
The Complete Mitochondrial Genome of Hyotissa hyotis (Bivalvia: Gryphaeidae) Reveals a Unique Gene Order within Ostreoidea





Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and DNA Extraction
2.2. Illumina Sequencing and Mitogenome Assembly
2.3. Mitogenome Annotation and Sequence Analysis
2.4. Gene Order Comparisons
2.5. Phylogenetic Analysis
3. Results and Discussion
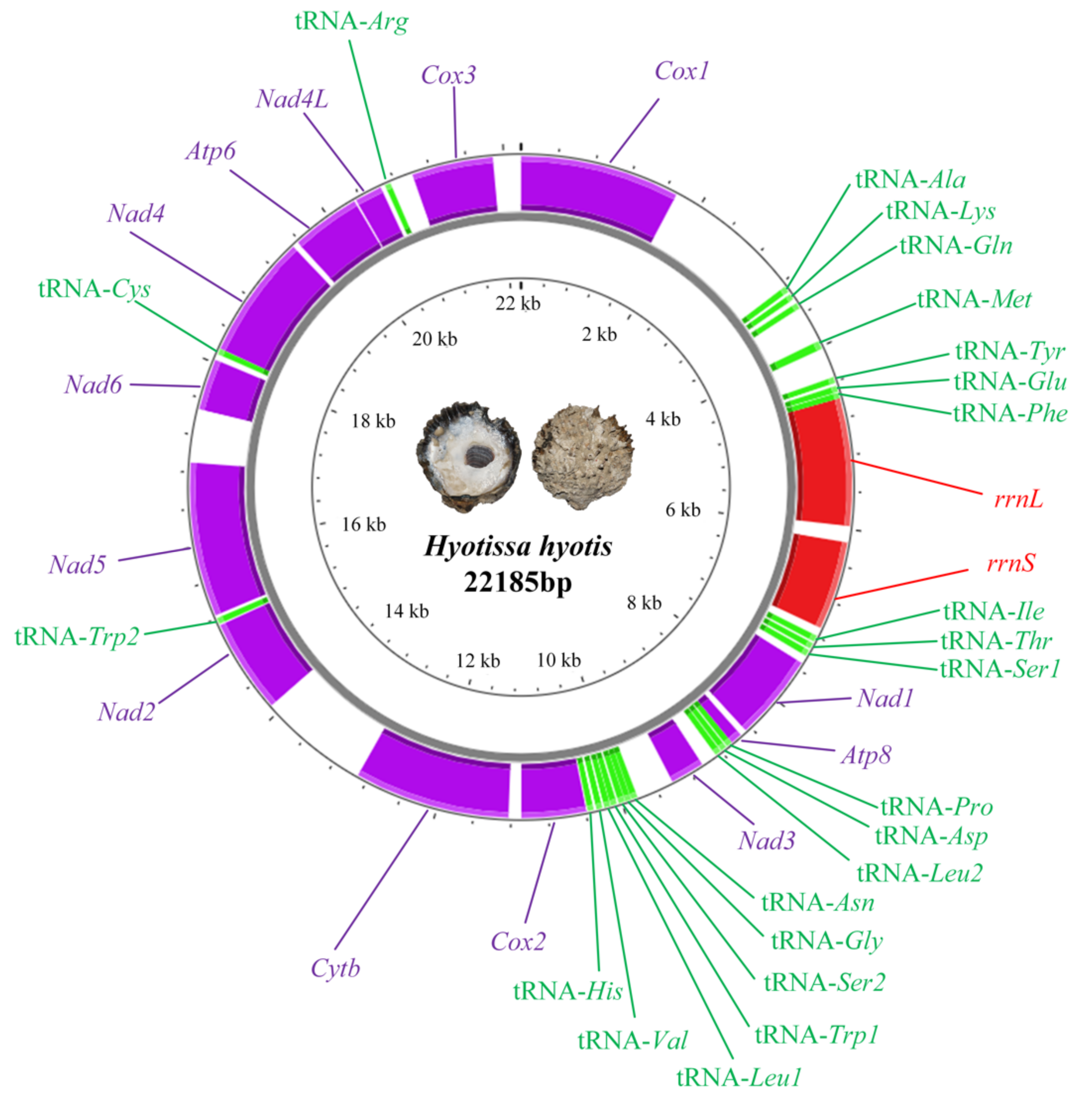
3.1. Genome Organization of Hyotissa Hyotis
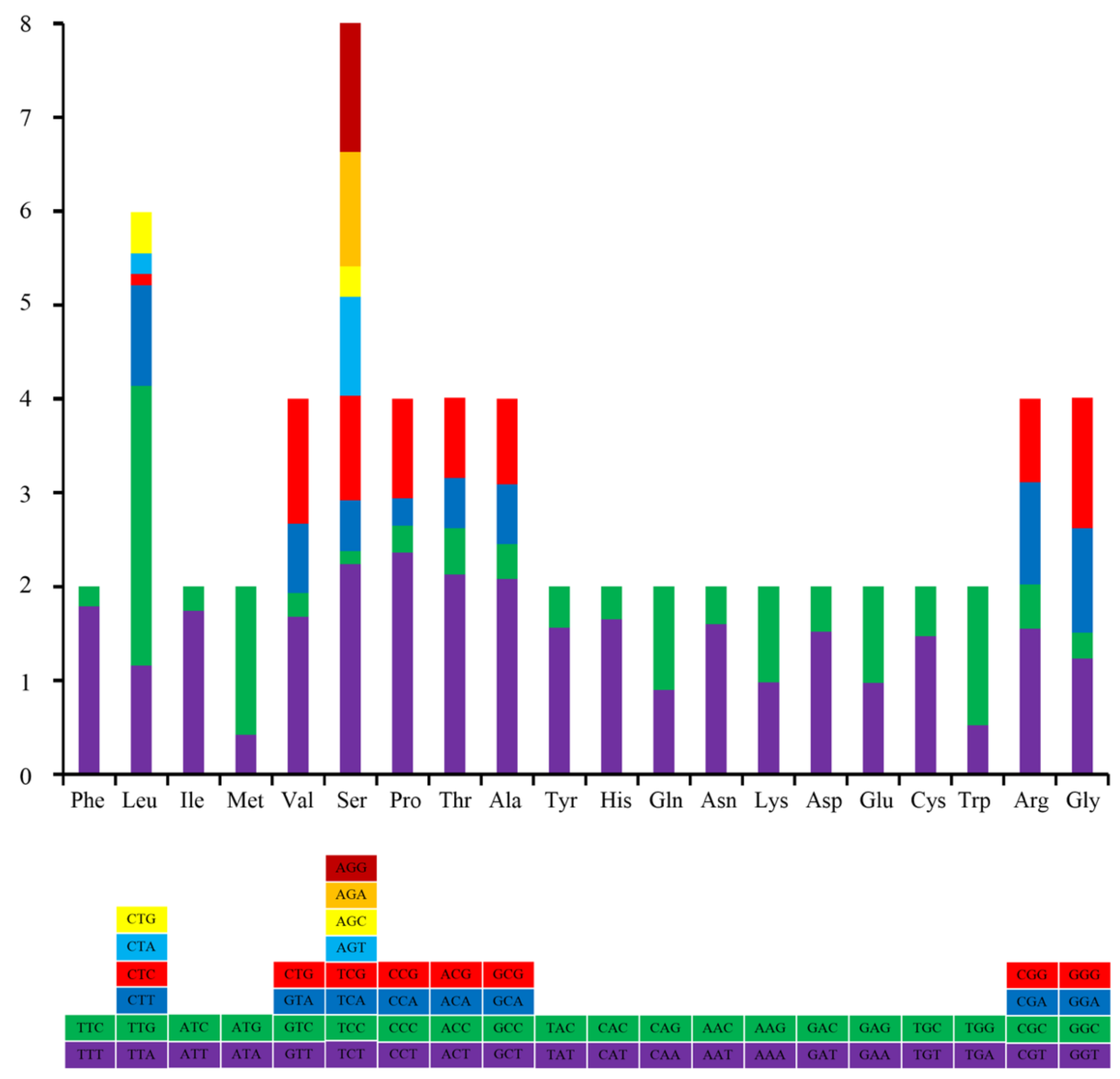
3.2. Protein Coding Genes (PCGs)
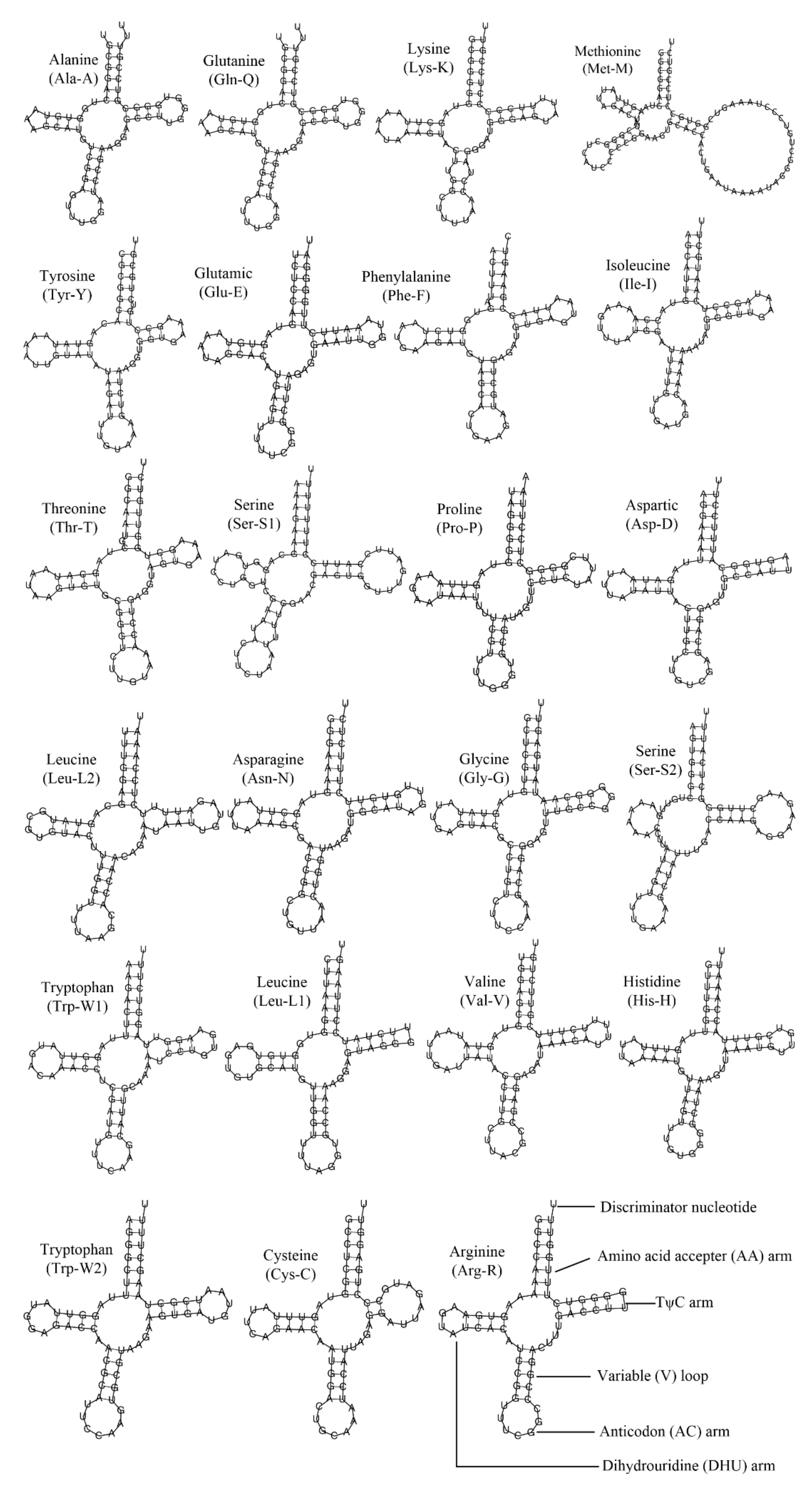
3.3. Ribosomal RNA and Transfer RNA Genes
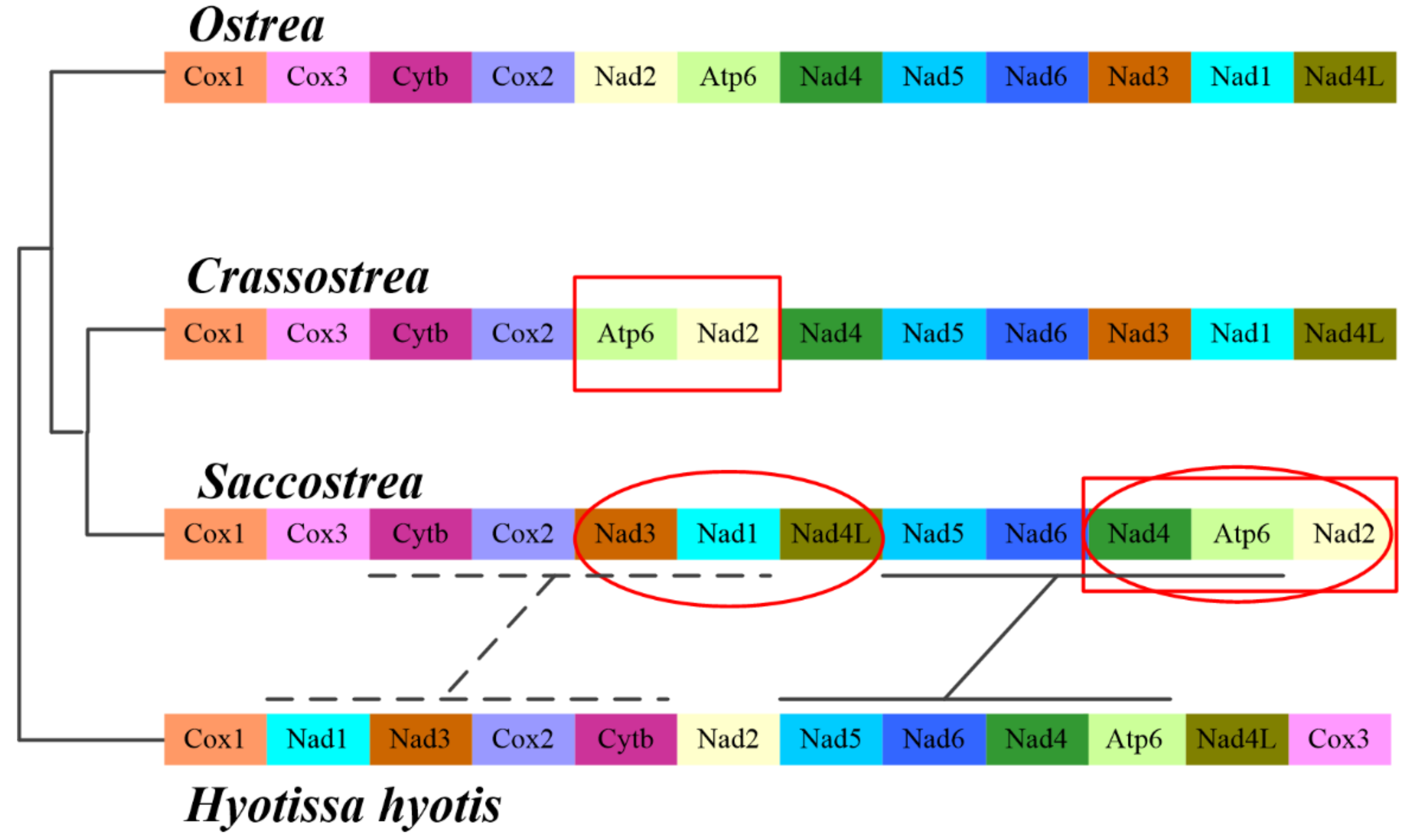
3.4. Gene Rearrangement
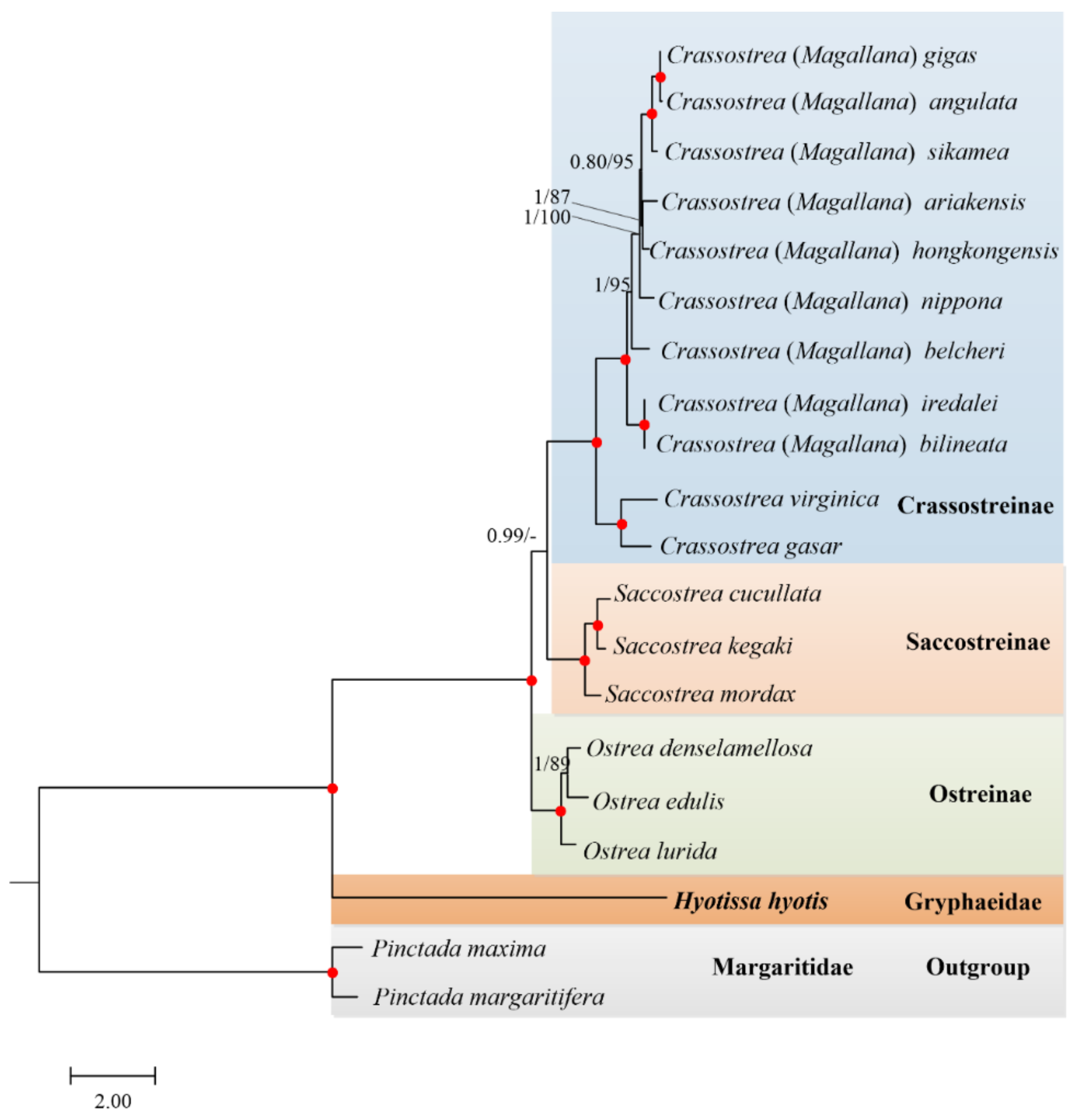
3.5. Phylogenetic Relationships
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Pett, W. Animal mitochondrial DNA as we do not know it: Mt-genome organization and evolution in nonbilaterian lineages. Genome Biol. Evol. 2016, 8, 2896–2913. [Google Scholar] [CrossRef] [PubMed]
- Bridge, D.; Cunningham, C.W.; Schierwater, B.; Desalle, R.; Buss, L.W. Class-level relationships in the phylum Cnidaria: Evidence from mitochondrial genome structure. Proc. Natl. Acad. Sci. USA 1992, 89, 8750–8753. [Google Scholar] [CrossRef] [PubMed]
- Kayal, E.; Bentlage, B.; Collins, A.G.; Kayal, M.; Pirro, S.; Lavrov, D.V. Evolution of linear mitochondrial genomes in medusozoan cnidarians. Genome Biol. Evol. 2012, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Pett, W.; Voigt, O.; Wörheide, G.; Forget, L.; Lang, B.F.; Kayal, E. Mitochondrial DNA of Clathrina clathrus (Calcarea, Calcinea): Six linear chromosomes, fragmented rRNAs, tRNA editing, and a novel genetic code. Mol. Biol. Evol. 2013, 30, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Li, Y.; Kocot, K.M.; Yang, Y.; Qi, L.; Li, Q.; Halanych, K.M. Mitogenomics reveals phylogenetic relationships of Arcoida (Mollusca, Bivalvia) and multiple independent expansions and contractions in mitochondrial genome size. Mol. Phylogenet. Evol. 2020, 150, 106857. [Google Scholar] [CrossRef]
- Sun, S.; Li, Q.; Kong, L. Relaxation of Selective Constraint on the Ultra-Large Mitochondrial Genomes of Arcidae (Mollusca: Bivalvia). J. Ocean Univ. China 2021, 20, 1157–1166. [Google Scholar] [CrossRef]
- Abalde, S.; Tenorio, M.J.; Afonso, C.M.; Uribe, J.E.; Echeverry, A.M.; Zardoya, R. Phylogenetic relationships of cone snails endemic to Cabo Verde based on mitochondrial genomes. BMC Evol. Biol. 2017, 17, 231. [Google Scholar] [CrossRef]
