
Comparative Analysis of Blood Transcriptome in the Yangtze Finless Porpoise (Neophocaena asiaeorientalis)


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Blood Sampling
2.2. RNA Extraction and Transcriptomesequencing
2.3. Raw Data Processing, Assembling and FPKM Calculation
2.4. Functional Unigene Annotation
2.5. DEGs between the Male and Female’s Blood Transcriptome
2.6. Validation of the Potential Function Genes by qRT-PCR
2.7. Data Deposition
3. Results and Discussion
3.1. Transcriptome Sequencing Output, Assembly and Annotation
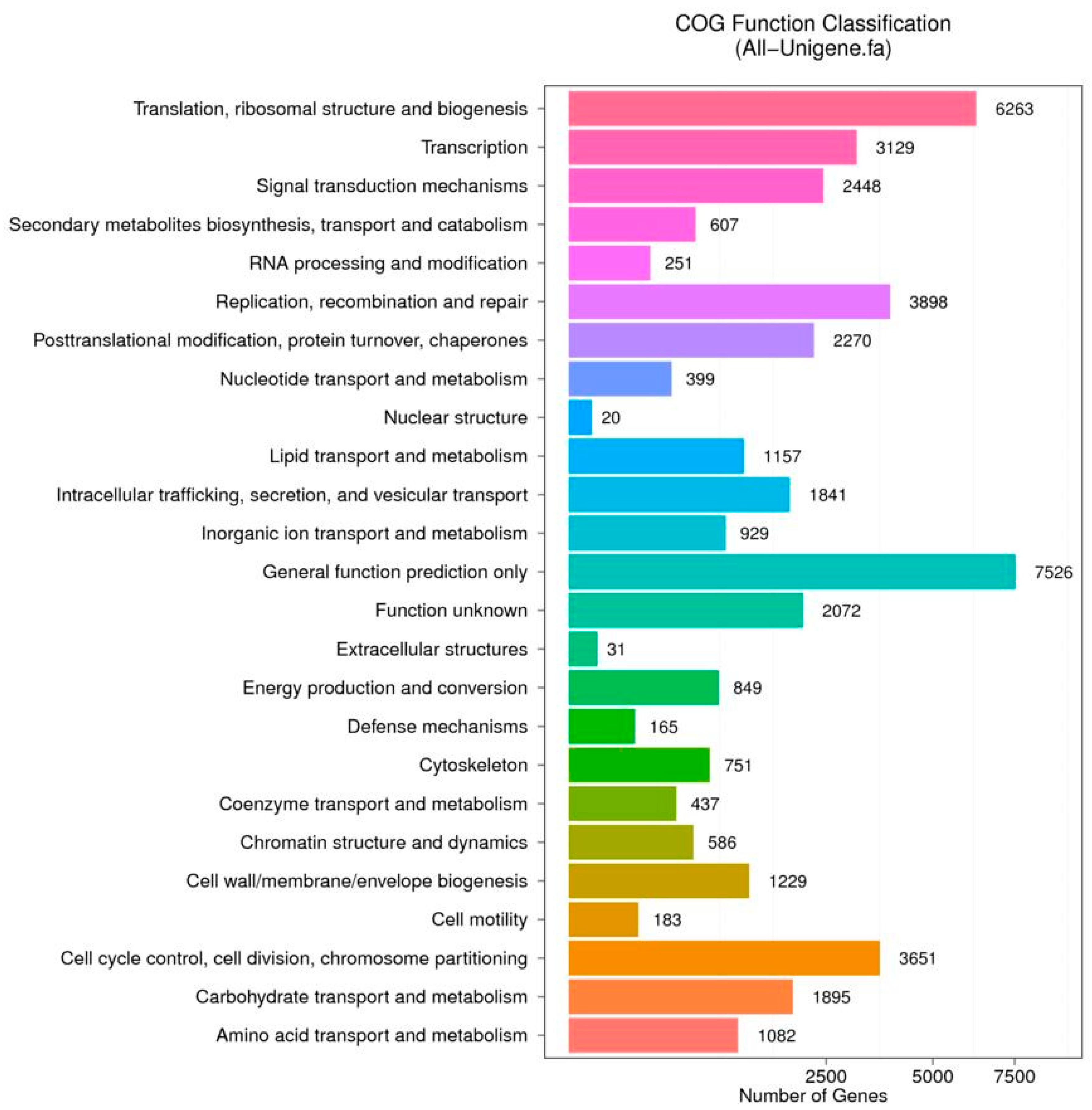
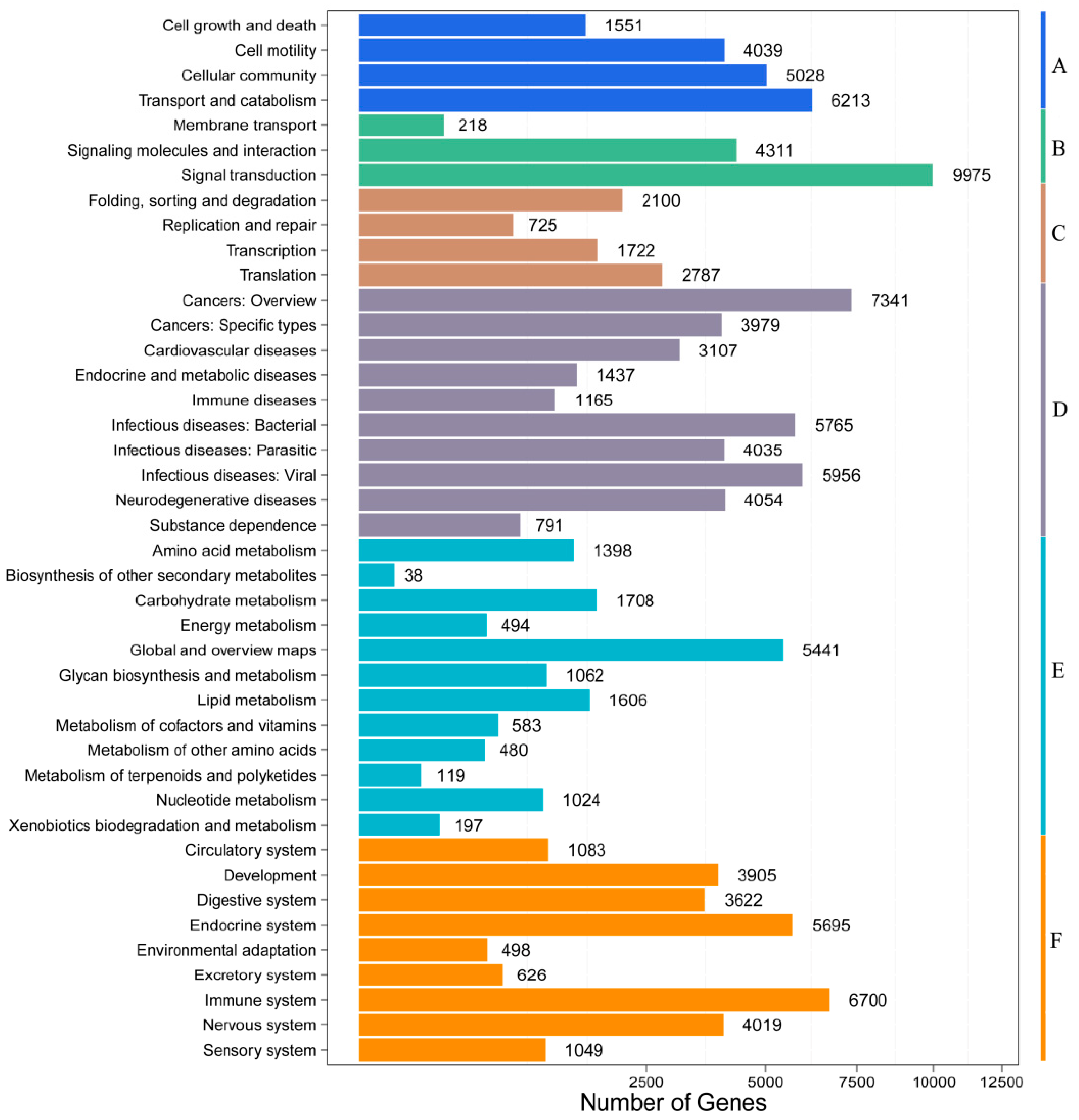
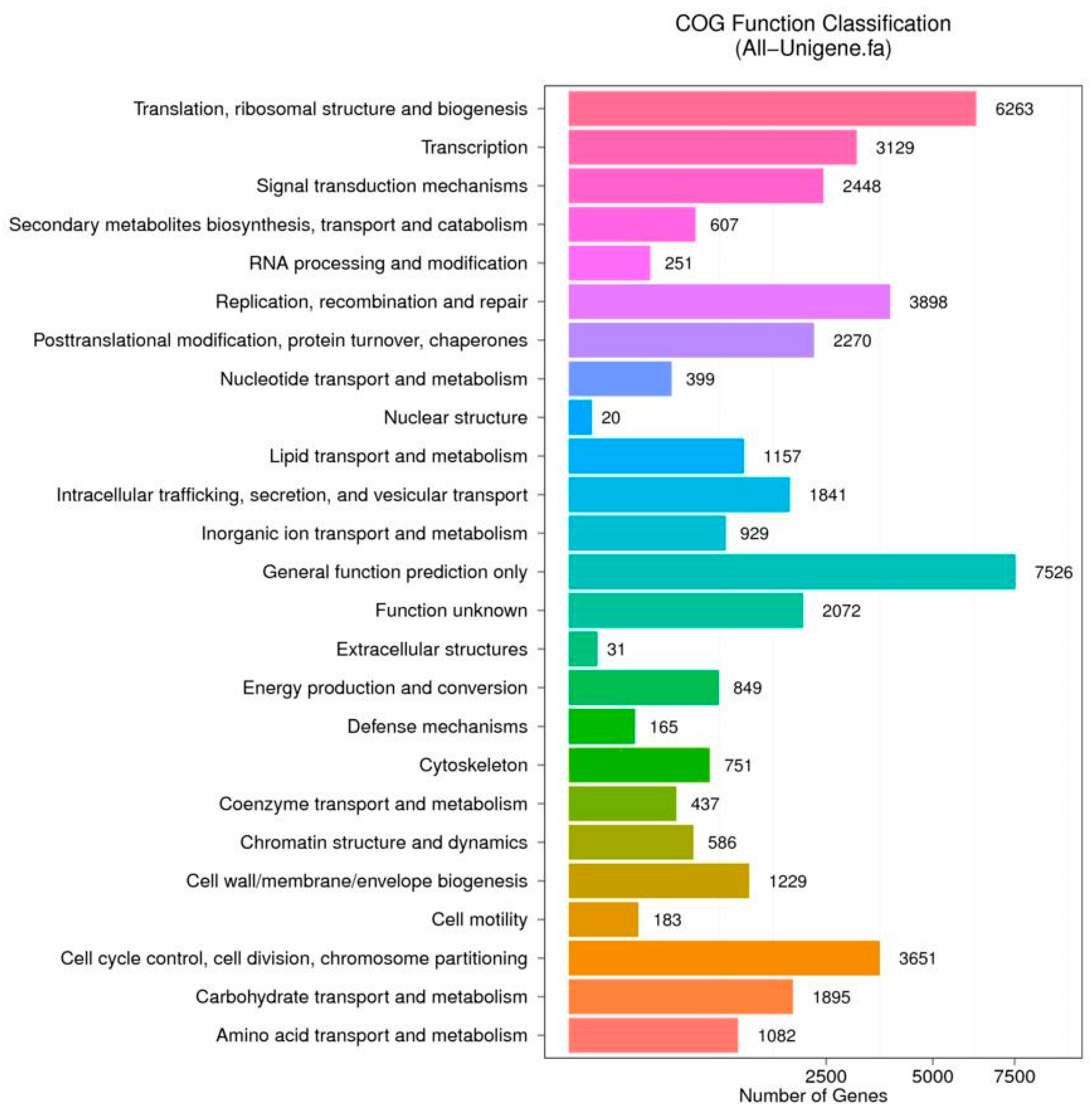
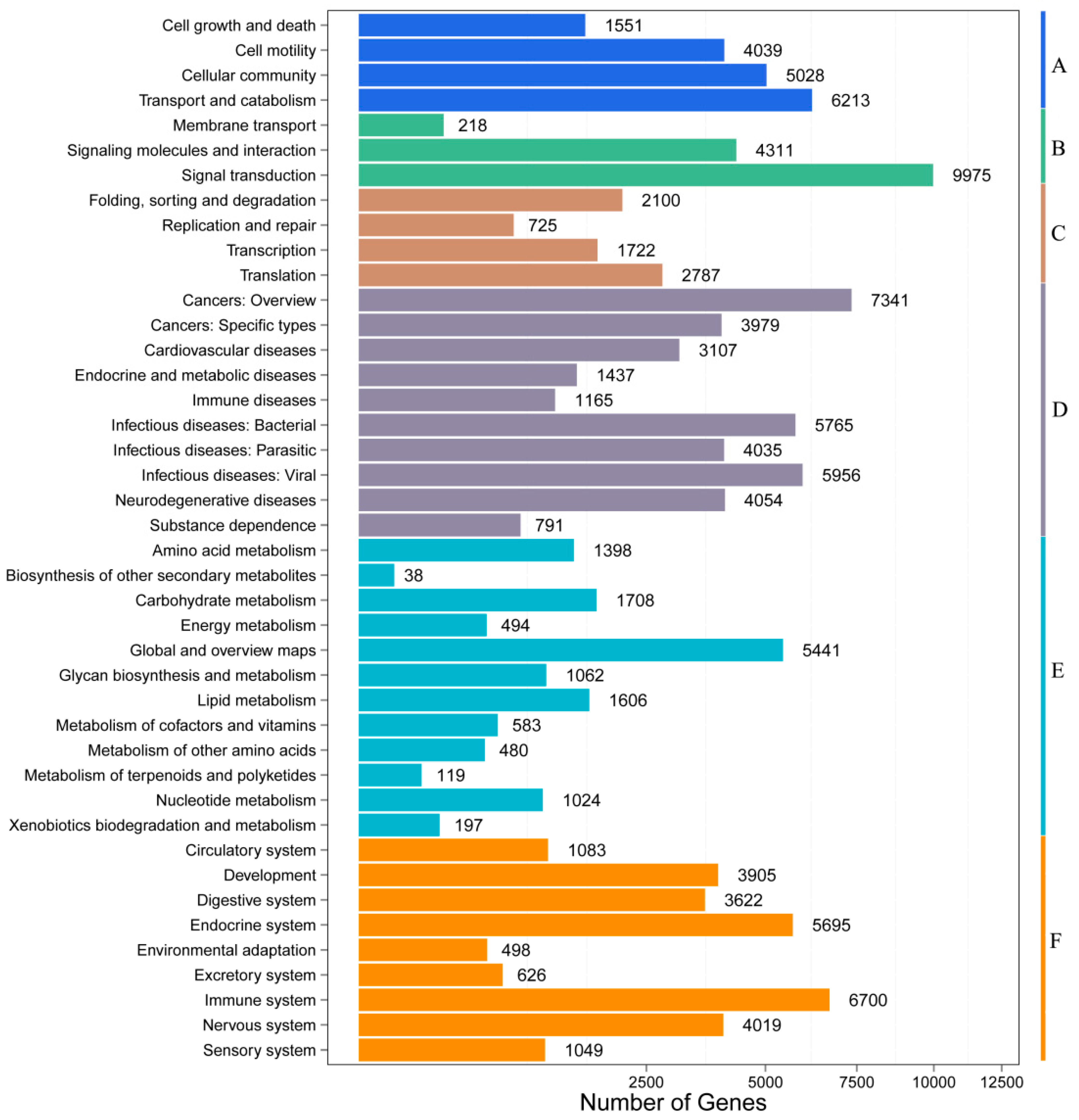
3.2. Functional Annotation and Pathway Assignment
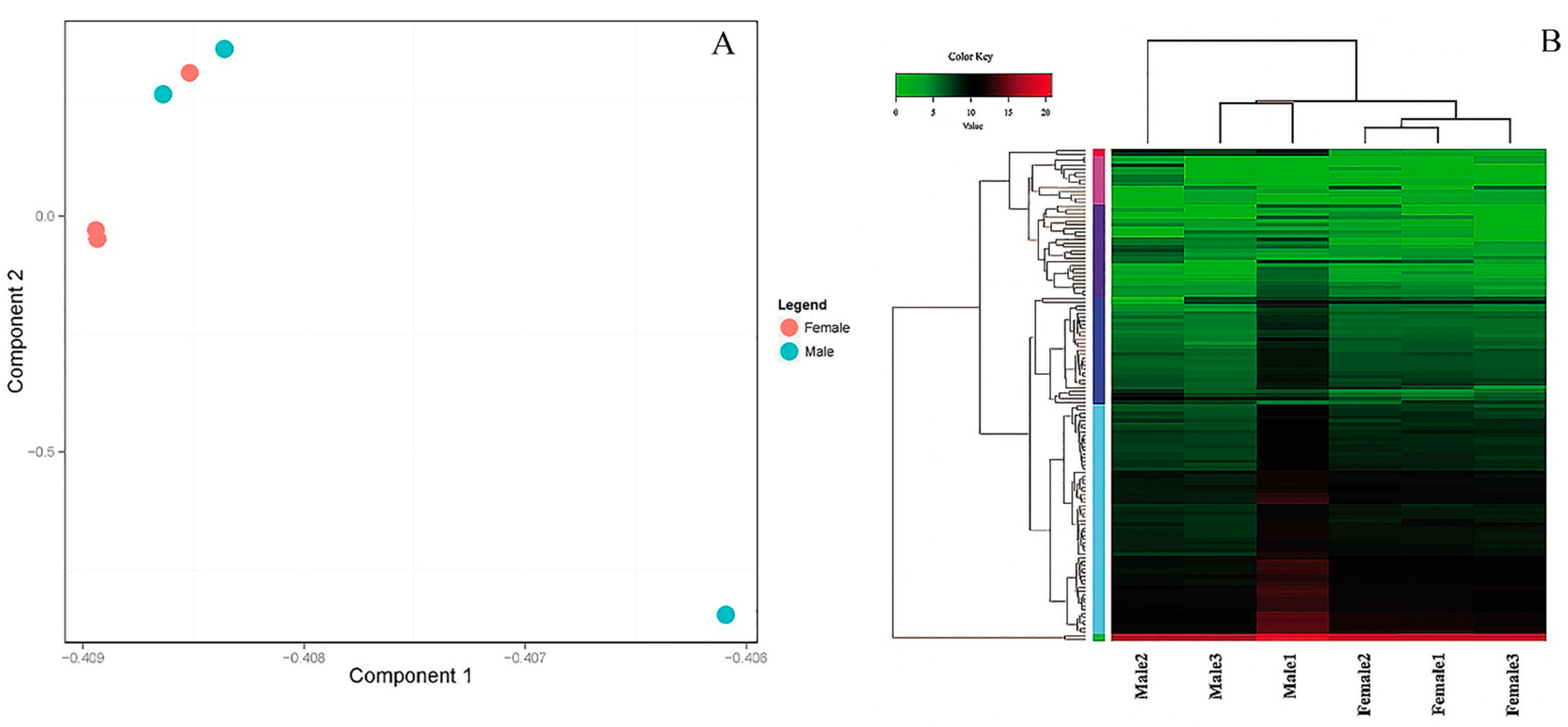
