
Transcriptome Analysis of the Immune Process of Golden Pompano (Trachinotus ovatus) Infected with Streptococcus agalactiae

 ,
,  , ,
, , 
Abstract

1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Fish, Bacterial Challenge, and Sampling
2.3. RNA Extraction and Sequencing
2.4. Differential Gene Expression Analysis
2.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analyses
2.6. Verification Result via qPCR
2.7. Statistical Analysis
3. Results
3.1. Transcriptome Sequencing and Raw Read Data Analysis
3.2. DEG Analysis during S. agalactiae Infection Stress
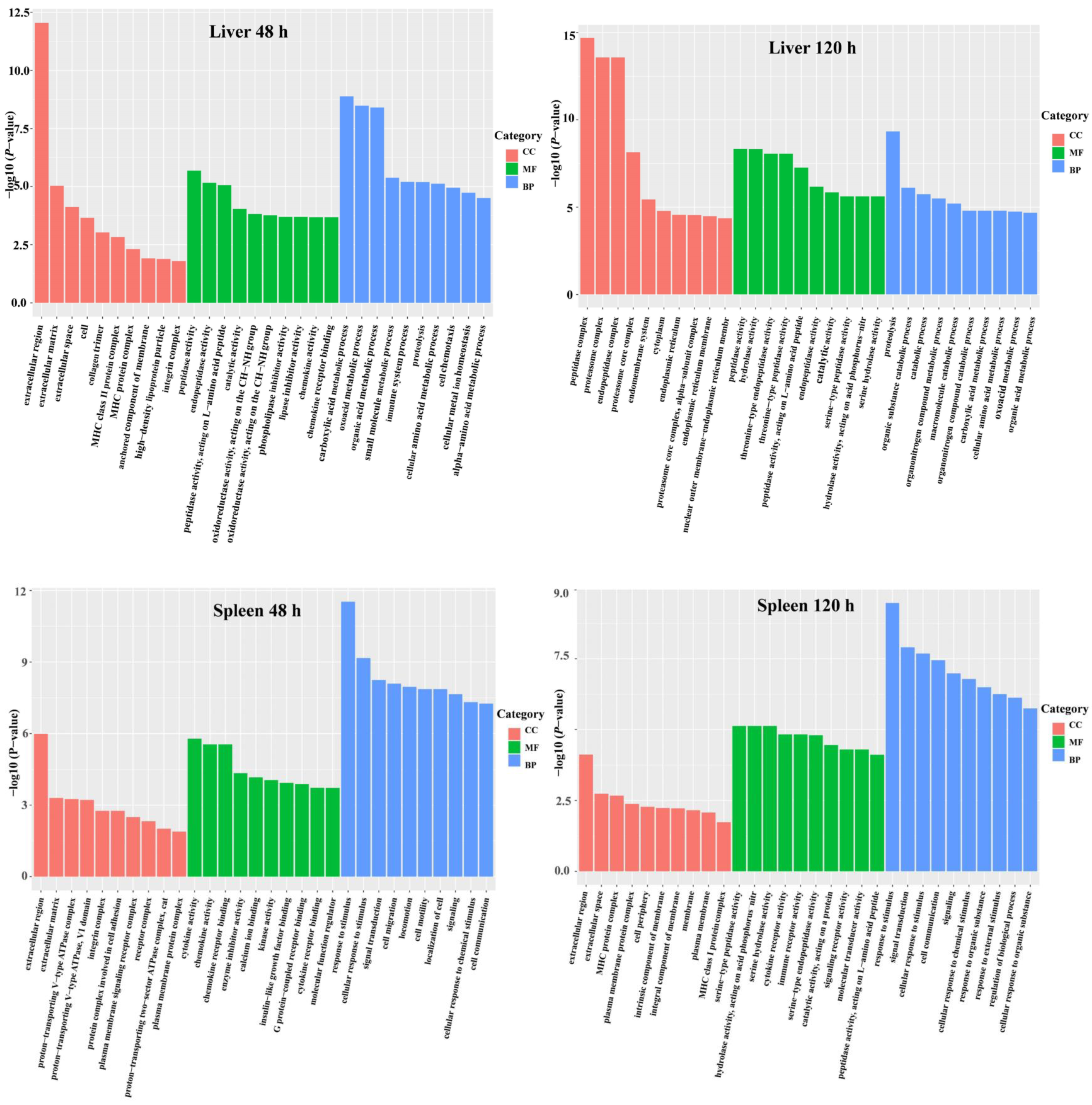
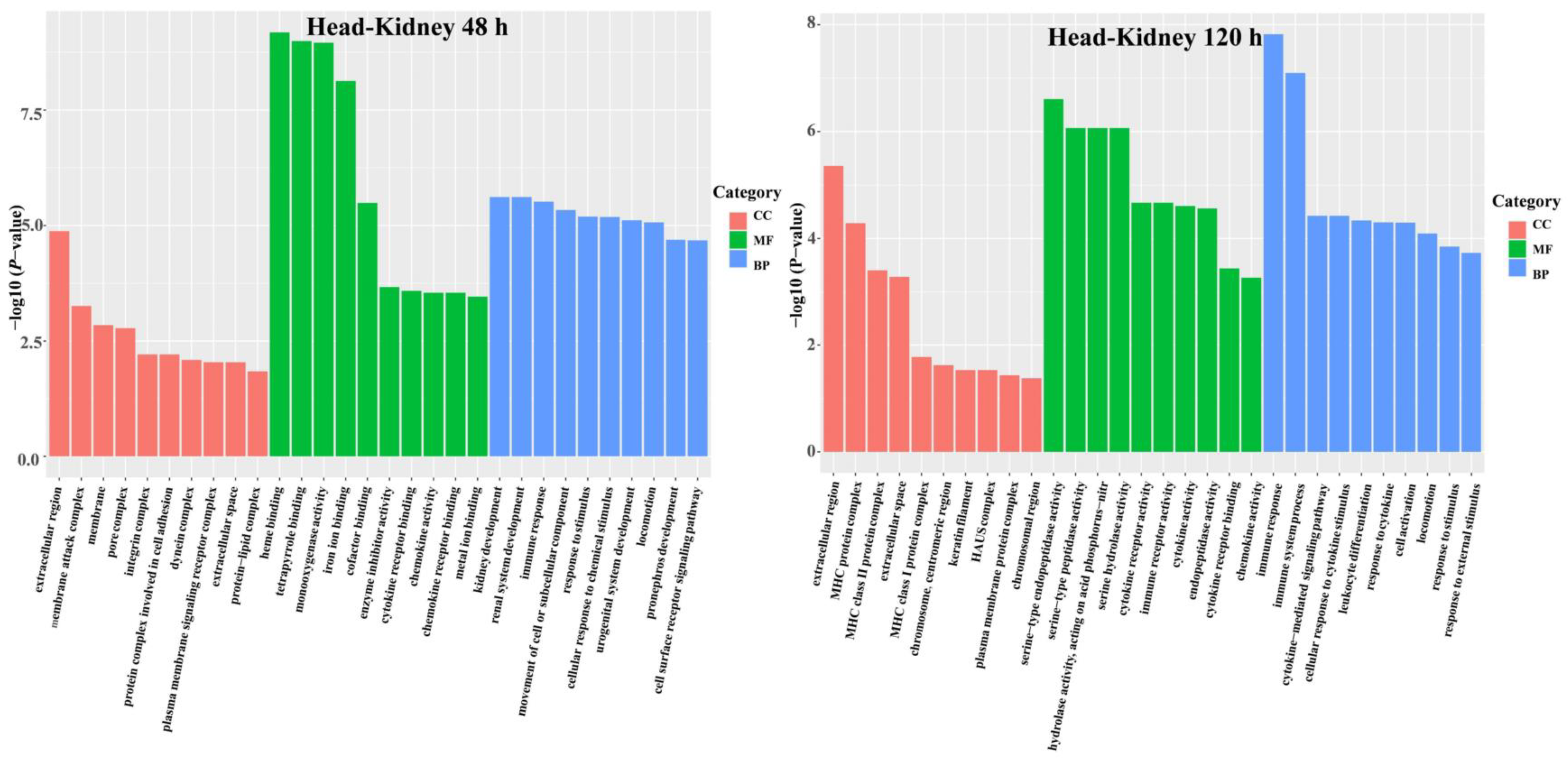
3.3. DEG Functional Analysis
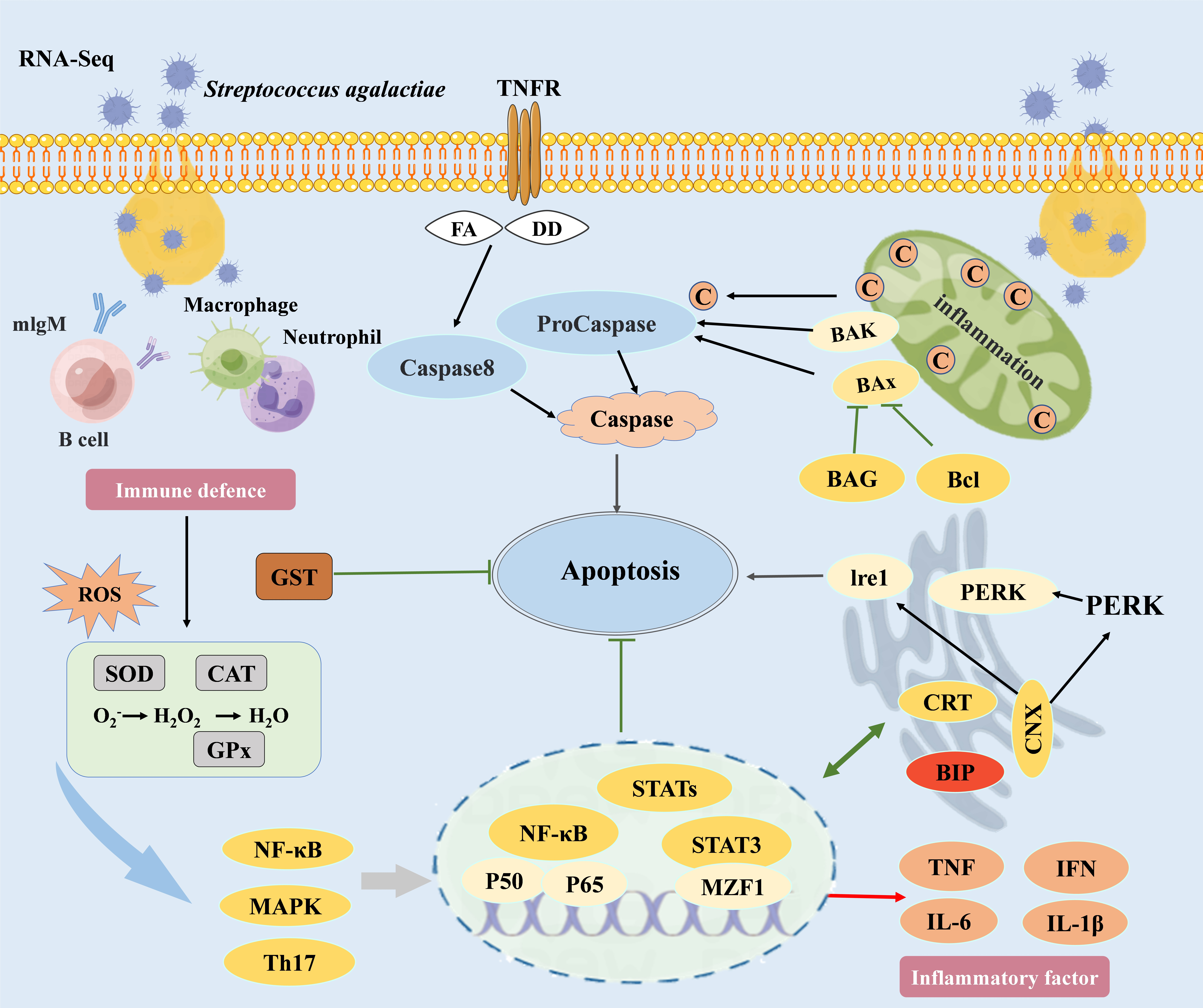
3.4. Inflammation-Related Genes against S. agalactiae Infection
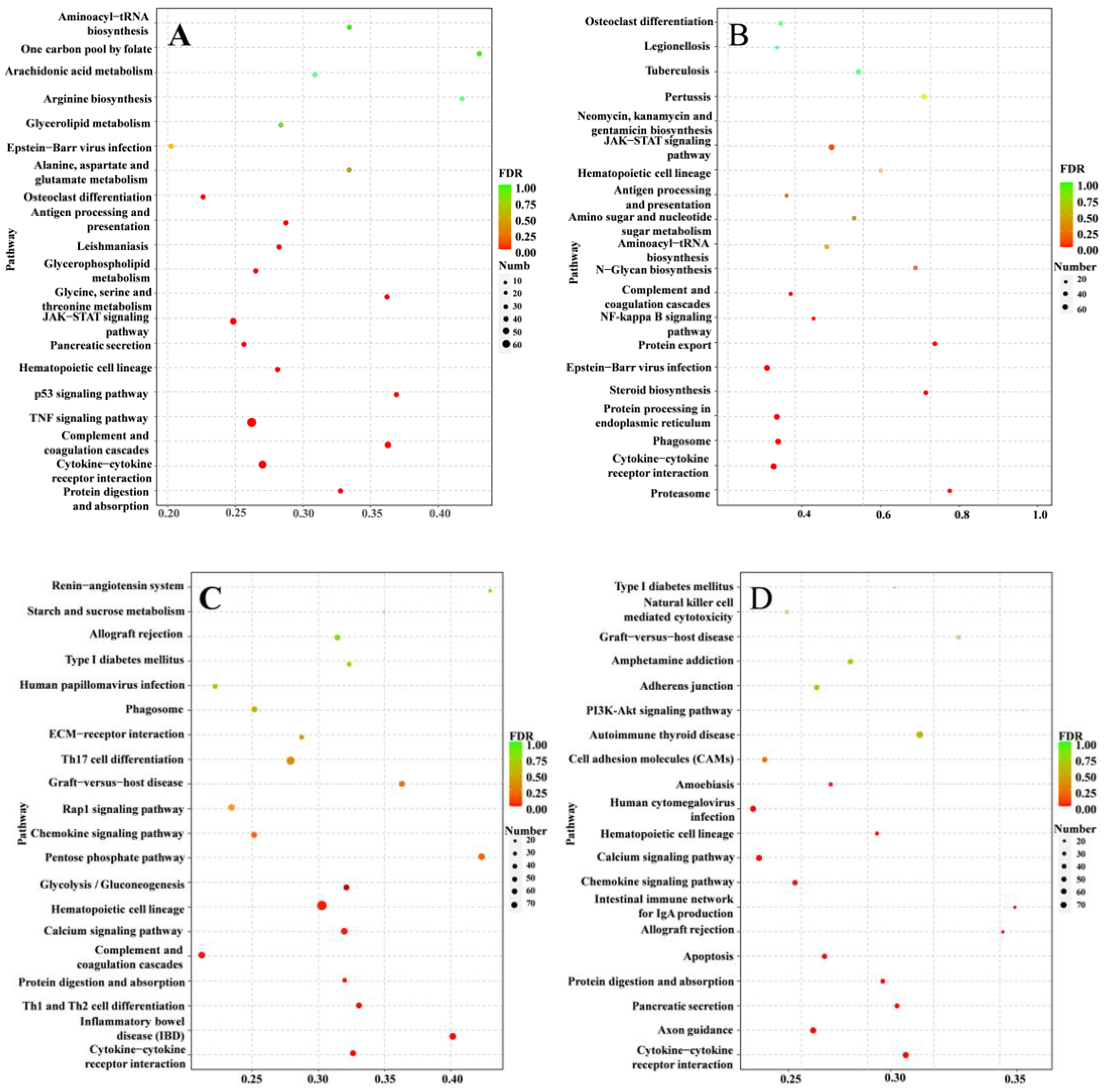
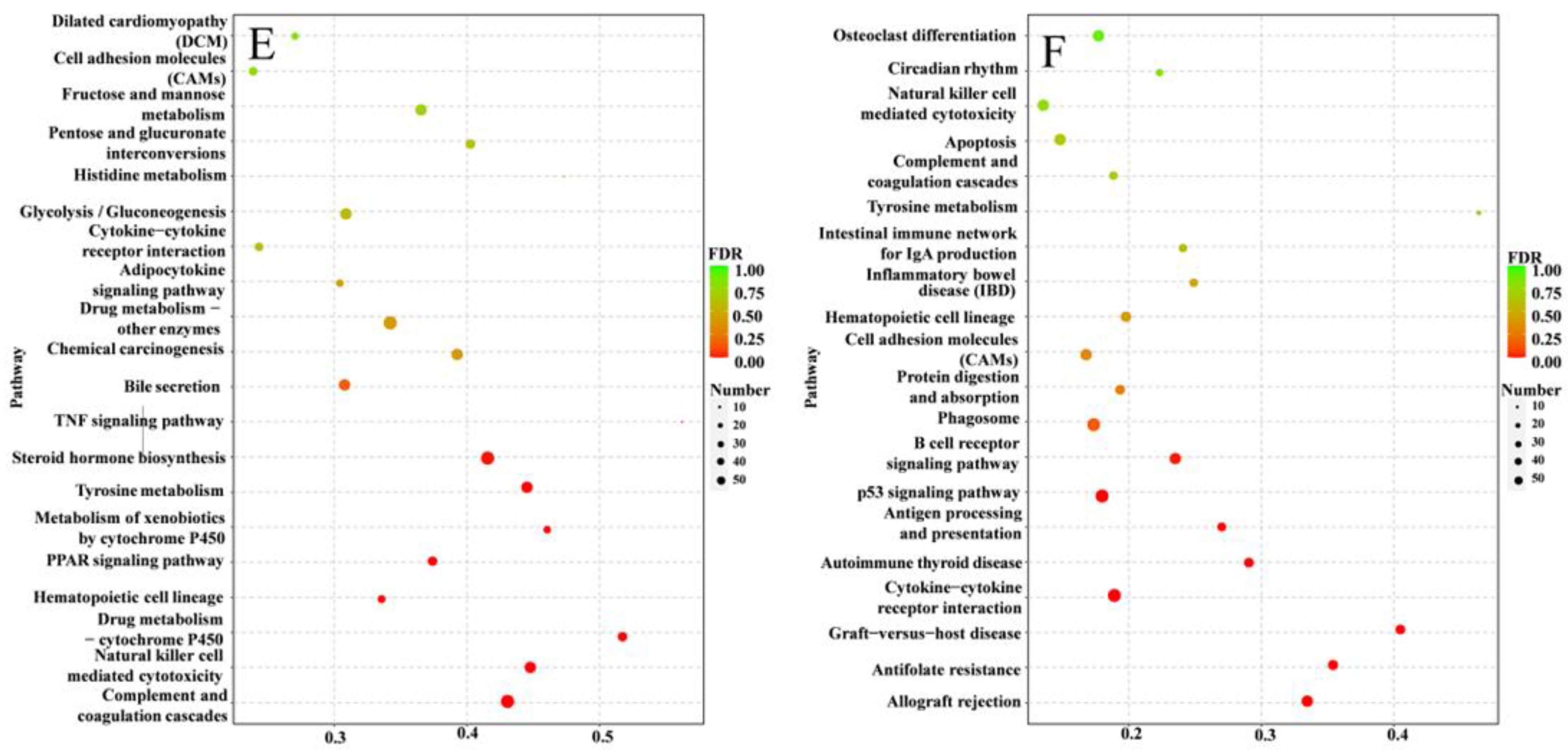
3.5. Inflammation-Related Pathways against S. agalactiae Infection
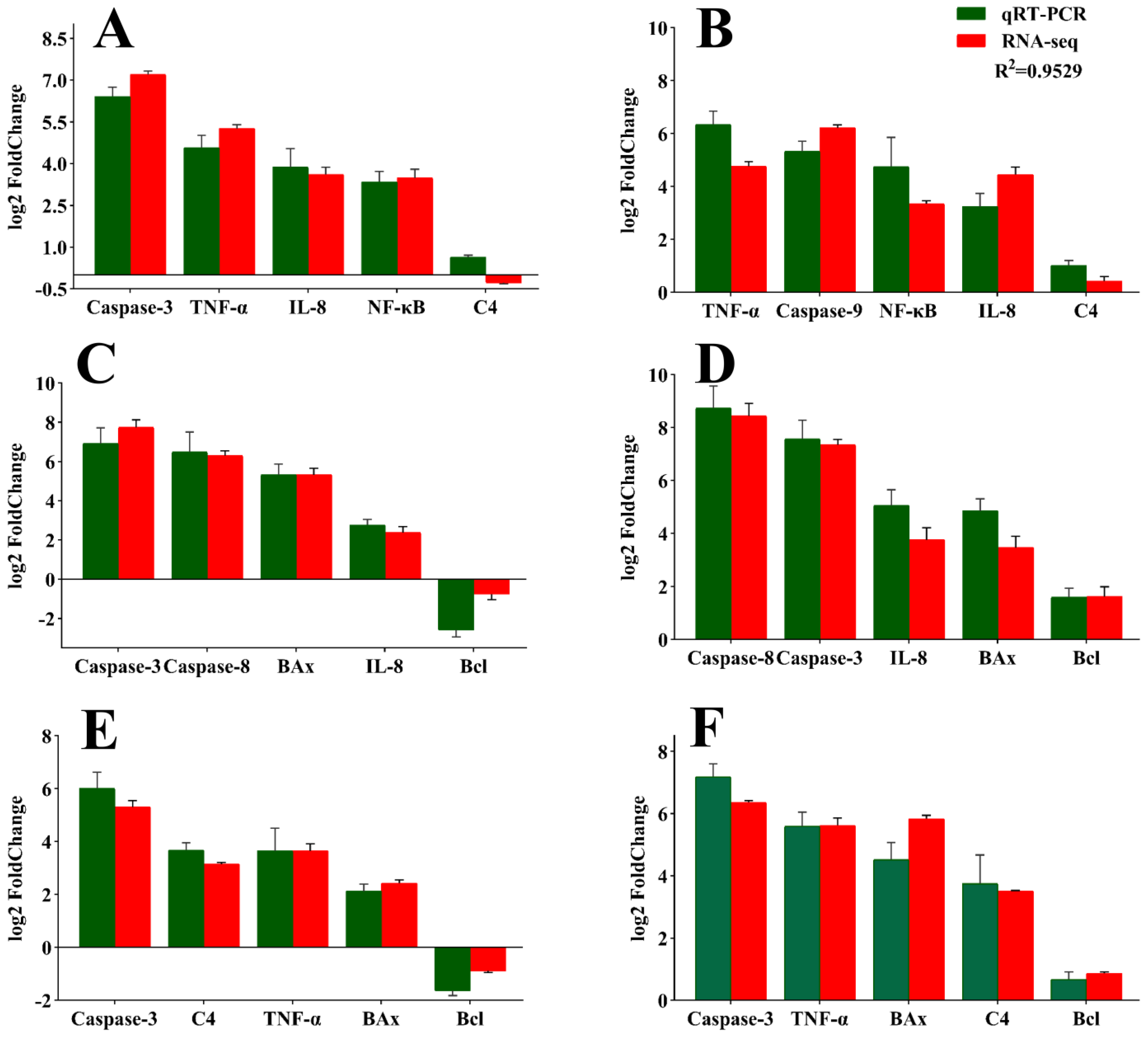
3.6. Validation of RNA Sequencing Data Results via qPCR
4. Discussion
4.1. Dynamic Immune Response of Golden Pompano against S. agalactiae
4.2. Changes in the Expression of Inflammation-Related DEGs in S. agalactiae Infection
4.3. Changes in the Expression of the JAK-STAT Signaling Pathway against S. agalactiae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, M.J.; Guo, H.Y.; Liu, B.; Zhu, K.C.; Guo, L.; Liu, B.S.; Zhang, N.; Yang, J.W.; Jiang, S.G.; Zhang, D.C. Gill oxidative damage caused by acute ammonia stress was reduced through the HIF-1α/NF-κb signaling pathway in golden pompano (Trachinotus ovatus). Ecotoxicol. Environ. Saf. 2021, 222, 112504. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Mo, Z.; Wang, Z.; Xu, J.; Li, Y.; Dan, X.; Li, A. Isolation and pathogenicity of Streptococcus iniae in offshore cage-cultured Trachinotus ovatus in China. Aquaculture 2018, 492, 247–252. [Google Scholar] [CrossRef]
- Qian, X.; Ba, Y.; Zhuang, Q.; Zhong, G. RNA-Seq technology and its application in fish transcriptomics. OMICS 2014, 18, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Angelini, C.; De Feis, I.; Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. J. BioMed. Biotechnol. 2010, 2010, 853–916. [Google Scholar] [CrossRef]
- Romero, A.; Saraceni, P.R.; Pereiro, P.; Tomás, J.; Merino, S.; Figueras, A.; Novoa, B. Development of a zebrafish larvae infection model to study virulence factors of A. hydrophila. Fish Shellfish. Immunol. 2016, 55, 85–86. [Google Scholar] [CrossRef]