- Abalde, S.; Tenorio, M.J.; Afonso, C.M.; Zardoya, R. Mitogenomic phylogeny of cone snails endemic to Senegal. Mol. Phylogenet. Evol. 2017, 112, 79–87. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, Q.; Yu, H.; Kong, L. The complete mitochondrial genomes of six heterodont bivalves (Tellinoidea and Solenoidea): Variable gene arrangements and phylogenetic implications. PLoS ONE 2012, 7, e32353. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Lee, Y.; Kwak, H.; Shin, J.; Kim, S.C.; Kim, T.; Park, J.K. A mitochondrial genome phylogeny of Mytilidae (Bivalvia: Mytilida). Mol. Phylogenet. Evol. 2019, 139, 106533. [Google Scholar] [CrossRef]
- Uribe, J.E.; Irisarri, I.; Templado, J.; Zardoya, R. New patellogastropod mitogenomes help counteracting long-branch attraction in the deep phylogeny of gastropod mollusks. Mol. Phylogenet. Evol. 2019, 133, 12–23. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M., Jr.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef]
- Ghiselli, F.; Gomes-dos-Santos, A.; Adema, C.M.; Lopes-Lima, M.; Sharbrough, J.; Boore, J.L. Molluscan mitochondrial genomes break the rules. Phil. Trans. R. Soc. B 2021, 376, 20200159. [Google Scholar] [CrossRef]
- Breton, S.; Beaupre, H.D.; Stewart, D.T.; Hoeh, W.R.; Blier, P.U. The unusual system of doubly uniparental inheritance of mtDNA: Isn’t one enough? Trends Genet. 2007, 23, 465–474. [Google Scholar] [CrossRef]
- Burzyński, A.; Zbawicka, M.; Skibinski, D.O.; Wenne, R. Evidence for recombination of mtDNA in the marine mussel Mytilus trossulus from the Baltic. Mol. Biol. Evol. 2003, 20, 388–392. [Google Scholar] [CrossRef]
- Ladoukakis, E.D.; Zouros, E. Direct evidence for homologous recombination in mussel (Mytilus galloprovincialis) mitochondrial DNA. Mol. Biol. Evol. 2001, 18, 1168–1175. [Google Scholar] [CrossRef]
- Gusman, A.; Lecomte, S.; Stewart, D.T.; Passamonti, M.; Breton, S. Pursuing the quest for better understanding the taxonomic distribution of the system of doubly uniparental inheritance of mtDNA. PeerJ 2016, 4, e2760. [Google Scholar] [CrossRef]
- Lubośny, M.; Śmietanka, B.; Arculeo, M.; Burzyński, A. No evidence of DUI in the Mediterranean alien species Brachidontes pharaonis (P. Fisher, 1870) despite mitochondrial heteroplasmy. Sci. Rep. 2022, 12, 8569. [Google Scholar] [CrossRef]
- Serb, J.M.; Lydeard, C. Complete mtDNA sequence of the North American freshwater mussel, Lampsilis ornata (Unionidae): An examination of the evolution and phylogenetic utility of mitochondrial genome organization in Bivalvia (Mollusca). Mol. Biol. Evol. 2003, 20, 1854–1866. [Google Scholar] [CrossRef] [PubMed]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Ahn, D.H. Complete mitochondrial genome of the antarctic soft-shelled clam, Laternula elliptica (Bivalvia; Laternulidae). Mitochondrial DNA B 2015, 26, 2. [Google Scholar] [CrossRef]