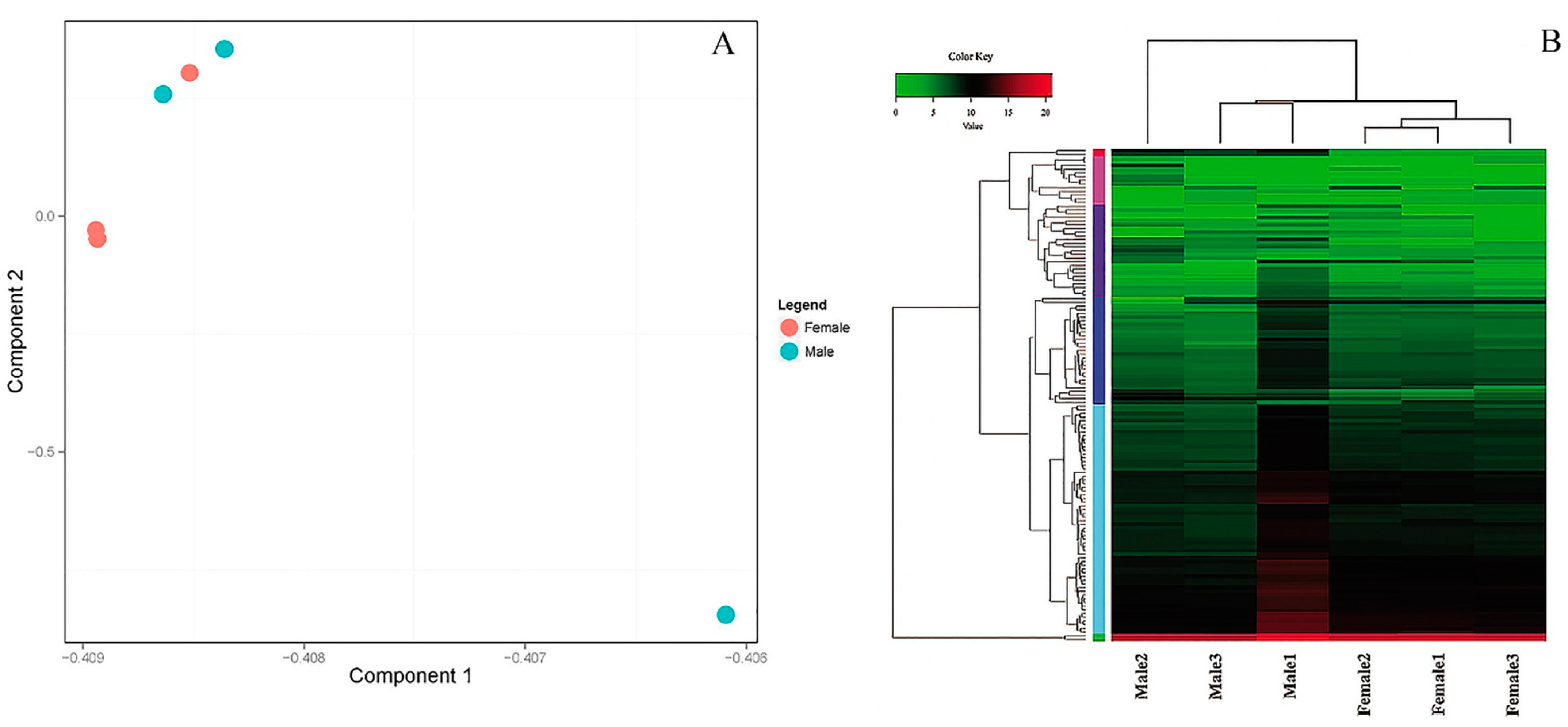
3.3. PCA Plot, Heatmap and Clustering Diagram Analysis
3.4. Differential Gene Expression Patterns
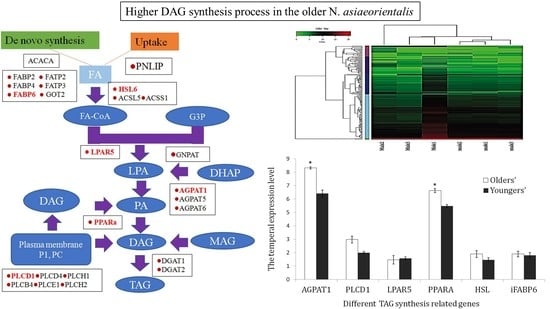
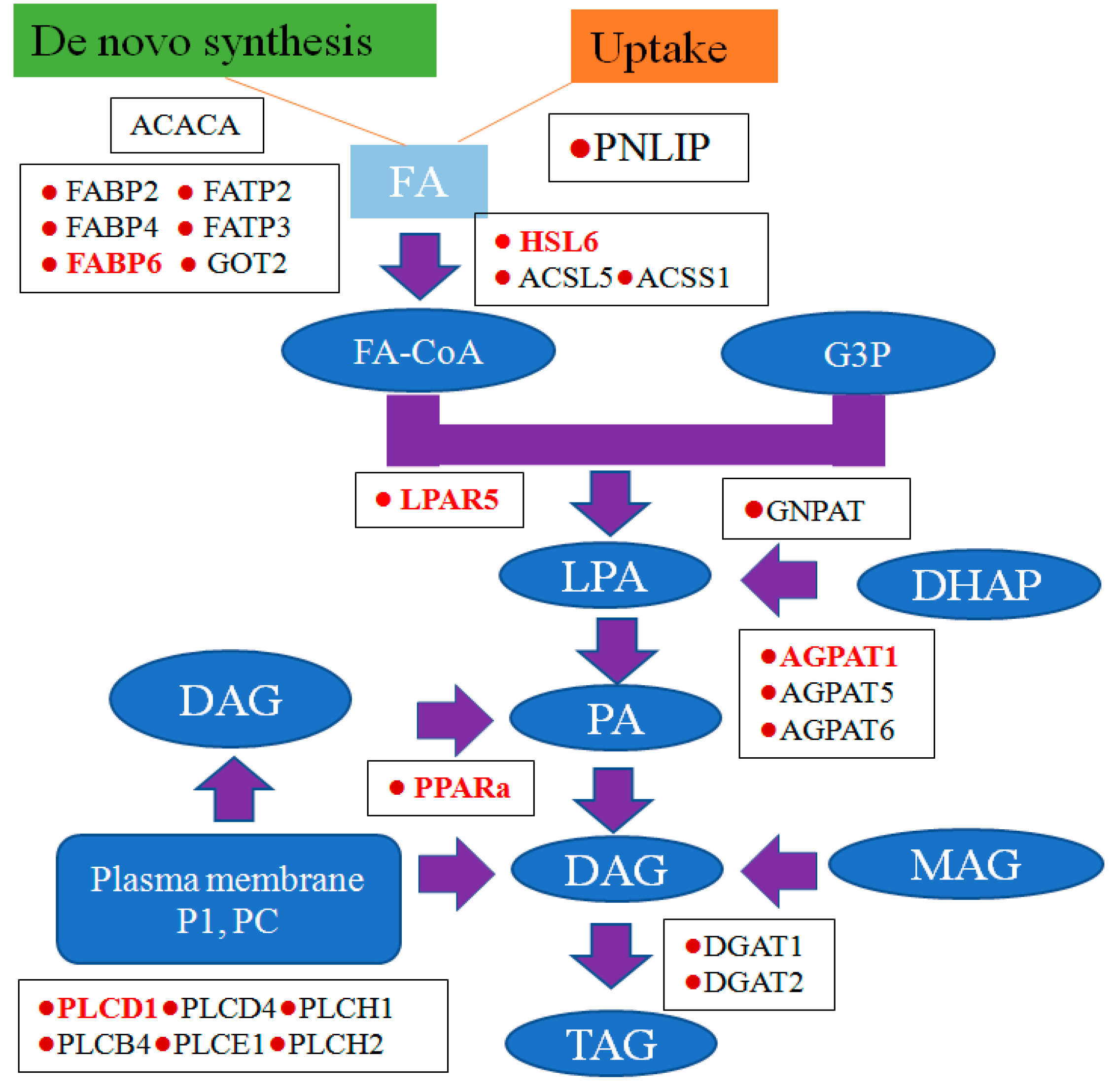
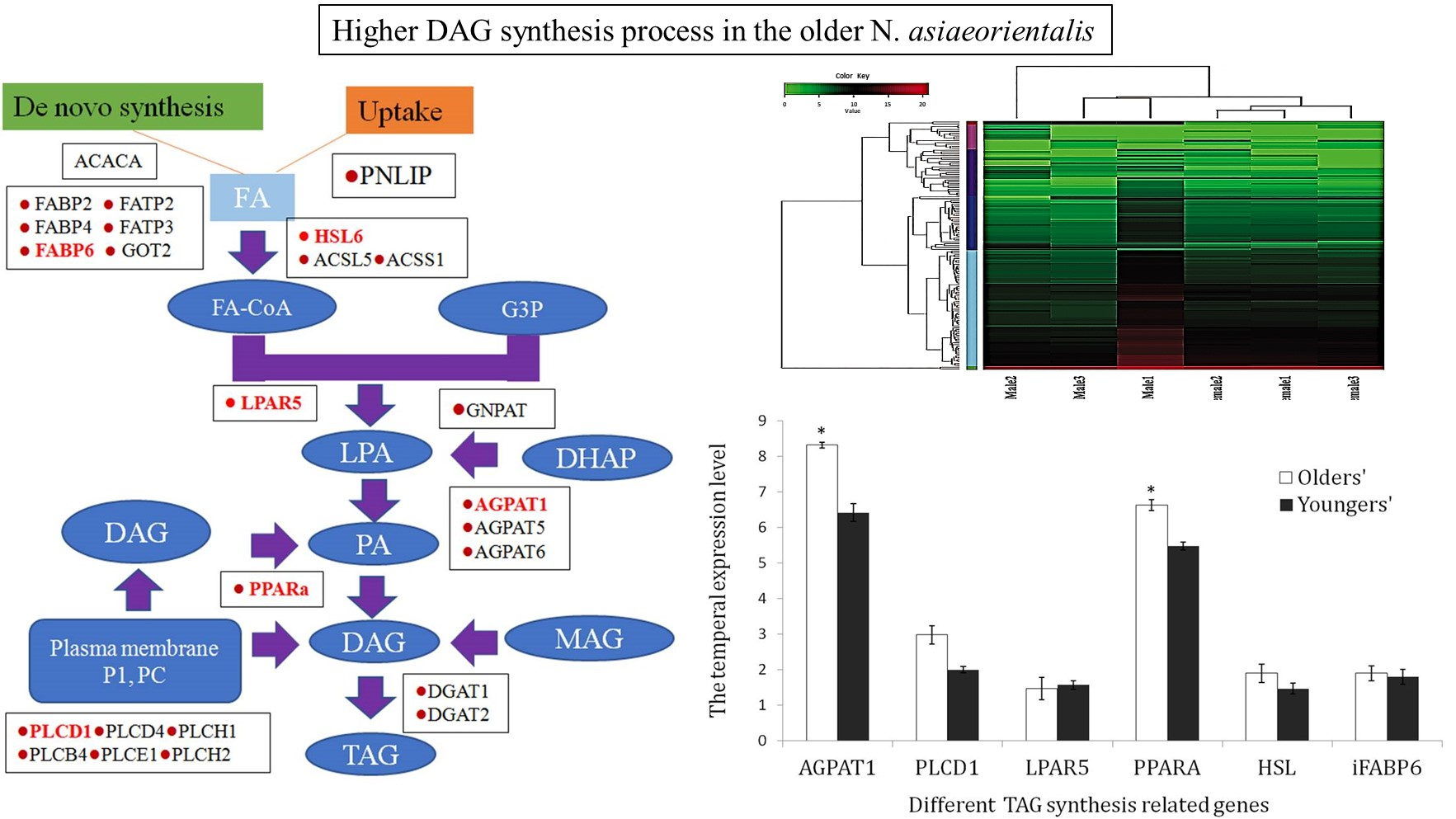