- Maekawa, S.; Wang, P.C.; Chen, S.C. Comparative study of immune reaction against bacterial infection from transcriptome analysis. Front. Immunol. 2019, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Yoon, D.; Lim, Y.; Roh, H.J.; Kim, S.; Park, C.I.; Kim, H.S.; Cha, H.J.; Choi, Y.H.; Kim, D.H. Co-expression network analysis of spleen transcriptome in rock bream (Oplegnathus fasciatus) naturally infected with rock bream iridovirus (RBIV). Int. J. Mol. Sci. 2020, 21, 1707. [Google Scholar] [CrossRef]
- Xu, D.; Han, P.; Xia, L.; Gan, J.; Xu, Q. A comparative transcriptome analysis focusing on immune responses of Asian swamp eel following infection with Aeromonas hydrophila. Aquaculture 2021, 539, 736655. [Google Scholar] [CrossRef]
- Song, L.; Dong, X.; Hu, G. Transcriptome analysis of red sea bream (Pagrus major) head kidney and spleen infected by Vibrio anguillarum. Aquac. Rep. 2021, 21, 100789. [Google Scholar] [CrossRef]
- Han, C.; Li, Q.; Chen, Q.; Zhou, G.; Huang, J.; Zhang, Y. Transcriptome analysis of the spleen provides insight into the immunoregulation of Mastacembelus armatus under Aeromonas veronii infection. Fish Shellfish. Immunol. 2019, 88, 272–283. [Google Scholar] [CrossRef]
- Long, M.; Zhao, J.; Li, T.; Tafalla, C.; Zhang, Q.; Wang, X.; Gong, X.; Shen, Z.; Li, A. Transcriptomic and proteomic analyses of splenic immune mechanisms of rainbow trout (Oncorhynchus mykiss) infected by Aeromonas salmonicida subsp. salmonicida. J. Proteom. 2015, 122, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, X.; Xu, J.; Pei, X.; Wu, Z.; Wang, T.; Yin, S. iTRAQ analysis of liver immune-related proteins from darkbarbel catfish (Pelteobagrus vachelli) infected with Edwardsiella ictaluri. Fish Shellfish. Immunol. 2019, 87, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Mugimba, K.K.; Byarugaba, D.K.; Mutoloki, S.Ø.; Evensen, H.M.; Munang’Andu, H.M. Challenges and solutions to viral diseases of finfish in Marine Aquaculture. Pathogens 2021, 10, 673. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.K.; Paoletti, L.C.; Glaser, P.; Dua, M.; Sharma, P.K.; Grandi, G.; Rappuoli, R. Group B Streptococcus: Global incidence and vaccine development. Nat. Rev. Microbiol. 2006, 4, 932–942. [Google Scholar] [CrossRef]
- Lee, I.P.A.; Andam, C.P. Frequencies and characteristics of genome-wide recombination in Streptococcus agalactiae, Streptococcus pyogenes, and Streptococcus suis. Sci. Rep. 2022, 12, 1515. [Google Scholar] [CrossRef]