- Sun, S.E.; Li, Q.; Kong, L.; Yu, H. Complete mitochondrial genomes of Trisidos kiyoni and Potiarca pilula: Varied mitochondrial genome size and highly rearranged gene order in Arcidae. Sci. Rep. 2016, 6, 33794. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Jiang, F.; Guo, X.; Liu, B. Unusual conservation of mitochondrial gene order in Crassostrea oysters: Evidence for recent speciation in Asia. BMC Evol. Biol. 2010, 10, 394. [Google Scholar] [CrossRef]
- Guo, X.; Li, C.; Wang, H.; Xu, Z. Diversity and evolution of living oysters. J. Shellfish Res. 2018, 37, 755–771. [Google Scholar] [CrossRef]
- Bouchet, P.; Rocroi, J.P.; Bieler, R.; Carter, J.G.; Coan, E.V. Nomenclator of bivalve families with a classification of bivalve families. Malacologia 2010, 52, 1–184. [Google Scholar] [CrossRef]
- Bayne, B.L. Biology of Oysters; Academic Press: London, UK, 2017; p. 860. [Google Scholar]
- Danic-Tchaleu, G.; Heurtebise, S.; Morga, B.; Lapègue, S. Complete mitochondrial DNA sequence of the European flat oyster Ostrea edulis confirms Ostreidae classification. BMC Res. Notes 2011, 4, 400. [Google Scholar] [CrossRef]
- Volatiana, J.A.; Fang, S.; Kinaro, Z.O.; Liu, X. Complete mitochondrial DNA sequences of Saccostrea mordax and Saccostrea cucullata: Genome organization and phylogeny analysis. Mitochondrial DNA A 2016, 27, 3024–3025. [Google Scholar] [CrossRef]
- Yu, H.; Kong, L.; Li, Q. Complete mitochondrial genome of Ostrea denselamellosa (Bivalvia, Ostreidae). Mitochondrial DNA A 2016, 27, 711–712. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; He, J.; Lin, Z.; Xue, Q. The complete mitochondrial genome of Crassostrea hongkongensis from East China Sea indicates species’ range may extend northward. Mol. Biol. Rep. 2022, 49, 1631–1635. [Google Scholar] [CrossRef]
- Gastineau, R.; Nguyễn, Đ.H.; Lemieux, C.; Turmel, M.; Tremblay, R.; Nguyễn, V.D.; Widowati, I.; Witkowski, A.; Mouget, J.L. The complete mitochondrial DNA of the tropical oyster Crassostrea belcheri from the Cần Giò’mangrove in Vietnam. Mitochondrial DNA B 2018, 3, 462–463. [Google Scholar] [CrossRef]
- Cavaleiro, N.P.; Solé-Cava, A.M.; Melo, C.M.; de Almeida, L.G.; Lazoski, C.; Vasconcelos, A.T.R. The complete mitochondrial genome of Crassostrea gasar (Bivalvia: Ostreidae). Mitochondrial DNA A 2016, 27, 2939–2940. [Google Scholar] [CrossRef]
- Yu, H.; Li, Q. Complete mitochondrial DNA sequence of Crassostrea nippona: Comparative and phylogenomic studies on seven commercial Crassostrea species. Mol. Biol. Rep. 2012, 39, 999–1009. [Google Scholar] [CrossRef]
