Abstract
The Chinese razor clam (Sinonovacula constricta), a bivalve species widely distributed in estuaries and mudflats, is often exposed to extreme environmental and microbial stresses. Histones are fundamental components of chromatin and play an important role in innate immunity, as demonstrated by its antimicrobial activities in clams. However, little attention has been paid to histones in bivalves. To fill this gap, we investigated the genomic distribution, structural characteristics, conserved motifs, and phylogenetic relationships of histones in S. constricta. A total of 114 histone genes were detected in the S. constricta genome, which were divided into 25 types in phylogenetic analysis. Among them, partial histones exhibited a tissue-dependent expression pattern, indicating that they may be involved in sustaining the homeostasis of organs/tissues in adult S. constricta. Furthermore, mRNA expression of certain histones changed significantly in S. constricta when infected with Vibrio parahaemolyticus, suggesting that histones play a role in the immune defense of S. constricta. All together, this study on histone genes in S. constricta not only greatly expands our knowledge of histone function in the clam, but also histone evolution in molluscs.
1. Introduction
Histones in eukaryotes are basically structural proteins of nucleosomes, which are responsible for the packing and compaction of DNA in the cell nucleus, thereby forming chromatin fiber [1,2,3]. Based on their structure and function, the histone gene family has been classified into two major groups: core histones comprised of H2A, H2B, H3, and H4, and linker histones comprised of H1 [4,5,6,7]. Histone variants, which have been distinguished from canonical histones, exhibit unique expression, chromosomal localization, and species-distribution patterns, and have acquired divergent properties during evolution [8,9]. Increasing evidence has revealed the significant contribution of histone variants to gene regulation and nucleosome function [10,11].
Generally, histone genes with high copy numbers are structured into clusters. These genes contain no introns and lack polyadenylation in their corresponding mRNAs, but contain a unique 3′ end structure. Conversely, the histone variants encoded by polyadenylated mRNAs are usually distributed independently and do contain introns. Canonical histones have a particular stem-loop structure located at the 3′ untranslated region, which is generated by cleavage using a machinery involving U7 snRNP and protein factors such as the stem-loop binding protein (SLBP), as identified in both metazoans and protozoans [12]. Additionally, there is also a histone downstream element (HDE) downstream of the stem-loop structure directed by endonucleolytic cleavage. The specific subunits Lsm10 and Lsm11 of the U7 snRNP can interact with the HDE element and be involved in the processing of histone mRNA, while the SLBP probably stabilizes binding of the U7 snRNP to the HDE [13,14]. In contrast, the histone variants exhibit constant but low-level expression through the cell cycle, and are present in chromatin in a replication-independent way, which is accompanied by the specialized chaperone and remodeling proteins [15,16]. Notably, the histone variant genes contain introns that allow for alternative splicing in some cases at least, and are not clustered in the genome [2,17].
Substantial evidence has shown that histones possess antibacterial activity and play a vital role in the innate immunity of animals, although the primary role of histones is as the dominant protein components of chromatin [18]. For example, histone H1 act as an important antimicrobial protein in the liver, intestine, and stomach of Atlantic salmon Salmo salar [19]. The recombinant histones H2A and H4 showed strong antibacterial activity in freshwater prawn Macrobrachium rosenbergii and African clawed frog Xenopus laevis against Escherichia coli, Vibrio anguillarum and Micrococcus luteus at micromolar concentrations [20]. In addition, high expression levels of core histones H2A, H2B, H3, and H4 were found in hemocytes of Pacific white shrimp Litopenaeus vannamei when responding to bacterial stress [21]. In molluscs, the histone H2A homologous to buforin I found in scallop Chlamys farreri and disk abalone Haliotis discus discus, inhibits the growth of Gram-positive and Gram-negative bacteria and yeast [22,23], and the histones H2B and H4 in eastern oyster Crassostrea virginica can defend against a variety of pathogenic bacteria [24].
With the completion of genome sequencing projects, investigations have been carried out to explore the histone gene family in a variety of species, such as C. farreri [25], grooved razor shell Solen marginatus [26], mediterranean mussel Mytilus galloprovincialis [1], the parasite Schistosoma japonicum [27], and the clam Lucina pectinata [28]. Studies on S. japonicum have demonstrated that there are totally 38 histone genes, and they are divided into 5 subfamilies and distributed on 25 scaffolds [27]. Due to their wide distribution and characteristics in eukaryotes, the histones are deemed to represent a model system for studying multigene families with their organization, structure, and expression, which are of interest in phylogenetic and evolutionary studies [29]. Recently, completion of the razor clam genome sequencing project [30] offers a reliable genome. Furthermore, abundant transcriptome datasets for razor clam enable systematic characterization of the crucial histone gene family.
