
Scale Development-Related Genes Identified by Transcriptome Analysis


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Zebrafish Maintenance
2.2. Alizarin Red Staining and Observation
2.3. RNA Isolation and Sequencing
2.4. Weighted Gene Co-Expression Analysis (WGCNA) of the Sequencing Data
2.5. Differentially Expressed Gene Analysis and Functional Enrichment
2.6. Quantitative Real-Time PCR Validation of RNA-Seq Data
3. Results
3.1. Scale Development in Zebrafish
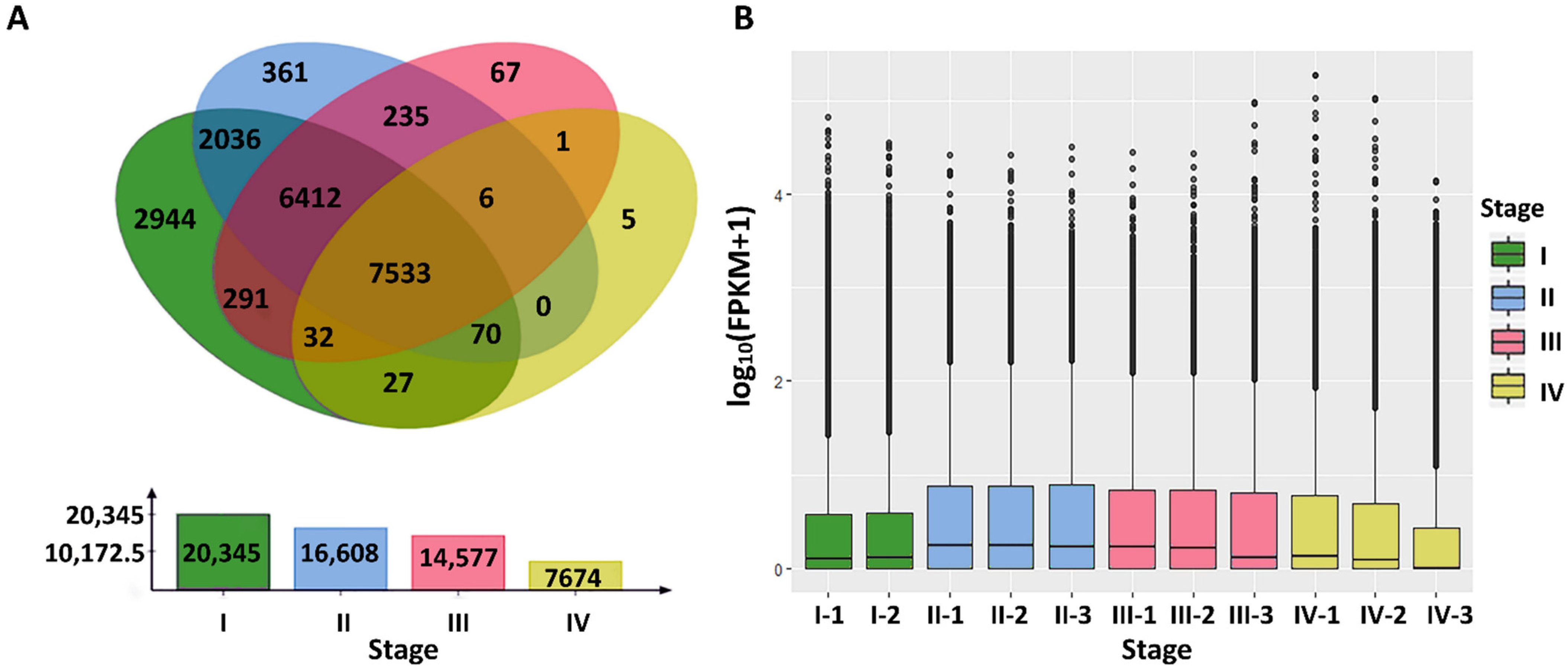
3.2. Gene Expression Quantification and Analysis of DEGs
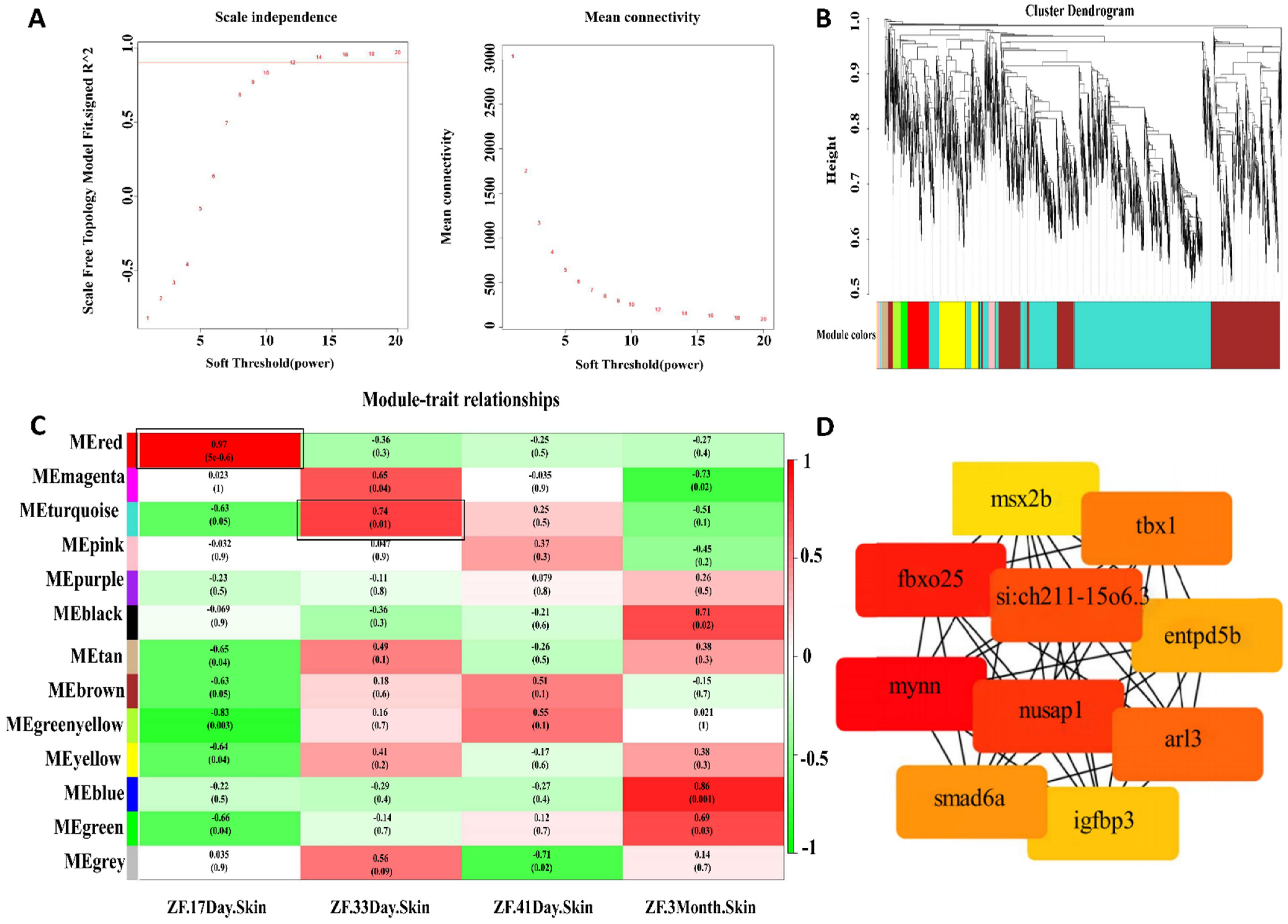
3.3. WGCNA Analysis and Hub-Genes Screening
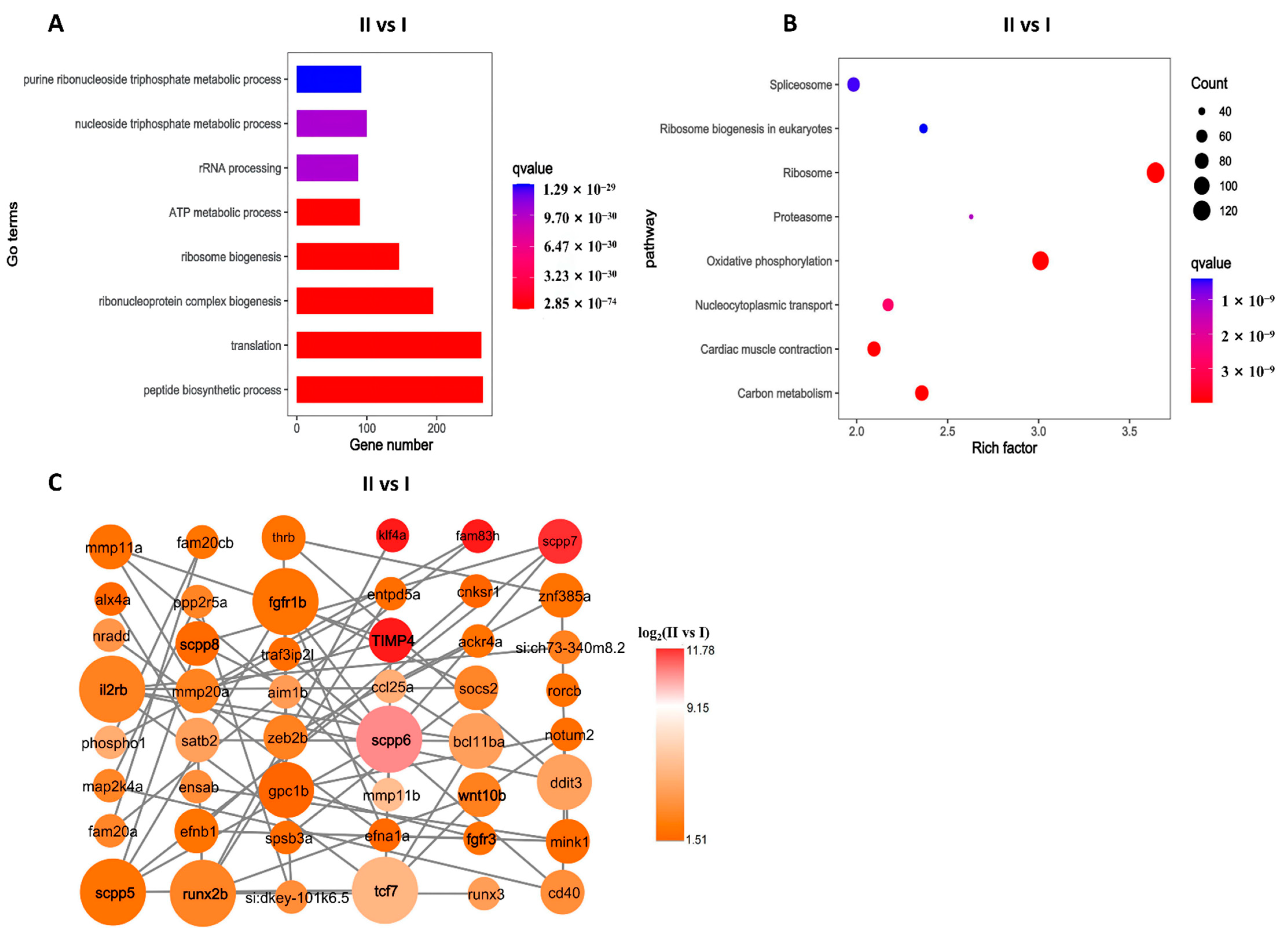
3.4. GO and KEGG Pathway Enrichment Analysis
3.5. Gene-Act Network
3.6. Key Genes and Pathways Related to Scale Development
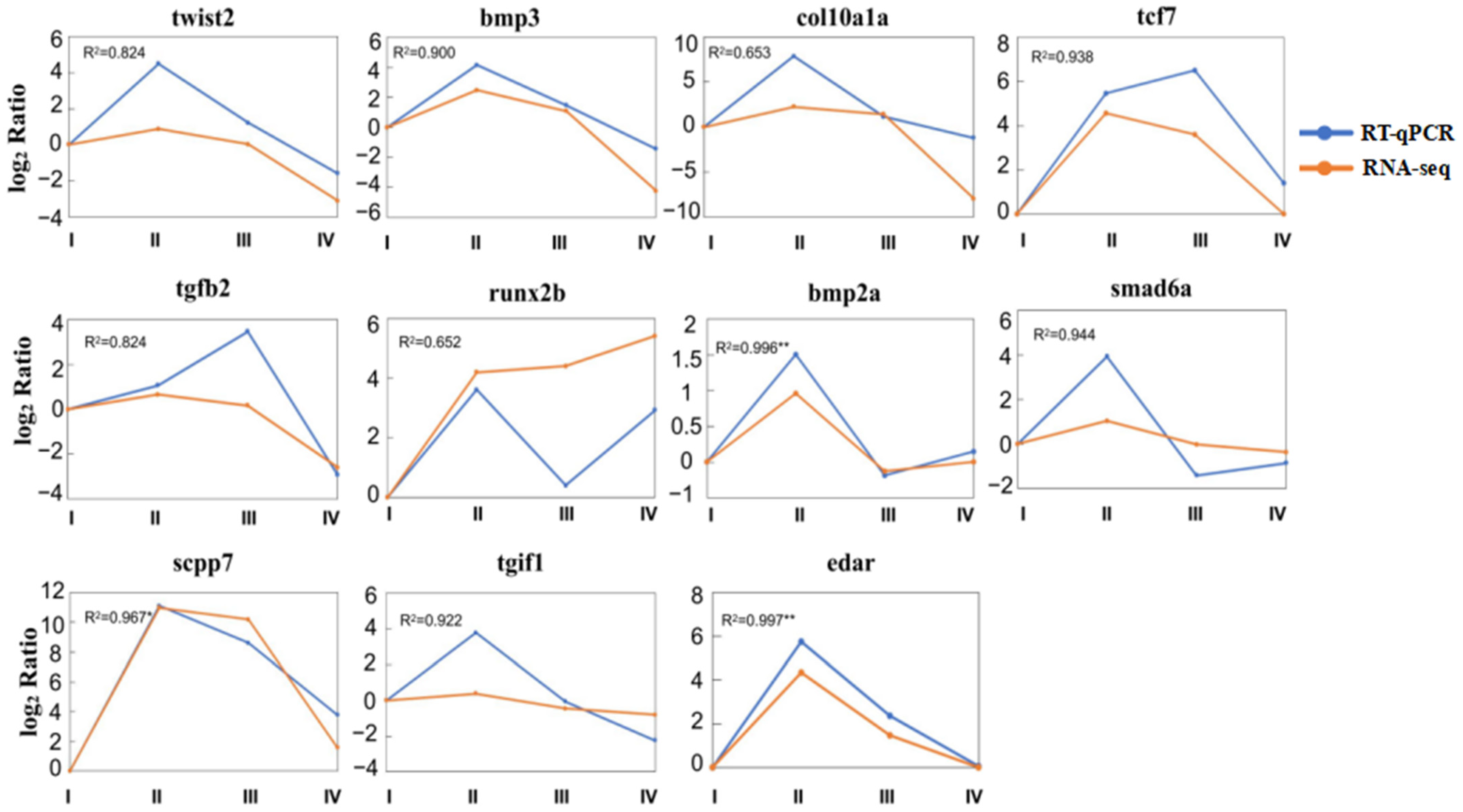
3.7. qPCR Analysis Verifications
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sire, J.-Y.; Allizard, F.; Babiar, O.; Bourguignon, J.; Quilhac, A. Scale development in zebrafish (Danio rerio). J. Anat. 1997, 190, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Able, K.W.; Sakowicz, G.P.; Lamonaca, J.C. Scale formation in selected fundulid and cyprinodontid fishes. Ichthyol. Res. 2008, 56, 1. [Google Scholar] [CrossRef]
- Zylberberg, L.; Géraudie, J.; Meunier, F.J. Biomineralisation in the integumental skeleton of the living lower vertebrates. Bone-Bone Metab. Miner. 1992, 4, 171–224. [Google Scholar]
- Meunier, F.J. Les Tissus Osseux des Ostéichthyens. In Structure, Genése, Croissance et Évolution; Institut d’ethnologie du Museum National d’histoire Naturelle: Paris, France, 1983. [Google Scholar]