- Wu, X.; Xu, X.; Yu, Z.; Wei, Z.; Xia, J. Comparison of seven Crassostrea mitogenomes and phylogenetic analyses. Mol. Phylogenet. Evol. 2010, 57, 448–454. [Google Scholar] [CrossRef]
- Milbury, C.A.; Gaffney, P.M. Complete mitochondrial DNA sequence of the eastern oyster Crassostrea virginica. Mar. Biotechnol. 2005, 7, 697–712. [Google Scholar] [CrossRef]
- Yu, H.; Li, Q. Mutation and selection on the wobble nucleotide in tRNA anticodons in marine bivalve mitochondrial genomes. PLoS ONE 2011, 6, e16147. [Google Scholar] [CrossRef]
- Xiao, S.; Wu, X.; Li, L.; Yu, Z. Complete mitochondrial genome of the Olympia oyster Ostrea lurida (Bivalvia, Ostreidae). Mitochondrial DNA 2015, 26, 471–472. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; Li, L.; Yu, Z. A unique tRNA gene family and a novel, highly expressed ORF in the mitochondrial genome of the silver-lip pearl oyster, Pinctada maxima (Bivalvia: Pteriidae). Gene 2012, 510, 22–31. [Google Scholar] [CrossRef]
- Bieler, R.; Mikkelsen, P.M.; Lee, T.; Foighil, D.O’. Discovery of the Indo-Pacific oyster Hyotissa hyotis (Linnaeus, 1758) in the Florida Keys (Bivalvia: Gryphaeidae). Molluscan Res. 2004, 24, 149–159. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Irwin, A.R.; Strong, E.E.; Kano, Y.; Harper, E.M.; Williams, S.T. Eight New Mitogenomes Clarify the Phylogenetic Relationships of Stromboidea Within the Caenogastropod Phylogenetic Framework. Mol. Phylogenet. Evol. 2021, 158, 107081. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis Across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A Comparative Genomics Tool for Circular Genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian Phylogenetic Inference Under Mixed Models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; Li, L.; Xu, X.; Xia, J.; Yu, Z. New features of Asian Crassostrea oyster mitochondrial genomes: A novel alloacceptor tRNA gene recruitment and two novel ORFs. Gene 2012, 507, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Breton, S.; Stewart, D.T.; Hoeh, W.R. Characterization of a mitochondrial ORF from the gender-associated mtDNAs of Mytilus spp. (Bivalvia: Mytilidae): Identification of the “missing” ATPase 8 gene. Mar. Genom. 2010, 3, 11–18. [Google Scholar] [CrossRef]
- Grande, C.; Templado, J.; Zardoya, R. Evolution of gastropod mitochondrial genome arrangements. BMC Evol. Biol. 2008, 8, 61. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, H.; Qi, L.; Kong, L.; Li, Q. Complete mitochondrial genomes of two toxin-accumulated Nassariids (Neogastropoda: Nassariidae: Nassarius) and their implication for phylogeny. Int. J. Mol. Sci. 2020, 21, 3545. [Google Scholar] [CrossRef]