The razor clam (Sinonovacula constricta) is a mariculture bivalve mollusc in China with economic importance because of its delicious taste and high nutritional value [30,31]. This species is widely distributed in the intertidal region and can be vulnerable to bacterial stresses during its lifetime, especially from the pathogenic Vibrios. V. parahaemolyticus is a curved, rod-shaped, and Gram-negative bacterium found in the sea, which has been reported to cause adverse effects such as decreasing growth, weakened immunity, and even outbreaks of mass mortality of shellfish [30,32]. Notably, bivalve species have no adaptive immune defense mechanism and rely on innate immunity [33]. In this regard, it is of great significance to characterize the innate immunity of S. constricta, which can contribute to sustainable development of its production industry. Given the important role of histones in the innate immunity of animals, we identified and characterized the genome-wide histone repertoire in S. constricta. A systematic analysis of histone genes was performed, including their gene copy numbers, gene structure, conserved motifs, genomic organization, and phylogenetic relationships. In addition, we investigated the expression profiles of histone genes in different organs/tissues and under stress of V. parahaemolyticus in S. constricta. Our findings elucidate the potential function of histone genes in S. constricta, which enhance our understanding of innate immunity in bivalves.
2. Materials and Methods
2.1. Genome Resource
The genome sequence data of S. constricta [30] was used for this study. Other genome sequence data for Homo sapiens (GCA_000001405.28), Xenopus laevis (GCA_001663975), Drosophila melanogaster (GCA_000001215.4), Branchiostoma belcheri (GCA_001625305.1), Crassostrea virginica (GCA_002022765.4), Crassostrea gigas (GCA_902806645.1), Tegillarca granosa (GCA_013375625.1), Pecten maximus (GCA_902652985.1), Mizuhopecten yessoensis (GCA_002113885.2), Octopus bimaculoides (GCA_001194135.1), Strongylocentrotus purpuratus (GCA_000002235.4), and Caenorhabditis elegans (GCA_000002985.3) were downloaded from the NCBI genome database (https://www.ncbi.nlm.nih.gov/genome/, accessed on 21 January 2006).
2.2. Genome-Wide Identification of Histone Genes
To identify histone genes in S. constricta, the known full-length histone amino acid sequences of C. virginica and P. maximus, which were retrieved from NCBI, were first used as initial query sequences to conduct a BLASTP search against the S. constricta genome with a cut-off E-value of 1 × 10−10 [34]. Then, those retained sequences that significantly matched known histone genes were further classified into corresponding groups (H1, H2A, H2B, H3, H4) based on the best BLAST hits. Subsequently, all candidate histone genes were compared back to the NCBI non-redundant database (BLASTX), and we removed manually spurious sequences. Last, to confirm and filter uncertain histone proteins, the conserved domains of candidate histone genes were predicted by the SMART tool (http://smart.embl-heidelberg.de, accessed on 26 October 2020) and CDSearch (https://www.ncbi.nlm.nih.gov/structure/bwrpsb/bwrpsb.cgi, accessed on 1 July 2004). Simultaneously, open reading frames (ORFs) were predicted using ORF-Finder (https://indra.mullins.microbiol.washington.edu/sms2/orf_find.html, accessed on 13 December 2006). The identified sequences were used for further analysis. Similar procedures were carried out to retrieve histone genes in the genomes of H. sapiens, X. laevis, D. melanogaster, B. belcheri, C. virginica, C. gigas, T. granosa, P. maximus, O. bimaculoides, S. purpuratus, and C. elegans.
2.3. Sequence Analysis and Chromosomal Localization of Histone Genes in S. constricta
Physicochemical parameters, including the molecular weight (kDa), isoelectric point (pI) and grand average of hydropathicity (GRAVY) of each gene product, were calculated using ExPASy (http://www.expasy.org/tools/, accessed on 1 July 2020) with default parameters. The gene structures were drawn by Tbtools [35]. The conserved motifs were predicted using the online program MEME (http://meme-suite.org/tools/meme, accessed on 8 February 2021) with motif length of 6–50 bp, and the maximum number of motifs was 15 [36]. The exact location of the stop codon in the histone coding sequence was determined by the gff3 annotation file of the S. constricta genome. Then, 300 nt downstream of the stop codon was extracted and aligned to identify the stem-loop structure and purine-rich element of the histone gene, and finally visualized by Jalview. Additionally, BLASTX was used to search for homologues of SLBP, Lsm10, Lsm11, U7 snRNP, and Symplekin proteins in the S. constricta genome, while SMART was used to identify the protein domains. Each histone gene was mapped to the respective chromosomes using the TBtools software [35], and the percent identity matrix of nucleotide sequences between duplicate genes was calculated by Clustal Omega.
2.4. Classification of Histone Genes
To classify all the newly retrieved histone genes into their respective groups, we constructed an unrooted phylogenetic tree using known histone genes by MrBayes 3.2.6 [37]. All histone genes in S. constricta were sequentially named based on the phylogenetic tree, number of copies, and chromosomal position.