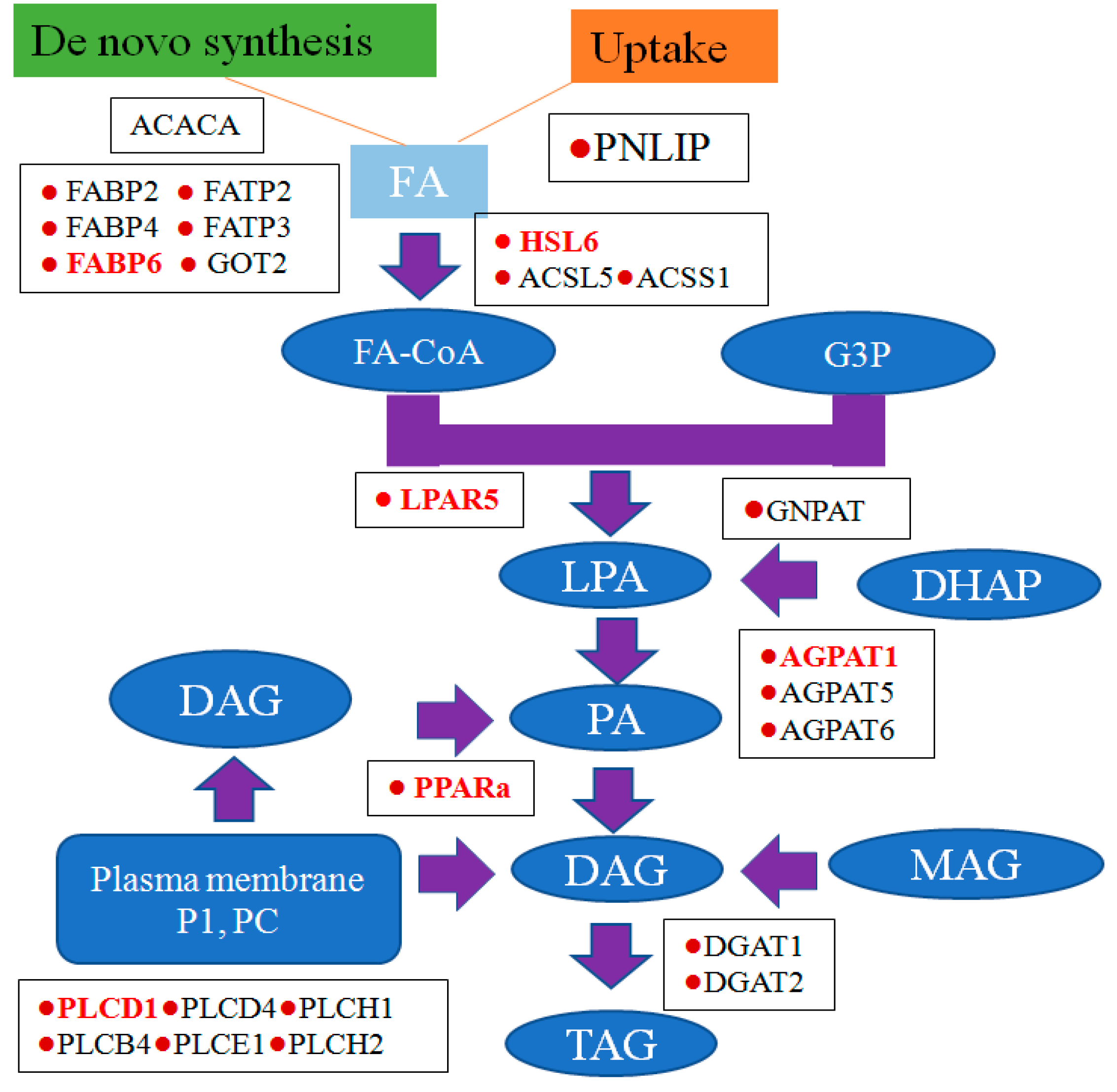
3.5. Potential Role of Triacylglycerol (TAG) Synthesis
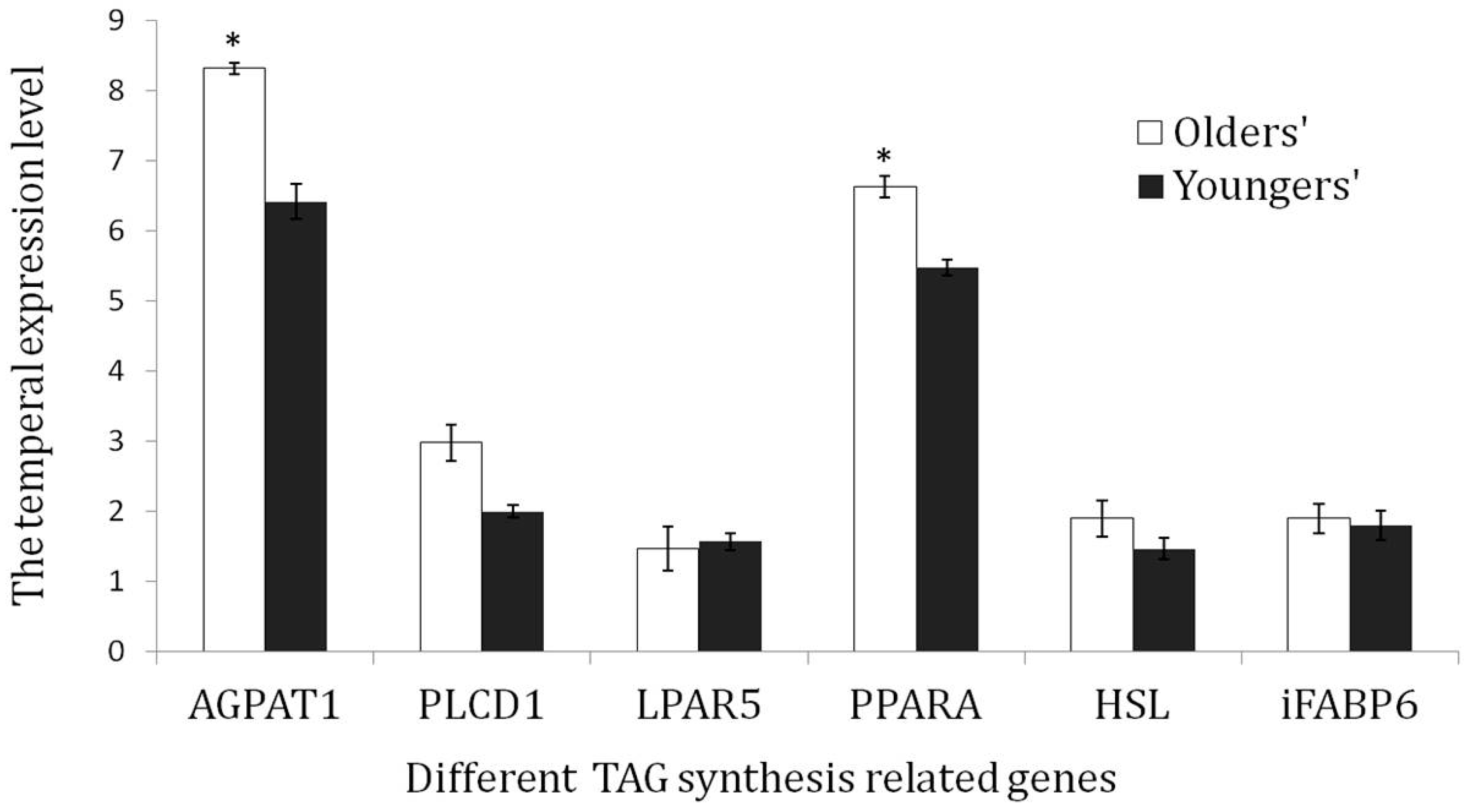
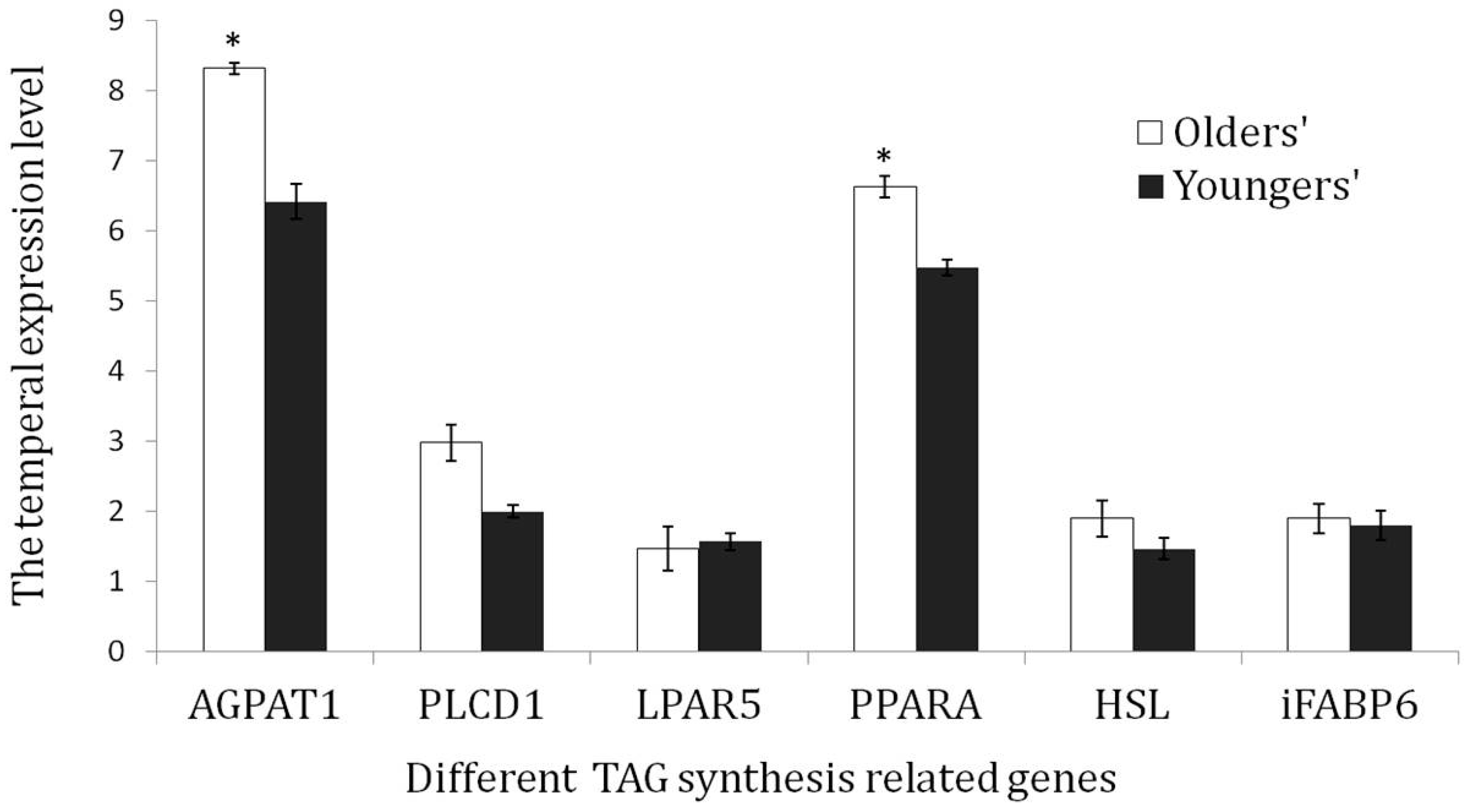
3.6. Validation of the TAG Synthesis-Related Genes via qRT-PCR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, D.; Turvey, S.T.; Zhao, X.; Mei, Z. Neophocaena asiaeorientalis ssp. Asiaeorientalis. IUCN Red List Threat. Species 2013, e.T43205774A45893487. Available online: https://www.iucnredlist.org/species/43205774/45893487 (accessed on 1 January 2022).