- Li, F.; Zheng, J.; Ma, Q.; Gu, Z.; Wang, A.; Yang, Y.; Liu, C. Phylogeny of Strombidae (Gastropoda) Based on Mitochondrial Genomes. Front. Mar. Sci. 2022, 9, 930910. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Liu, H.; Kong, L.; Yu, H.; Liu, S.; Li, Q. Phylogeny of Veneridae (Bivalvia) based on mitochondrial genomes. Zool. Scr. 2021, 50, 58–70. [Google Scholar] [CrossRef]
- Sun, S.; Kong, L.; Yu, H.; Li, Q. The complete mitochondrial DNA of Tegillarca granosa and comparative mitogenomic analyses of three Arcidae species. Gene 2015, 557, 61–70. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal Mitochondrial DNA: Structure and Evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In Comparative Genomics: Empirical and Analytical Approaches to Gene Order Dynamics, Map Alignment and the Evolution of Gene Families; Sankoff, D., Nadeau, J.H., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 133–147. [Google Scholar]
- Stenzel, H.B. Oysters. In Treatise on Invertebrate Paleontology. Part N. Mollusca 6: Bivalvia; Moore, R.C., Ed.; Geological Society of America and University of Kansas Press: Lawrence, KS, USA, 1971; Volume 3, pp. 953–1224. [Google Scholar]
- Harry, H.W. Synopsis of the supraspecific classification of living oysters (Bivalvia: Gryphaeidae and Ostreidae). Veliger 1985, 28, 121–158. [Google Scholar]
- Li, C.; Kou, Q.; Zhang, Z.; Hu, L.; Huang, W.; Cui, Z.; Liu, Y.; Ma, P.; Wang, H. Reconstruction of the evolutionary biogeography reveal the origins and diversification of oysters (Bivalvia: Ostreidae). Mol. Phylogenet. Evol. 2021, 164, 107268. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Q.; Kong, L.; Yu, H.; Zheng, X. Identifying the true oysters (Bivalvia, Ostreidae) with mitochondrial phylogeny and distance-based DNA barcoding. Mol. Ecol. Resour. 2011, 11, 820–830. [Google Scholar] [CrossRef]
- Salvi, D.; Macali, A.; Mariottini, P. Molecular phylogenetics and systematics of the bivalve family Ostreidae based on rRNA sequence-structure models and multilocus species tree. PLoS ONE 2014, 9, e108696. [Google Scholar] [CrossRef]
- Pfeiffer, J.M.; Breinholt, J.W.; Page, L.M. Unioverse: A phylogenomic resource for reconstructing the evolution of freshwater mussels (Bivalvia, Unionoida). Mol. Phylogenet. Evol. 2019, 137, 114–126. [Google Scholar] [CrossRef]