- Zeng, Y.F.; Zeng, Y.F.; Zeng, Y.F.; Zeng, Y.F.; Chen, C.M.; Chen, C.M.; Li, X.Y.; Chen, J.J.; Chen, J.J.; Chen, J.J.; et al. Development of a droplet digital PCR method for detection of Streptococcus agalactiae. BMC Microbiol. 2020, 20, 179. [Google Scholar] [CrossRef]
- Causey, D.R.; Pohl MA, N.; Stead, D.A.; Martin SA, M.; Secombes, C.J.; Macqueen, D.J. High-throughput proteomic profiling of the fish liver following bacterial infection. BMC Genom. 2018, 19, 717. [Google Scholar] [CrossRef]
- Huang, J.; Chen, W.; Wang, Q.; Zhang, Y.; Liu, Q.; Yang, D. Iso-Seq assembly and functional annotation of full-length transcriptome of turbot (Scophthalmus maximus) during bacterial infection. Mar. Genom. 2022, 63, 100954. [Google Scholar] [CrossRef]
- Ding, F.F.; Li, C.H.; Chen, J. Molecular characterization of the NK-lysin in a teleost fish, Boleophthalmus pectinirostris: Antimicrobial activity and immunomodulatory activity on monocytes/macrophages. Fish Shellfish. Immunol. 2019, 92, 256–264. [Google Scholar] [CrossRef]
- Ye, H.; Xiao, S.; Wang, X.; Wang, Z.; Zhang, Z.; Zhu, C.; Hu, B.; Lv, C.; Zheng, S.; Luo, H. Characterization of Spleen Transcriptome of Schizothorax prenanti during Aeromonas hydrophila Infection. Mar. Biotechnol. 2018, 20, 246–256. [Google Scholar] [CrossRef]
- Roubach, R.; Gomes, L.C.; Leão Fonseca, F.A.; Val, A.L. Eugenol as an efficacious anaesthetic for tambaqui, Colossoma macropomum (Cuvier). Aquac. Res. 2005, 36, 1056–1061. [Google Scholar] [CrossRef]
- Loureiro, S.; Amorim, A.; Cainé, L.; Silva, B.; Gomes, I. Evaluation of two DNA/RNA co-extraction methods for organism fluid identification in forensics. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 250–252. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Liu, M.J.; Guo, H.Y.; Zhu, K.C.; Liu, B.S.; Liu, B.; Guo, L.; Zhang, N.; Yang, J.W.; Jiang, S.G.; Zhang, D.C. Effects of acute ammonia exposure and recovery on the antioxidant response and expression of genes in the Nrf2-Keap1 signaling pathway in the juvenile golden pompano (Trachinotus ovatus). Aquat. Toxicol. 2021, 240, 105969. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Luo, G.; Sun, Y.; Huang, L.; Su, Y.; Zhao, L.; Qin, Y.; Xu, X.; Yan, Q. Time-resolved dual RNA-seq of tissue uncovers Pseudomonas plecoglossicida key virulence genes in host-pathogen interaction with Epinephelus coioides. Environ. Microbiol. 2020, 22, 677–693. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liu, M.J.; Guo, H.Y.; Zhu, K.C.; Liu, B.; Liu, B.S.; Zhang, N.; Zhang, D.C. ROS Induced by Streptococcus agalactiae Activate Inflammatory Responses via the TNF-α/NF-κB Signaling Pathway in Golden Pompano Trachinotus ovatus (Linnaeus, 1758). Antioxidants 2022, 11, 1809. [Google Scholar] [CrossRef]