2.5. Sequence Alignment and Phylogenetic Analysis
The multiple sequence alignment was produced using MAFFT 7.471 [38] with the FFT-NS-i algorithm for the full-length amino acid sequences and Gblocks [39] was used to delete gaps. The result of multiple alignment was converted to Nexus format through MEGA X for further analyses. Bayesian Inference (BI) phylogenetic analysis was conducted by MrBayes 3.2.6 [40]. Four Markov chains were run for 5 × 107 generations, with sampling performed every 2000 generations, to yield a posterior probability distribution of 104 trees. The first 25% of the trees were discarded as burn-in when compiling summary statistics and consensus trees. Finally, the alignments and Bayesian tree were visualized using Jalview version 2.11.1.0 and iTOL, respectively [41]. In total, the phylogenetic analysis involved 219 amino acids across 6 animals, including S. constricta, C. gigas, P. maximus, H. sapiens, C. elegans, and S. purpuratus.
2.6. Expression Profiles Assessed by Transcriptomics and qRT-PCR
To further characterize the histone genes’ expression patterns in S. constricta, the RPKM (reads per kilo per million reads) values were acquired from the public RNA-seq datasets of different organs/tissues of adult razor clam. Six organs/tissues, including gill, foot, adductor muscle, hepatopancreas, mantle and siphon, were obtained from our laboratory (accession numbers: gillSorry for the mistakes. We revised all the tables’ order, SRR2162883; foot, SRR2162887; adductor muscle, SRR2162892; hepatopancreas, SRR2162895; mantle, SRR2162898; siphon, SRR2162902). To visualize the gene expression patterns, heatmaps were developed using the heatmap package [35] under the R environment. Quantitative real-time PCR (qRT-PCR) was performed to measure histone genes’ expression under V. parahaemolyticus challenge with a final concentration of 1 × 108 cfu/mL. The 18S rRNA was used as the housekeeping gene to normalize the gene expression level. The primers for qRT-PCR are shown in Table 1. Differences were considered significant at p < 0.05.

Table 1.
Histone primers of S. constricta for qRT-PCR.
3. Results
3.1. Genome-Wide Identification and Genomic Distribution of Histone Genes in S. constricta
A total of 114 histone genes were detected from whole-genome sequences of S. constricta (Table 2). The details on gene types, genomic distribution, and other characteristics of histone genes are displayed in Table 2, Figure 1, and Supplementary Table S1. In order to exactly describe histone genes, each member was named following three steps. First, each gene name was started with H1, H2A, H2B, H3, or H4 to distinguish five major groups based on their sequence similarity. Second, the members in each group belonging to different types in which the similarities were not 100% were designated “.1”, “.2”, e.g., Sc H1.2 and H1.5 at ctg4484 and Hic_sam_1, respectively. Last, the members whose sequence identities were 100% were classified into same type and then numbered by “−1”, “−2” according to the genomic location, e.g., ScH2A.1-1 and ScH2A.1-2 at ctg6665 (Figure 1). As shown in Table 2, the razor clam histone genes were divided into 25 types in total. We identified seven histone types in H1 group, which consisted of five histone H1-delta (ScH1.1, ScH1.2, ScH1.3, ScH1.4, and ScH1.5), one oocyte-specific (ScH1oo), and one replication-independent variants (ScH1.0). In the H2A group, two types (H2A.1 and H2A.2) showed significant similarity to canonical H2A, and the remaining three variant types were matched to ScH2A.V, ScH2A.1macro, and ScH2Asperm, respectively. The H2B group comprised four types and showed a high degree of similarity to the canonical forms, while the H3 group consisted of a maximum of eight types. Notably, S. constricta had only one H4 type, which was highly consistent with most organisms, but did not display variants for H4. It is worth noting that a large portion of histone types were encoded by multiple copies in the genome of S. constricta. Further, we counted all copies of these histone genes. The results showed that H1 (ScH1.1, ScH1.3, ScH1.4), ScH2A.1, ScH2B.1, ScH3.1, and ScH4 included (4, 7, 2), 19, 22, 19, and 23 copies, respectively (Table 2).
Table 2.
Types of S. constricta histones.
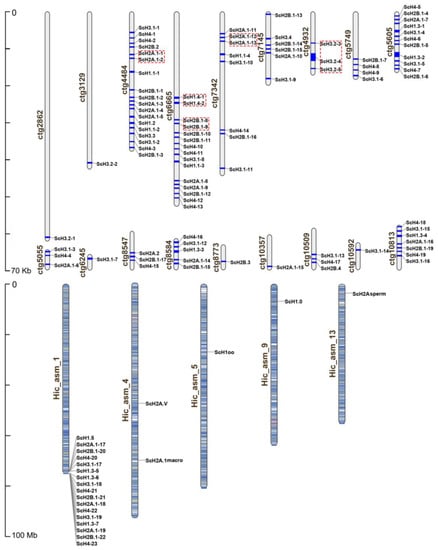
Figure 1.