- Salvi, D.; Mariottini, P. Molecular taxonomy in 2D: A novel ITS2 rRNA sequence-structure approach guides the description of the oysters’ subfamily Saccostreinae and the genus Magallana (Bivalvia: Ostreidae). Zool. J. Linn. Soc.-Lond. 2017, 179, 263–276. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Z.; Guo, X. Differences in the rDNA-bearing Chromosome divide the Asian-Pacific and Atlantic species of Crassostrea (Bivalvia, Mollusca). Biol. Bull. 2004, 206, 46–54. [Google Scholar] [CrossRef]
- Bayne, B.L.; Ahrens, M.; Allen, S.K.; D’auriac, M.A.; Backeljau, T.; Beninger, P.; Bohn, R.; Boudry, P.; Davis, J.; Green, T.; et al. The proposed dropping of the genus Crassostrea for all Pacific cupped oysters and its replacement by a new genus Magallana: A dissenting view. J. Shellfish Res. 2017, 36, 545–547. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| New Mitochondrial Genome | ||||
| Family | Species | Length (bp) | Sampling Time | Accession No. OP151093 |
| Gryphaeidae | Hyotissa hyotis | 22,185 | May, 2022 | |
| GenBank Mitochondrial Genome | ||||
| Family | Species | Length (bp) | Accession No. | Reference |
| Ostreidae | Crassostrea hongkongensis | 18,617 | MZ337404 | Liu et al. (2022) [32] |
| Ostreidae | Crassostrea bilineata | 22,420 | MT985154 | Arshad et al. (2020) |
| Ostreidae | Crassostrea belcheri | 21,020 | MH051332 | Gastineau et al. (2018) [33] |
| Ostreidae | Crassostrea gasar | 17,685 | KR856227 | Cavaleiro et al. (2016) [34] |
| Ostreidae | Crassostrea nippona | 20,030 | HM015198 | Yu and Li (2012) [35] |
| Ostreidae | Crassostrea iredalei | 22,446 | FJ841967 | Wu et al. (2010) [36] |
| Ostreidae | Crassostrea ariakensis | 18,414 | EU672835 | Ren et al. (2010) [25] |
| Ostreidae | Crassostrea sikamea | 18,243 | EU672833 | Ren et al. (2010) [25] |
| Ostreidae | Crassostrea angulata | 18,225 | EU672832 | Ren et al. (2010) [25] |
| Ostreidae | Crassostrea gigas | 18,225 | EU672831 | Ren et al. (2010) [25] |
| Ostreidae | Crassostrea virginica | 17,244 | AY905542 | Milbury and Gaffney (2005) [37] |
| Ostreidae | Ostrea denselamellosa | 16,277 | HM015199 | Yu and Li (2011) [38] |
| Ostreidae | Ostrea edulis | 16,320 | JF274008 | Danic-Tchaleu et al. (2011) [29] |
| Ostreidae | Ostrea lurida | 16,344 | KC768038 | Xiao et al. (2015) [39] |
| Ostreidae | Saccostrea mordax | 16,532 | FJ841968 | Wu et al. (2009) |
| Ostreidae | Saccostrea cucullata | 16,396 | KP967577 | Volatiana et al. (2016) [30] |
| Ostreidae | Saccostrea kegaki | 16,260 | KX065089 | Hsiao (2016) |
| Margaritidae | Pinctada margaritifera | 15,680 | HM467838 | Wu et al. (2010) |
| Margaritidae | Pinctada maxima | 16,994 | GQ452847 | Wu et al. (2012) [40] |
| Gene | Strand | Location | Size (bp) | Start Codon | Stop Codon | Intergenic Nucleotides |