- Gates, K.; Sandoval-Castillo, J.; Bernatchez, L.; Beheregaray, L.B. De novo transcriptome assembly and annotation for the desert rainbowfish (Melanotaenia splendida tatei) with comparison with candidate genes for future climates. Mar. Genom. 2017, 35, 63–68. [Google Scholar] [CrossRef]
- Chou, C.H.; Huang, H.Y.; Huang, W.C.; Hsu S da Hsiao C der Liu, C.Y.; Chen, Y.H.; Liu, Y.C.; Huang, W.Y.; Lee, M.L.; Chen, Y.C.; Huang, H.D. The aquatic animals’ transcriptome resource for comparative functional analysis. BMC Genom. 2018, 19, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Mola, S.; Foisy, S.; Boucher, G.; Major, F.; Beauchamp, C.; Karaky, M.; Goyette, P.; Lesage, S.; Rioux, J.D. A transcriptome-based approach to identify functional modules within and across primary human immune cells. PLoS ONE 2020, 15, e0233543. [Google Scholar] [CrossRef]
- Li, B.J.; Jiang, D.L.; Meng, Z.N.; Zhang, Y.; Zhu, Z.X.; Lin, H.R.; Xia, J.H. Genome-wide identification and differentially expression analysis of lncRNAs in tilapia 06 Biological Sciences 0604 Genetics. BMC Genom. 2018, 19, 2–12. [Google Scholar] [CrossRef]
- Anitha, A.; Gupta, Y.R.; Deepa, S.; Ningappa, M.; Rajanna, K.B.; Senthilkumaran, B. Gonadal transcriptome analysis of the common carp, Cyprinus carpio: Identification of differentially expressed genes and SSRs. Gen. Comp. Endocrinol. 2019, 279, 67–77. [Google Scholar] [CrossRef]
- Wu, X.M.; Cao, L.; Hu, Y.W.; Chang, M.X. Transcriptomic characterization of adult zebrafish infected with Streptococcus agalactiae. Fish Shellfish. Immunol. 2019, 94, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, G.R.; Ji, W.; Shi, Z.C.; Ma, X.F.; Luo, Z.L.; Wei, K.J. The Dynamic Immune Response of Yellow Catfish (Pelteobagrus fulvidraco) Infected With Edwardsiella ictaluri Presenting the Inflammation Process. Front. Immunol. 2021, 12, 625928. [Google Scholar] [CrossRef]
- Schneebauer, G.; Dirks, R.P.; Pelster, B. Anguillicola crassus infection affects mRNA expression levels in gas gland tissue of European yellow and silver eel. PLoS ONE 2017, 12, e0183128. [Google Scholar] [CrossRef]
- Li, C.; Jiang, J.; Xie, J.; Yang, W.; Wang, Y. Transcriptome profiling and differential expression analysis of the immune-related genes during the acute phase of infection with Mycobacterium marinum in the goldfish (Carassius auratus L.). Aquaculture 2021, 533, 736198. [Google Scholar] [CrossRef]