- Cooper, J.A. Scale Development as Related to Growth of Juvenile Black Crappie, Pomoxis nigromaculatus Lesueur. Trans. Am. Fish. Soc. 1971, 100, 570–572. [Google Scholar] [CrossRef]
- Huysseune, A.; Sire, J. Evolution of patterns and processes in teeth and tooth-related tissues in non-mammalian vertebrates. Eur. J. Oral Sci. 1998, 106, 437–481. [Google Scholar] [CrossRef]
- Zhu, D.; Ortega, C.F.; Motamedi, R.; Szewciw, L.; Vernerey, F.; Barthelat, F. Structure and Mechanical Performance of a “Modern” Fish Scale. Adv. Eng. Mater. 2011, 14, B185–B194. [Google Scholar] [CrossRef]
- Sire, J.-Y. Development and fine structure of the bony scutes in Corydoras arcuatus (Siluriformes, callichthyidae). J. Morphol. 1993, 215, 225–244. [Google Scholar] [CrossRef]
- Schneider, P.; Street, S.L.; Gaide, O.; Hertig, S.; Tardivel, A.; Tschopp, J.; Runkel, L.; Alevizopoulos, K.; Ferguson, B.M.; Zonana, J. Mutations Leading to X-linked Hypohidrotic Ectodermal Dysplasia Affect Three Major Functional Domains in the Tumor Necrosis Factor Family Member Ectodysplasin-A. J. Biol. Chem. 2001, 276, 18819–18827. [Google Scholar] [CrossRef] [Green Version]
- Sehring, I.M.; Jahn, C.; Weidinger, G. Zebrafish fin and heart: What’s special about regeneration? Curr. Opin. Genet. Dev. 2016, 40, 48–56. [Google Scholar] [CrossRef]
- Motamedi, M.; Zeinali, F.; Soltanian, S. Expression Patterns of Three Regulatory Genes in Caudal Fin Regeneration of the Euryhaline Killifish, Aphanius hormuzensis (Teleostei: Aphaniidae). Iran. J. Sci. Technol. Trans. A Sci. 2019, 43, 2115–2122. [Google Scholar] [CrossRef]
- Aman, A.J.; Fulbright, A.N.; Parichy, D.M. Wnt/β-catenin regulates an ancient signaling network during zebrafish scale development. eLife 2018, 7, 37001. [Google Scholar] [CrossRef] [PubMed]