- Zhou, X.; Guang, X.; Sun, D.; Xu, S.; Li, M.; Seim, I.; Jie, W.; Yang, L.; Zhu, Q.; Xu, J.; et al. Population genomics of finless porpoises reveal an incipient cetacean species adapted to freshwater. Nat. Commun. 2018, 9, 1276. [Google Scholar] [CrossRef] [Green Version]
- Mei, Z.; Zhang, X.; Huang, S.-L.; Zhao, X.; Hao, Y.; Zhang, L.; Qian, Z.; Zheng, J.; Wang, K.; Wang, D. The yangtze finless porpoise: On an accelerating path to extinction? Biol. Conserv. 2014, 172, 117–123. [Google Scholar] [CrossRef]
- Kimura, S.; Akamatsu, T.; Li, S.; Dong, L.; Wang, K.; Wang, D.; Arai, N. Seasonal changes in the local distribution of yangtze finless porpoises related to fish presence. Mar. Mammal Sci. 2012, 28, 308–324. [Google Scholar] [CrossRef]
- Wang, D. Population status, threats and conservation of the yangtze finless porpoise. Chin. Sci. Bull. 2009, 54, 3473. [Google Scholar] [CrossRef] [Green Version]
- Isobe, T.; Oshihoi, T.; Hamada, H.; Nakayama, K.; Yamada, T.K.; Tajima, Y.; Amano, M.; Tanabe, S. Contamination status of pops and bfrs and relationship with parasitic infection in finless porpoises (Neophocaena phocaenoides) from seto inland sea and omura bay, Japan. Mar. Pollut. Bull. 2011, 63, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Barlow, J.; Taylor, B.L.; Pitman, R.L.; Wang, K.; Wei, Z.; Stewart, B.S.; Turvey, S.T.; Akamatsu, T.; Reeves, R.R.; et al. Abundance and conservation status of the yangtze finless porpoise in the yangtze river, china. Biol. Conserv. 2008, 141, 3006–3018. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Qian, Z.; Ruan, Y.; Hao, Y.; Dong, W.; Li, K.; Mei, Z.; Wang, K.; Wu, C.; Wu, J.; et al. First evaluation of legacy persistent organic pollutant contamination status of stranded yangtze finless porpoises along the yangtze river basin, china. Sci. Total Environ. 2020, 710, 136446. [Google Scholar] [CrossRef]
- Muka, S.; Zarpentine, C. Cetacean conservation and the ethics of captivity. Biol. Conserv. 2021, 262, 109303. [Google Scholar] [CrossRef]
- Ju, J.; Yang, M.; Xu, S.; Zhou, K.; Yang, G. High level population differentiation of finless porpoises (Neophocaena phocaenoides) in chinese waters revealed by sequence variability of four nuclear introns. Mol. Biol. Rep. 2012, 39, 7755–7762. [Google Scholar] [CrossRef]
- Spitz, J.; Becquet, V.; Rosen, D.A.S.; Trites, A.W. A nutrigenomic approach to detect nutritional stress from gene expression in blood samples drawn from steller sea lions. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2015, 187, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.-S.; Cho, Y.S.; Guang, X.; Kang, S.G.; Jeong, J.-Y.; Cha, S.-S.; Oh, H.-M.; Lee, J.-H.; Yang, E.C.; Kwon, K.K.; et al. Minke whale genome and aquatic adaptation in cetaceans. Nat. Genet. 2014, 46, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.Y.; Kwon, M.G.; Jung, S.-H.; Park, M.A.; Kim, D.-W.; Cho, W.S.; Park, J.W.; Son, M.-H. Rna-seq transcriptome analysis of the olive flounder (paralichthys olivaceus) kidney response to vaccination with heat-inactivated viral hemorrhagic septicemia virus. Fish Shellfish Immunol. 2017, 62, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Mancia, A. Chapter 11—new technologies for monitoring marine mammal health. In Marine Mammal Ecotoxicology; Fossi, M.C., Panti, C., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 291–320. [Google Scholar]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Liu, Y. Soapdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 2047–2217X. [Google Scholar] [CrossRef]
- Grabherr, M.; Haas, B.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I. Trinity: Reconstructing a full-length transcriptome without a genome from rna-seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. Tigr gene indices clustering tools (tgicl): A software system for fast clustering of large est datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from rna-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The cog database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The kegg resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. Ebseq: An empirical bayes hierarchical model for inference in rna-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2−δδct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Mancia, A.; Ryan, J.C.; Van Dolah, F.M.; Kucklick, J.R.; Rowles, T.K.; Wells, R.S.; Rosel, P.E.; Hohn, A.A.; Schwacke, L.H. Machine learning approaches to investigate the impact of pcbs on the transcriptome of the common bottlenose dolphin (tursiops truncatus). Mar. Environ. Res. 2014, 100, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Shen, H.; Liu, J.; Xie, F.; Zhang, W.; Mai, K. High level of dietary soybean oil depresses the growth and anti-oxidative capacity and induces inflammatory response in large yellow croaker larimichthys crocea. Fish Shellfish Immunol. 2018, 77, 465–473. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Z.; Xu, S.; Ren, W.; Zhou, K.; Yang, G. ‘Obesity’ is healthy for cetaceans? Evidence from pervasive positive selection in genes related to triacylglycerol metabolism. Sci. Rep. 2015, 5, 14187. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Hayashi, Y.; Nemoto-Sasaki, Y.; Ito, M.; Oka, S.; Tanikawa, T.; Waku, K.; Sugiura, T. Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog. Lipid Res. 2014, 53, 18–81. [Google Scholar] [CrossRef]