- Hossini, A.M.; Quast, A.S.; Plötz, M.; Grauel, K.; Exner, T.; Küchler, J.; Stachelscheid, H.; Eberle, J.; Rabien, A.; Makrantonaki, E.; et al. PI3K/AKT signaling pathway is essential for survival of induced pluripotent stem cells. PLoS ONE 2016, 11, e0154770. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M. Dancing to the tune of chemokines. Nat. Immunol. 2001, 2, 129–134. [Google Scholar] [CrossRef]
- Jiang, Y.; Feng, S.; Zhang, S.; Liu, H.; Feng, J.; Mu, X.; Sun, X.; Xu, P. Transcriptome signatures in common carp spleen in response to Aeromonas hydrophila infection. Fish Shellfish. Immunol. 2016, 57, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and non-canonical aspects of JAK-STAT signaling: Lessons from interferons for cytokine responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Ezeonwumelu, I.J.; Garcia-Vidal, E.; Ballana, E. JAK-STAT pathway: A novel target to tackle viral infections. Viruses 2021, 13, 2379. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Hulpiau, P.; Saeys, Y.; Bertrand MJ, M.; Vandenabeele, P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol. 2016, 26, 721–732. [Google Scholar] [CrossRef]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef]
- Chazaud, B. Inflammation and Skeletal Muscle Regeneration: Leave It to the Macrophages! Trends Immunol. 2020, 41, 481–492. [Google Scholar] [CrossRef]
- Pérez-Figueroa, E.; Álvarez-Carrasco, P.; Ortega, E.; Maldonado-Bernal, C. Neutrophils: Many Ways to Die. Front. Immunol. 2021, 12, 631821. [Google Scholar] [CrossRef]
- Ribeiro, A.B.; de Barcellos-Filho PC, G.; Franci, C.R.; Menescal-de-Oliveira, L.; Saia, R.S. Pro-inflammatory cytokines, IL-1β and TNF-α, produce persistent compromise in tonic immobility defensive behaviour in endotoxemia guinea-pigs. Acta Physiol. 2016, 218, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.; Nguyen, D.H.; Satkunendrarajah, K.; Branch, D.R.; Fehlings, M.G. Evaluating the role of IL-11, a novel cytokine in the IL-6 family, in a mouse model of spinal cord injury. J. Neuroinflam. 2012, 9, 2–12. Available online: http://www.jneuroinflammation.com/content/9/1/134 (accessed on 5 October 2022). [CrossRef] [PubMed]
- Tang, W.; Geba, G.P.; Zheng, T.; Ray, P.; Homer, R.J.; Kuhn, C.; Flavell, R.A.; Elias, J.A. Targeted expression of IL-11 in the murine airway causes lymphocytic inflammation, bronchial remodeling, and airways obstruction. J. Clin. Investig. 1996, 98, 2845–2853. [Google Scholar] [CrossRef]