- Streisinger, G.; Walker, C.; Dower, N.; Knauber, D.; Singer, F. Production of clones of homozygous diploid zebra fish. Nature 1981, 291, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Akimenko, M.; Johnson, S.; Westerfield, M.; Ekker, M. Differential induction of four msx homeobox genes during fin development and regeneration in zebrafish. Development 1995, 121, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Kuwahara, Y.; Kondo, M.; Naruse, K.; Mitani, H.; Wakamatsu, Y.; Ozato, K.; Asakawa, S.; Shimizu, N.; Shima, A. The medaka rs-3 locus required for scale development encodes ectodysplasin-A receptor. Curr. Biol. 2001, 11, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Chen, C.-F.; Foley, J.; Tang, P.-C.; Li, A.; Jiang, T.X.; Wu, P.; Widelitz, R.B.; Chuong, C.M. Development, Regeneration, and Evolution of Feathers. Annu. Rev. Anim. Biosci. 2015, 3, 169–195. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, Z.; Chen, D.; Zhang, B.; Tian, R.-R.; Wu, J.; Zhang, Y.; Xu, K.; Yang, L.-M.; Cheng, C.; et al. Annotation and cluster analysis of spatiotemporal- and sex-related lncRNA expression in rhesus macaque brain. Genome Res. 2017, 27, 1608–1620. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sharpe, P. Fish scale development: Hair today, teeth and scales yesterday? Curr. Biol. 2001, 11, R751–R752. [Google Scholar] [CrossRef] [Green Version]
- Francillon-Vieillot, H.; de Buffrénil, V.; Castanet, J.; Géraudie, J.; Meunier, F.; Sire, J.Y.; Zylberberg, L.; de Ricqlès, A. Microstructure and Mineralization of Vertebrate Skeletal Tissues; American Geophysical Union: Washington, DC, USA, 2013; pp. 175–234. [Google Scholar]