- Springer, M.S.; Guerrero-Juarez, C.F.; Huelsmann, M.; Collin, M.A.; Danil, K.; McGowen, M.R.; Oh, J.W.; Ramos, R.; Hiller, M.; Plikus, M.V.; et al. ; et al. Genomic and anatomical comparisons of skin support independent adaptation to life in water by cetaceans and hippos. Curr. Biol. 2021, 31, 2124–2139.e3. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (ppars): Tissue distribution of ppar-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Körbes, A.P.; Kulcheski, F.R.; Margis, R.; Margis-Pinheiro, M.; Turchetto-Zolet, A.C. Molecular evolution of the lysophosphatidic acid acyltransferase (lpaat) gene family. Mol. Phylogenetics Evol. 2016, 96, 55–69. [Google Scholar] [CrossRef]
- Hammond, G.R.V.; Balla, T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 746–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Brocker, C.N.; Yan, T.; Xie, C.; Krausz, K.W.; Xiang, R.; Gonzalez, F.J. Corrigendum to “metabolic adaptation to intermittent fasting is independent of peroxisome proliferator-activated receptor alpha”. Mol. Metab. 2018, 9, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-E.; Herr, D.R.; Chun, J. Lysophosphatidic acid (lpa) receptors: Signaling properties and disease relevance. Prostaglandins Other Lipid Mediat. 2010, 91, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, H.; Souza, S.C.; Zhang, H.-H.; Strissel, K.J.; Christoffolete, M.A.; Kovsan, J.; Rudich, A.; Kraemer, F.B.; Bianco, A.C.; Obin, M.S.; et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 15837–15844. [Google Scholar] [CrossRef] [Green Version]
- Fisher, E.; Grallert, H.; Klapper, M.; Pfäfflin, A.; Schrezenmeir, J.; Illig, T.; Boeing, H.; Döring, F. Evidence for the thr79met polymorphism of the ileal fatty acid binding protein (fabp6) to be associated with type 2 diabetes in obese individuals. Mol. Genet. Metab. 2009, 98, 400–405. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | No. of Raw Reads (Mb) | Length (bp) | No. of Bases (Gb) | No. of Clean Reads (Mb) | Clean Reads Q20 | Clean Reads Q30 | Clean Reads Ratio (%) |
|---|---|---|---|---|---|---|---|
| Female 1 | 50.86 | 150 | 7.63 | 48.14 | 96.87 | 93.07 | 94.66 |
| Female 2 | 50.64 | 150 | 7.60 | 47.99 | 96.93 | 93.17 | 94.77 |
| Female 3 | 43.48 | 150 | 6.52 | 40.44 | 96.81 | 92.98 | 93.03 |
| Male 1 | 47.91 | 150 | 7.19 | 44.76 | 97.08 | 93.45 | 93.42 |
| Male 2 | 43.97 | 150 | 6.60 | 42.02 | 96.88 | 93.11 | 95.56 |
| Male 3 | 44.10 | 150 | 6.62 | 41.40 | 96.77 | 92.91 | 93.88 |
| Primers Name | Sequence(5’–3’) | Tm | Products |
|---|---|---|---|
| 1-acylglycerol-3-phosphate O-acyltransferase 1 | |||
| AGPAT1-F AGPAT1-R | AGGTTCCCATTGTTCCCATAGTCAT CCTCCAGGCTTCTTCAGATAGTCAC | 64.2 62.8 | 238 bp |
| phospholipase C, delta 1, transcript variant 2 | |||
| PLCD1-F PLCD1-R | AAGGACAACAAGATGAACTTCAAGGAG AGACCACTAACTGATCCACCGACAG | 63.8 63.6 | 249 bp |
| lysophosphatidic acid receptor5 | |||
| LPAR5-F LPAR5-R | TCTGAGTCTTGACTGGGACTACGTT CACCATCCCTGGGTATTCTCATCTC | 61.4 65.0 | 194 bp |
| peroxisome proliferator-activated receptor α | |||
| PPARA-F PPARA-R | CCCTGAACTGGTTGACTGTGCTTTA CTTGTACTGGCTTGGCTAGGATAGG | 64.7 63.2 | 173 bp |
| Hormone-sensitive lipase | |||
| HSL-F HSL-R | CGCAAGATGATATGAGAATAACTGC CCTCTGTGAAATGGGTACAAGAAGA | 60.5 61.8 | 221 bp |
| fatty acid binding protein 6, ileal | |||
| iFABP6-F iFABP6-R | AAGACTATCTCAGAGGTGCAGCAGG CACGATCTCCGCAGTGTAGTTGTAG | 63.4 63.2 | 210 bp |
| β-actin | |||
| β-actin-F β-actin-R | GAATCTTGTGGCATCCACGAAACTA GATGATCTTGATCTTCATCGTGCTG | 64.5 62.8 | 183 bp |
| Sample | Total Number | Total Length | Mean Length | N50 | N70 | N90 | GC (%) |
|---|---|---|---|---|---|---|---|
| Female 1 | 89,766 | 88,972,821 | 991 | 2320 | 1059 | 323 | 50.91 |
| Female 2 | 86,000 | 82,641,300 | 960 | 2250 | 1003 | 314 | 51.13 |
| Female 3 | 97,485 | 94,156,799 | 965 | 2251 | 996 | 316 | 51.15 |
| Male 1 | 37,907 | 32,026,539 | 844 | 1593 | 778 | 308 | 52.47 |
| Male 2 | 102,959 | 100,765,677 | 978 | 2363 | 1030 | 317 | 50.35 |
| Male 3 | 101,594 | 98,586,378 | 970 | 2341 | 1019 | 314 | 50.96 |
| Annotated Databases | Annotated Genes (Female/Male) | Annotated Percentage | Total Annotated Genes/Percentage |
|---|---|---|---|
| All annotated unigenes | 109,043/110,352 | 46.26%/44.84% | 119,039 (78.72%) |
| Nr | 49,794/48,801 | 45.66%/44.22% | 61,071 (40.39%) |
| SwissProt | 43,277/42,212 | 39.69%/38.25% | 52,607 (34.79%) |
| KEGG | 39,796/38,732 | 36.50%/35.10% | 48,051 (31.78%) |
| COG | 19,206/18,724 | 17.61%/16.97% | 23,017 (15.22%) |
| GO | 5564/5480 | 5.10%/4.97% | 6563 (4.34%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, D.-A.; Liu, K.; Xu, D.-P.; Wang, Y.-P.; Xu, P. Comparative Analysis of Blood Transcriptome in the Yangtze Finless Porpoise (Neophocaena asiaeorientalis). Fishes 2022, 7, 61. https://doi.org/10.3390/fishes7020061
Fang D-A, Liu K, Xu D-P, Wang Y-P, Xu P. Comparative Analysis of Blood Transcriptome in the Yangtze Finless Porpoise (Neophocaena asiaeorientalis). Fishes. 2022; 7(2):61. https://doi.org/10.3390/fishes7020061
Chicago/Turabian StyleFang, Di-An, Kai Liu, Dong-Po Xu, Yin-Ping Wang, and Pao Xu. 2022. "Comparative Analysis of Blood Transcriptome in the Yangtze Finless Porpoise (Neophocaena asiaeorientalis)" Fishes 7, no. 2: 61. https://doi.org/10.3390/fishes7020061
APA StyleFang, D.-A., Liu, K., Xu, D.-P., Wang, Y.-P., & Xu, P. (2022). Comparative Analysis of Blood Transcriptome in the Yangtze Finless Porpoise (Neophocaena asiaeorientalis). Fishes, 7(2), 61. https://doi.org/10.3390/fishes7020061