|---|---|---|---|---|---|---|
| Cox1 | H | 1–1725 | 1725 | ATG | TAG | 1528 |
| tRNA-Ala | H | 3254–3319 | 66 | 31 | ||
| tRNA-Gln | H | 3351–3413 | 63 | 51 | ||
| tRNA-Lys | H | 3465–3531 | 67 | 403 | ||
| tRNA-Met | H | 3935–4025 | 91 | 325 | ||
| tRNA-Tyr | H | 4351–4414 | 64 | 36 | ||
| tRNA-Glu | H | 4451–4516 | 66 | 8 | ||
| tRNA-Phe | H | 4525–4587 | 63 | 0 | ||
| rrnL | H | 4588–5966 | 1379 | 175 | ||
| rrnS | H | 6142–7099 | 958 | 93 | ||
| tRNA-Ile | H | 7193–7260 | 68 | 22 | ||
| tRNA-Thr | H | 7283–7345 | 63 | 20 | ||
| tRNA-Ser1 | H | 7366–7435 | 70 | 90 | ||
| Nad1 | H | 7526–8460 | 935 | ATG | TA- | 71 |
| Atp8 | H | 8532–8654 | 123 | ATG | missing | 1 |
| tRNA-Pro | H | 8656–8720 | 65 | 13 | ||
| tRNA-Asp | H | 8734–8797 | 64 | 10 | ||
| tRNA-Leu2 | H | 8808–8873 | 66 | 177 | ||
| Nad3 | H | 9051–9417 | 367 | TTT | T-- | 408 |
| tRNA-Asn | H | 9826–9892 | 67 | 4 | ||
| tRNA-Gly | H | 9897–9961 | 65 | 3 | ||
| tRNA-Ser2 | H | 9965–10034 | 70 | 21 | ||
| tRNA-Trp1 | H | 10056–10122 | 67 | 7 | ||
| tRNA-Leu1 | H | 10130–10194 | 65 | 20 | ||
| tRNA-Val | H | 10215–10281 | 67 | 29 | ||
| tRNA-His | H | 10311–10375 | 65 | 15 | ||
| Cox2 | H | 10391–11089 | 699 | TTG | TAA | 138 |
| Cytb | H | 11228–12910 | 1683 | ATG | TAA | 1191 |
| Nad2 | H | 14102–15121 | 1020 | ATT | TAG | 13 |
| tRNA-Trp2 | H | 15135–15201 | 67 | 35 | ||
| Nad5 | H | 15237–16901 | 1667 | AAG | TAA | 578 |
| Nad6 | H | 17480–18034 | 555 | ATG | TAA | 64 |
| tRNA-Cys | H | 18099–18162 | 64 | 2 | ||
| Nad4 | H | 18165–19508 | 1344 | GTG | TAA | 66 |
| Atp6 | H | 19575–20324 | 750 | ATG | TAA | 20 |
| Nad4L | H | 20345–20638 | 294 | TAT | TAA | 52 |
| tRNA-Arg | H | 20691–20753 | 63 | 252 | ||
| Cox3 | H | 21006–21884 | 879 | ATA | TAA | 301 |
| Feature | (A+T)% | AT skew | GC skew |
|---|---|---|---|
| Whole genome | 59.2 | −0.20 | 0.33 |
| PCGs | 59.4 | −0.29 | 0.34 |
| PCGs1 | 57.3 | −0.09 | 0.38 |
| PCGs2 | 59.9 | −0.34 | 0.08 |
| PCGs3 | 61.0 | −0.42 | 0.57 |
| Atp6 | 59.8 | −0.23 | 0.40 |
| Atp8 | 60.1 | −0.30 | 0.14 |
| Cox1 | 58.6 | −0.28 | 0.29 |
| Cox2 | 59.0 | −0.25 | 0.37 |
| Cox3 | 59.3 | −0.31 | 0.30 |
| Cytb | 60.5 | −0.20 | 0.27 |
| Nad1 | 59.5 | −0.36 | 0.36 |
| Nad2 | 59.1 | −0.30 | 0.39 |
| Nad3 | 58.4 | −0.43 | 0.50 |
| Nad4 | 61.6 | −0.33 | 0.35 |
| Nad4L | 58.5 | −0.33 | 0.43 |
| Nad5 | 59.1 | −0.29 | 0.39 |
| Nad6 | 57.7 | −0.24 | 0.39 |
| tRNAs | 57.9 | −0.12 | 0.25 |
| rrnS | 53.7 | 0.11 | 0.11 |
| rrnL | 58.8 | −0.02 | 0.20 |
| LNR1 | 64.8 | −0.08 | 0.16 |
| LNR2 | 51.7 | −0.21 | 0.52 |
| Amino Acid | Codon | Count (RSCU) | Amino | Codon | Count (RSCU) |