- Garg, V.; Yamagishi, C.; Huab, T.; Kathiriyaab, I.S.; Yamagishiab, H.; Srivastava, D. Tbx1, a DiGeorge Syndrome Candidate Gene, Is Regulated by Sonic Hedgehog during Pharyngeal Arch Development. Dev. Biol. 2001, 235, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S.; et al. TBX1 Is Responsible for Cardiovascular Defects in Velo-Cardio-Facial/DiGeorge Syndrome. Cell 2001, 104, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Carroll, T.J.; McMahon, A.P. Sonic hedgehog regulates proliferation and differentiation of mesenchymal cells in the mouse metanephric kidney. Development 2002, 129, 5301–5312. [Google Scholar] [CrossRef]
- Vrijens, K.; Lin, W.; Cui, J.; Farmer, D.; Low, J.; Pronier, E.; Zeng, F.-Y.; Shelat, A.A.; Guy, K.; Taylor, M.R.; et al. Identification of Small Molecule Activators of BMP Signaling. PLoS ONE 2013, 8, e59045. [Google Scholar] [CrossRef] [Green Version]
- Thorimbert, V.; König, D.; Marro, J.; Ruggiero, F.; Jazwinska, A. Bone morphogenetic protein signaling promotes morphogenesis of blood vessels, wound epidermis, and actinotrichia during fin regeneration in zebrafish. FASEB J. 2015, 29, 4299–4312. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Sinha, N.R.; Siddiqui, S.; Mohan, R.R. Role of 5′TG3′-interacting factors (TGIFs) in Vorinostat (HDAC inhibitor)-mediated Corneal Fibrosis Inhibition. Mol. Vis. 2015, 21, 974–984. [Google Scholar] [PubMed]
- Liu, Z.; Liu, S.; Yao, J.; Bao, L.; Zhang, J.; Li, Y.; Jiang, C.; Sun, L.; Wang, R.; Zhang, Y.; et al. The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 2016, 7, 11757. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Weiss, K.M. Mineralized tissue and vertebrate evolution: The secretory calcium-binding phosphoprotein gene cluster. Proc. Natl. Acad. Sci. USA 2003, 100, 4060–4065. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, K. The SCPP gene repertoire in bony vertebrates and graded differences in mineralized tissues. Dev. Genes Evol. 2009, 219, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, K. The SCPP Gene Family and the Complexity of Hard Tissues in Vertebrates. Cells Tissues Organs 2011, 194, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Amemiya, C.T. SCPP genes in the coelacanth: Tissue mineralization genes shared by sarcopterygians. J. Exp. Zool. Part B Mol. Dev. Evol. 2013, 322, 390–402. [Google Scholar] [CrossRef]
- Kawasaki, K.; Weiss, K. SCPP Gene Evolution and the Dental Mineralization Continuum. J. Dent. Res. 2008, 87, 520–531. [Google Scholar] [CrossRef]
- Kawasaki, K.; Weiss, K.M. Evolutionary genetics of vertebrate tissue mineralization: The origin and evolution of the secretory calcium-binding phosphoprotein family. J. Exp. Zool. Part B Mol. Dev. Evol. 2006, 306, 295–316. [Google Scholar] [CrossRef]
- Ouji, Y.; Yoshikawa, M.; Shiroi, A.; Ishizaka, S. Wnt-10b promotes differentiation of skin epithelial cells in vitro. Biochem. Biophys. Res. Commun. 2006, 342, 28–35. [Google Scholar] [CrossRef]
- Schultz, G.A.; Harvey, M.B.; Watson, A.J.; Arcellana-Panlilio, M.Y.; Jones, K.; Westhusin, M.E. Regulation of Early Embryonic Development by Growth Factors: Growth Factor Gene Expression in Cloned Bovine Embryos. J. Anim. Sci. 1996, 74, 50–57. [Google Scholar] [CrossRef]
- Lacome, D. Clinical dysmorphology beyond developmental genetics recent advances in some human developmental genes. Ann. Genet. 1995, 38, 137–144. [Google Scholar]