- Tie, H.M.; Jiang, W.D.; Feng, L.; Wu, P.; Liu, Y.; Kuang, S.Y.; Tang, L.; Zhou, X.Q. Dietary nucleotides in the diets of on-growing grass carp (Ctenopharyngodon idella) suppress Aeromonas hydrophila induced intestinal inflammation and enhance intestinal disease-resistance via NF-κB and TOR signaling. Aquaculture 2021, 533, 736075. [Google Scholar] [CrossRef]
- Chou, F.C.; Chen, H.Y.; Chen, H.H.; Lin, G.J.; Lin, S.H.; Sytwu, H.K. Differential modulation of IL-12 family cytokines in autoimmune islet graft failure in mice. Diabetologia 2017, 60, 2409–2417. [Google Scholar] [CrossRef]
- Nadeem, A.; Al-Harbi, N.O.; Alfardan, A.S.; Ahmad, S.F.; AlAsmari, A.F.; Al-Harbi, M.M. IL-17A-induced neutrophilic airway inflammation is mediated by oxidant-antioxidant imbalance and inflammatory cytokines in mice. Biomed. Pharmacother. 2018, 107, 1196–1204. [Google Scholar] [CrossRef]
- Laing, K.J.; Secombes, C.J. Chemokines. Dev. Comp. Immunol. 2004, 28, 443–460. [Google Scholar] [CrossRef]
- Korbecki, J.; Kojder, K.; Barczak, K.; Simińska, D.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Hypoxia alters the expression of CC chemokines and cc chemokine receptors in a tumor–a literature review. Int. J. Mol. Sci. 2020, 21, 5647. [Google Scholar] [CrossRef]
- Tian, C.; Chen, Y.; Ao, J.; Chen, X. Molecular characterization and bioactivity of a CXCL13 chemokine in large yellow croaker Pseudosciaena crocea. Fish Shellfish. Immunol. 2010, 28, 445–452. [Google Scholar] [CrossRef]
- Hong, S.; Li, R.; Xu, Q.; Secombes, C.J.; Wang, T. Two types of TNF-alpha exist in teleost fish: Phylogeny, expression, and bioactivity analysis of type-II TNF-alpha3 in rainbow trout Oncorhynchus mykiss. J. Immunol. 2013, 191, 5959–5972. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ao, J.; Ai, C.; Chen, X. Molecular and functional identification of three interleukin-17A/F (IL-17A/F) homologues in large yellow croaker (Larimichthys crocea). Dev. Comp. Immunol. 2016, 55, 221–232. [Google Scholar] [CrossRef]
- Zou, J.; Secombes, C.J. The function of fish cytokines. Biology 2016, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Robinson, M.W.; Roche, G.; Biron, C.A.; O’Farrelly, C.; Ryan, E.J. Immune Cell Profiling of IFN-λ Response Shows pDCs Express Highest Level of IFN-λR1 and Are Directly Responsive via the JAK-STAT Pathway. J. Interferon Cytokine Res. 2016, 36, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Liongue, C.; Sertori, R.; Ward, A.C. Evolution of Cytokine Receptor Signaling. J. Immunol. 2016, 197, 11–18. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Miyagi, T.; Gil, M.P.; Wang, X.; Louten, J.; Chu, W.M.; Biron, C.A. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 2007, 204, 2383–2396. [Google Scholar] [CrossRef]