|---|---|---|---|---|---|
| Phe | TTT | 289.0(1.78) | Ala | GCT | 134.0(2.08) |
| TTC | 35.0(0.22) | GCC | 24.0(0.37) | ||
| Leu | TTA | 91.0(1.16) | GCA | 41.0(0.64) | |
| TTG | 232.0(2.96) | GCG | 59.0(0.91) | ||
| CTT | 84.0(1.07) | Gly | GGT | 101.0(1.22) | |
| CTC | 9.0(0.11) | GGC | 24.0(0.29) | ||
| CTA | 18.0(0.23) | GGA | 91.0(1.10) | ||
| CTG | 36.0(0.46) | GGG | 115.0(1.39) | ||
| Ile | ATT | 193.0(1.74) | Arg | CGT | 40.0(1.52) |
| ATC | 29.0(0.26) | CGC | 12.0(0.46) | ||
| Met | ATA | 47.0(0.44) | CGA | 28.0(1.07) | |
| ATG | 169.0(1.56) | CGG | 25.0(0.95) | ||
| Val | GTT | 155.0(1.68) | Tyr | TAT | 135.0(1.56) |
| GTC | 25.0(0.27) | TAC | 38.0(0.44) | ||
| GTA | 68.0(0.74) | His | CAT | 70.0(1.65) | |
| GTG | 122.0(1.32) | CAC | 15.0(0.35) | ||
| Ser | TCT | 99.0(2.21) | Gln | CAA | 22.0(0.90) |
| TCC | 6.0(0.13) | CAG | 27.0(1.10) | ||
| TCA | 25.0(0.56) | Asn | AAT | 81.0(1.59) | |
| TCG | 49.0(1.09) | AAC | 21.0(0.41) | ||
| AGT | 47.0(1.05) | Lys | AAA | 81.0(0.98) | |
| AGC | 15.0(0.34) | AAG | 84.0(1.02) | ||
| AGA | 54.0(1.21) | Asp | GAT | 67.0(1.52) | |
| AGG | 63.0(1.41) | GAC | 21.0(0.48) | ||
| Pro | CCT | 82.0(2.33) | Glu | GAA | 65.0(0.97) |
| CCC | 11.0(0.31) | GAG | 69.0(1.03) | ||
| CCA | 11.0(0.31) | Cys | TGT | 77.0(1.47) | |
| CCG | 37.0(1.05) | TGC | 28.0(0.53) | ||
| Thr | ACT | 83.0(2.11) | Trp | TGA | 39.0(0.52) |
| ACC | 19.0(0.48) | TGG | 110.0(1.48) | ||
| ACA | 22.0(0.56) | ||||
| ACG | 33.0(0.84) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Fan, M.; Wang, S.; Gu, Z.; Wang, A.; Liu, C.; Yang, Y.; Liu, S. The Complete Mitochondrial Genome of Hyotissa hyotis (Bivalvia: Gryphaeidae) Reveals a Unique Gene Order within Ostreoidea. Fishes 2022, 7, 317. https://doi.org/10.3390/fishes7060317
Li F, Fan M, Wang S, Gu Z, Wang A, Liu C, Yang Y, Liu S. The Complete Mitochondrial Genome of Hyotissa hyotis (Bivalvia: Gryphaeidae) Reveals a Unique Gene Order within Ostreoidea. Fishes. 2022; 7(6):317. https://doi.org/10.3390/fishes7060317
Chicago/Turabian StyleLi, Fengping, Mingfu Fan, Shunshun Wang, Zhifeng Gu, Aimin Wang, Chunsheng Liu, Yi Yang, and Shikai Liu. 2022. "The Complete Mitochondrial Genome of Hyotissa hyotis (Bivalvia: Gryphaeidae) Reveals a Unique Gene Order within Ostreoidea" Fishes 7, no. 6: 317. https://doi.org/10.3390/fishes7060317
APA StyleLi, F., Fan, M., Wang, S., Gu, Z., Wang, A., Liu, C., Yang, Y., & Liu, S. (2022). The Complete Mitochondrial Genome of Hyotissa hyotis (Bivalvia: Gryphaeidae) Reveals a Unique Gene Order within Ostreoidea. Fishes, 7(6), 317. https://doi.org/10.3390/fishes7060317