- Daane, J.M.; Rohner, N.; Konstantinidis, P.; Djuranovic, S.; Harris, M.P. Parallelism and Epistasis in Skeletal Evolution Identified through Use of Phylogenomic Mapping Strategies. Mol. Biol. Evol. 2016, 33, 162–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohner, N.; Bercsényi, M.; Orbán, L.; Kolanczyk, M.E.; Linke, D.; Brand, M.; Nüsslein-Volhard, C.; Harris, M.P. Duplication of fgfr1 Permits Fgf Signaling to Serve as a Target for Selection during Domestication. Curr. Biol. 2009, 19, 1642–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Logan, C.Y.; Nusse, R. The Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [Green Version]
- Ducy, P.; Starbuck, M.; Priemel, M.; Shen, J.; Pinero, G.; Geoffroy, V.; Amling, M.; Karsenty, G. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999, 13, 1025–1036. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhou, Y.; Yang, C.; Fan, S.; Huang, L.; Zhou, T.; Wang, Q.; Zhao, R.; Tang, C.; Tao, M.; et al. Comparative analyses of hypothalamus transcriptomes reveal fertility-, growth-, and immune-related genes and signal pathways in different ploidy cyprinid fish. Genomics 2021, 113, 595–605. [Google Scholar] [CrossRef]
- Hu, P.; Liu, M.; Zhang, N.; Wang, J.; Niu, H.; Liu, Y.; Wu, Z.; Han, B.; Zhai, W.; Shen, Y.; et al. Global identification of the genetic networks and cis-regulatory elements of the cold response in zebrafish. Nucleic Acids Res. 2015, 43, 9198–9213. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Sun, Z.; Xu, H.; Song, N.; Gao, T. Transcriptome and co-expression network analyses reveal the regulatory pathways and key genes associated with temperature adaptability in the yellow drum (Nibea albiflora). J. Therm. Biol. 2021, 100, 103071. [Google Scholar] [CrossRef]
- Jacob, T.; Chakravarty, A.; Panchal, A.; Patil, M.; Ghodadra, G.; Sudhakaran, J.; Nuesslein-Volhard, C. Zebrafish twist2/dermo1 regulates scale shape and scale organization during skin development and regeneration. Cells Dev. 2021, 166, 203684. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Prime Sequence (5′→3′) | Product Length | Tm |