- Yao, K.Z.; Chen, Q.; Wu, Y.; Liu, F.; Chen, X.; Zhang, Y. Unphosphorylated STAT1 represses apoptosis in macrophages during Mycobacterium Tuberculosis infection. J. Cell Sci. 2017, 130, 1740–1751. [Google Scholar] [CrossRef]
- Nakayamada, S.; Poholek, A.C.; Lu, K.T.; Takahashi, H.; Kato, M.; Iwata, S.; Hirahara, K.; Cannons, J.L.; Schwartzberg, P.L.; Vahedi, G.; et al. Type I IFN Induces Binding of STAT1 to Bcl6: Divergent Roles of STAT Family Transcription Factors in the T Follicular Helper Cell Genetic Program. J. Immunol. 2014, 192, 2156–2166. [Google Scholar] [CrossRef]
- Miyawaki, A.; Iizuka, Y.; Sugino, H.; Watanabe, Y. IL-11 prevents IFN-γ-induced hepatocyte death through selective downregulation of IFN-γ/STAT1 signaling and ROS scavenging. PLoS ONE 2019, 14, e0211123. [Google Scholar] [CrossRef]
- Pfitzner, E.; Kliem, S.; Baus, D.; Litterst, C.M. The Role of STATs in Inflammation and Inflammatory Diseases. Curr. Pharm. Des. 2004, 10, 2839–2850. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Gibson, S.A.; Buckley, J.A.; Qin, H.; Benveniste, E.N. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin. Immunol. 2018, 189, 4–13. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequences (5′−3′) | Amplification Target |
|---|---|---|
| Caspase-3-F | GCTGCTCTACTGCTTCTGCCTGATG | qRT-PCR |
| Caspase-3-R | TGGCTGAGGATTGTGATGTTGCTG | |
| Caspase-8-F | GCAACAAAACAGCCATCCA | qRT-PCR |
| Caspase-8-R | GCAGGGGTAAAGGGTCATT | |
| TNF-α-F | GCTCCTCACCCACACCATCA | qRT-PCR |
| TNF-α-R | CCAAAGTAGACCTGCCCAGACT | |
| NF-κB-F | CGTGAGGTCAGCGAGCCAATG | qRT-PCR |
| NF-κB-R | ATGTGCCGTCTATCTTGTGGAATGG | |
| 1L-8-F | TGCATCACCACGGTGAAAAA | qRT-PCR |
| 1L-8-R | GCATCAGGGTCCAGACAAATC | |
| C4-F | TGGAGAAAAAGTTAAAGGGGC | qRT-PCR |
| C4-R | CAGGAAGGAAGTATGAGCGAGT | |
| Bax-F | ACTGAGCAGGAGCAGAAGGT | qRT-PCR |
| Bax-R | CTGAATCCAGCCCAAGAGA | |
| Bcl-F | GGACATCGTGGAGGATTACC | qRT-PCR |
| Bcl-R | GACCGTCATTAGGCTTTGC |
| Time | Liver | Spleen | Head Kidney | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Upregulated (%) | Downregulated (%) | Total | Upregulated (%) | Downregulated (%) | Total | Upregulated (%) | Downregulated (%) | Total | |
| 48 h | 1655 (67.28) | 805 (32.72) | 2460 | 1362 (43.40) | 1776 (56.60) | 3138 | 1203 (33.85) | 2351 (66.15) | 3554 |
| 120 h | 2059 (57.18) | 1542 (42.82) | 3601 | 2195 (47.54) | 2422 (52.46) | 4617 | 1026 (63.22) | 597 (36.78) | 1623 |
