt) | Contig Identity b, % | Contig Length, (nt) | Number of Contigs | Protein Product |
|---|---|---|---|---|---|---|---|---|
| North_KZ | Reovirales | unclassified Reovirales | Cimodo virus | 109–493 | 71.5–100 | 337–1500 | 26 | hypothetical protein |
| Nodamuvirales | Nodaviridae | Covert mortality nodavirus | 112–204 | 87.5–90.2 | 337–612 | 2 | RdRp | |
| Pavlodar | Reovirales | unclassified Reovirales | Cimodo virus | 106–153 | 69–100 | 302–459 | 14 | hypothetical protein |
| Picornavirales | Dicistroviridae | Cripavirus sp. | 100–308 | 65.6–67.5 | 304–922 | 3 | nonstructural polyprotein | |
| Picornaviridae | Picornaviridae sp. | 107–200 | 72.5–75.7 | 322–605 | 2 | RdRp | ||
| Almaty | Picornavirales | Dicistroviridae | Dicistroviridae sp. | 114–239 | 32.2–55.1 | 364–1626 | 4 | putative replicase |
| Stellavirales | Astroviridae | Microbat bastrovirus | 376 | 37.8 | 1107 | 1 | non-structural polyprotein | |
| Nelson Astrovirus-like 1 | 289 | 47.8 | 879 | 1 | hypothetical protein | |||
| Sobelivirales | Solemoviridae | Solemoviridae sp. | 392 | 44.1 | 4992 | 1 | hypothetical protein | |
| Tolivirales | Tombusviridae | Tombusviridae sp. | 232–679 | 37–41 | 788–6137 | 4 | putative replicase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, M.; Karamendin, K.; Mazhibayeva, Z.; Kassymbekov, Y.; Sabyrzhan, T.; Isbekov, K.; Assylbekova, S.; Kydyrmanov, A. Evaluation of the Viral Diversity of Artemia Cysts from Saline Lakes in Kazakhstan Using Viral Metagenomics Analysis. Fishes 2023, 8, 487. https://doi.org/10.3390/fishes8100487
Kumar M, Karamendin K, Mazhibayeva Z, Kassymbekov Y, Sabyrzhan T, Isbekov K, Assylbekova S, Kydyrmanov A. Evaluation of the Viral Diversity of Artemia Cysts from Saline Lakes in Kazakhstan Using Viral Metagenomics Analysis. Fishes. 2023; 8(10):487. https://doi.org/10.3390/fishes8100487
Chicago/Turabian StyleKumar, Marat, Kobey Karamendin, Zhanara Mazhibayeva, Yermukhammet Kassymbekov, Temirlan Sabyrzhan, Kuanysh Isbekov, Saule Assylbekova, and Aidyn Kydyrmanov. 2023. "Evaluation of the Viral Diversity of Artemia Cysts from Saline Lakes in Kazakhstan Using Viral Metagenomics Analysis" Fishes 8, no. 10: 487. https://doi.org/10.3390/fishes8100487
APA StyleKumar, M., Karamendin, K., Mazhibayeva, Z., Kassymbekov, Y., Sabyrzhan, T., Isbekov, K., Assylbekova, S., & Kydyrmanov, A. (2023). Evaluation of the Viral Diversity of Artemia Cysts from Saline Lakes in Kazakhstan Using Viral Metagenomics Analysis. Fishes, 8(10), 487. https://doi.org/10.3390/fishes8100487