|---|---|---|---|
| scpp7 | F: TTATTGCGCTCCGCAAGTG R:GATTTCAGAGGGTCTTGCTGC | 103 bp | 57 °C |
| twist2 | F:TGATAATGCCGAACGGACTGT R:GAATGTCCTTTGGCCACGTC | 100 bp | 58 °C |
| bmp3 | F:CTGATATCGGCTGGAGCGAG R:GGAAGGTTTCAGAGACTTTGGC | 104 bp | 57 °C |
| tcf7 | F: CTACGTGAGTGCTTTGGGCA R:CGCGGCATTTCTTTGGAGAG | 92 bp | 58 °C |
| tgif1 | F: TGCGCTCGATACTTCGTAACA R: GACATCGCCAAAACACCCTT | 139 bp | 59 °C |
| smad6 a | F:CCCAGGACTATTCAGATGCCA R:CGTCGTGAACTGGGTACAGG | 117 bp | 56 °C |
| tgfb2 | F: ACGCGCTTTGCAGGTATAGA R: AGACGGTATGATGGCAGCAG | 122 bp | 60 °C |
| il2 rb | F: ACAAGCTGGGAGATGGCAAA R: ATGACCGTCAGTTTTCGGCT | 138 bp | 57 °C |
| edar | F: TGCGGACACTGTTTACCAGG R:GTGTGGACCTCATGCACTCT | 121 bp | 57 °C |
| bmp2a | F:GAGCTTCCACCATGATGAATCTACA R:ACCAACTCCTCGTCTGGGAT | 105 bp | 60 °C |
| ZF-β-Actin | F: CACTGAGGCTCCCCTGAATC R: GGGTCACACCATCACCAGAG | 167 bp | 60 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Ji, F.; Jiang, S.; Wu, Z.; Xu, Q. Scale Development-Related Genes Identified by Transcriptome Analysis. Fishes 2022, 7, 64. https://doi.org/10.3390/fishes7020064
Zhang Z, Ji F, Jiang S, Wu Z, Xu Q. Scale Development-Related Genes Identified by Transcriptome Analysis. Fishes. 2022; 7(2):64. https://doi.org/10.3390/fishes7020064
Chicago/Turabian StyleZhang, Zhicong, Fengyu Ji, Shouwen Jiang, Zhichao Wu, and Qianghua Xu. 2022. "Scale Development-Related Genes Identified by Transcriptome Analysis" Fishes 7, no. 2: 64. https://doi.org/10.3390/fishes7020064
APA StyleZhang, Z., Ji, F., Jiang, S., Wu, Z., & Xu, Q. (2022). Scale Development-Related Genes Identified by Transcriptome Analysis. Fishes, 7(2), 64. https://doi.org/10.3390/fishes7020064