| Total | 3714 | 2347 | 6061 | 3557 | 4198 | 7775 | 2229 | 2948 | 5177 |
| Time | Liver | Spleen | Head Kidney | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Total DEGs | GO-Annotated DEGs | KEGG-Annotated DEGs | Total DEGs | GO-Annotated DEGs | KEGG-Annotated DEGs | Total DEGs | GO-Annotated DEGs | KEGG-Annotated DEGs | |
| 48 h | 323,646 | 39,011 | 6320 | 326,269 | 54,618 | 6445 | 326,546 | 54,228 | 6285 |
| 120 h | 327,857 | 59,650 | 4979 | 326,592 | 47,559 | 6121 | 316,992 | 24,039 | 3155 |
| Total | 651,503 | 98,661 | 11,299 | 652,861 | 102,177 | 12,566 | 643,538 | 78,267 | 9440 |
| Ko id | Term | Number of DEGs | p-Value |
|---|---|---|---|
| ko04630 | JAK-STAT signaling pathway | 77 | 0.000150446 |
| ko04064 | NF-kappa B signaling pathway | 72 | 0.000123683 |
| ko04060 | Cytokine–cytokine receptor interaction | 63 | 2.63 × 10−10 |
| ko04668 | TNF signaling pathway | 52 | 3.31 × 10−8 |
| ko04062 | Chemokine signaling pathway | 52 | 9.47 × 10−5 |
| ko04659 | Th17 cell differentiation | 35 | 0.000938016 |
| ko04610 | Complement and coagulation cascades | 33 | 1.90 × 10−9 |
| ko04010 | MAPK signaling pathway | 31 | 0.006077547 |
| ko04612 | Antigen processing and presentation | 29 | 4.36 × 10−5 |
| ko04658 | Th1 and Th2 cell differentiation | 26 | 0.004317579 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Guo, H.-Y.; Liu, M.-J.; Zhu, K.-C.; Liu, B.; Liu, B.-S.; Zhang, N.; Jiang, S.-G.; Zhang, D.-C. Transcriptome Analysis of the Immune Process of Golden Pompano (Trachinotus ovatus) Infected with Streptococcus agalactiae. Fishes 2023, 8, 52. https://doi.org/10.3390/fishes8010052
Gao J, Guo H-Y, Liu M-J, Zhu K-C, Liu B, Liu B-S, Zhang N, Jiang S-G, Zhang D-C. Transcriptome Analysis of the Immune Process of Golden Pompano (Trachinotus ovatus) Infected with Streptococcus agalactiae. Fishes. 2023; 8(1):52. https://doi.org/10.3390/fishes8010052
Chicago/Turabian StyleGao, Jie, Hua-Yang Guo, Ming-Jian Liu, Ke-Cheng Zhu, Bo Liu, Bao-Suo Liu, Nan Zhang, Shi-Gui Jiang, and Dian-Chang Zhang. 2023. "Transcriptome Analysis of the Immune Process of Golden Pompano (Trachinotus ovatus) Infected with Streptococcus agalactiae" Fishes 8, no. 1: 52. https://doi.org/10.3390/fishes8010052
APA StyleGao, J., Guo, H.-Y., Liu, M.-J., Zhu, K.-C., Liu, B., Liu, B.-S., Zhang, N., Jiang, S.-G., & Zhang, D.-C. (2023). Transcriptome Analysis of the Immune Process of Golden Pompano (Trachinotus ovatus) Infected with Streptococcus agalactiae. Fishes, 8(1), 52. https://doi.org/10.3390/fishes8010052