Chromosomal distribution of the S. constricta histone genes. The gene names on the right side of each chromosome and scaffold correspond to the approximate locations of each histone gene. Tandemly duplicated homologs are marked by red boxes. Ctg means scaffolds that are not mapped on chromosomes, while Hic_sam means chromosomes.
The gene structures of the canonical histone of razor clam proved extremely conserved and contained only one exon, but the exon number of corresponding variants exhibited significant variability (Table 2), which paralleled those of vertebrate histones. The analysis of physical and chemical properties showed that the amino acid lengths of histones ranged from 94 aa (ScH3.2-3) to 381 aa (ScH2A.1macro), while the molecular weights ranged from 10.61 kDa (ScH3.2-3) to 40.62 kDa (ScH2A.1macro). The predicted pI was 9.95 (ScH2A.1macro) ~11.45 (ScH3.2-3). Additionally, the value of grand average of hydropathy (GRAVY) varied from −0.159 (ScH2A.1macro) to −1.023 (ScH1.0), suggesting that all histone proteins were hydrophilic in S. constricta.
As shown in Figure 1, the 114 histone genes were located at 23 loci in total, among which only 22 histone genes were successfully mapped to 5 chromosomes. One possible explanation for this pattern was due to the inadequate assembly integrity of the S. constricta genome. Moreover, a majority of canonical histones were clustered on ctg4484 (18), Hic_asm_1 (17), ctg6665 (15), and ctg5605 (11), followed by ctg7342 (8), ctg10813 (7), ctg7145 (6), ctg8584 (5), and ctg5749 (4). In addition, the survey of gene duplication events indicated that tandem duplication occurred in five histone gene pairs (marked by red box) in S. constricta. Meanwhile, the comparison of nucleotide sequences showed that 100% sequence identity was observed in all gene pairs for ScH1.4, ScH2A.1 and ScH2B.1, while the sequence identity was higher than 94% for gene pair ScH3.2 (Supplementary Table S2). Collectively, the results supported the occurrence of tandem duplication rather than segmental duplication (Figure 1).
3.2. Phylogenetic Analysis of Histone Genes
To compare the dynamics that occurred in different types of histones in S. constricta, a multiple sequence alignment of putative proteins from molluscs and reference human sequences was implemented. The multiple sequence alignment showed that H3 and H4 were more highly conserved than H1, H2A, and H2B (Figure 2 and Supplementary Figures S1–S4). The two major regions, including those downstream of the N-terminal and upstream of the C-terminal, were hypervariable in core H2A and H2B (Figure 2). Notably, both H1 and H1oo showed great variability (Supplementary Figure S1). In contrast, variants of H2Asperm, H2A.V, and H2A.1macro were highly conserved among homologs rather than paralogs. Additionally, the N-terminal alanine (A), showed conserved amino acid sites among selected species, but was replaced by proline (P) in ScH2A.V, while numerous amino acid substitutions emerged in ScH2A.1macro (Supplementary Figure S2).

Figure 2.
Multiple sequence alignment of core H2A and H2B amino acid sequences from selected molluscs and human. Percent similarity is shown in blue, while the intensity of the color corresponds to the degree of similarity. The amino (N) and carboxy (C) terminal ends as well as the predicted secondary structure are shown above the alignment. Unique amino acids that varied in S. constricta in comparison to the other molluscs are marked by red ovals below the alignments.
As summarized in Table 3, we systematically counted the gene numbers of each histone group in different species including vertebrates and invertebrates. To make the statistics and classification more reliable, we double-checked the data using BI tree built with all counted histone genes (Supplementary Figure S5). Among all the selected species, fruit fly D. melanogaster and S. constricta had the greatest number of histone genes with n = 115 and n = 114, respectively. Intriguingly, with the exception of S. constricta and C. virginica, other molluscs had lower numbers of histone genes than vertebrates, indicating that the histone genes in S. constricta and C. virginica might have undergone species-specific expansions.
Table 3.
Number of histone genes for each group among Metazoans.
To explore the evolutionary relationships among six species, five phylogenetic trees of different histone groups (H1, H2A, H2B, H3, and H4) were constructed separately (Figure 3). For multi-copy types, only one sequence was selected as the representative due to the completely identical sequences (Supplementary Table S3). The results illustrated good agreement with the evolutionary relationships among the selected species. Compared to those in human, the histone genes in S. constricta had a closer relationship with those of Pacific oyster C. gigas, scallop P. maximus, nematode C. elegans, and urchin S. purpuratus, most of which were clustered into subtypes. These findings suggested that the histone genes were relatively conserved during evolution.
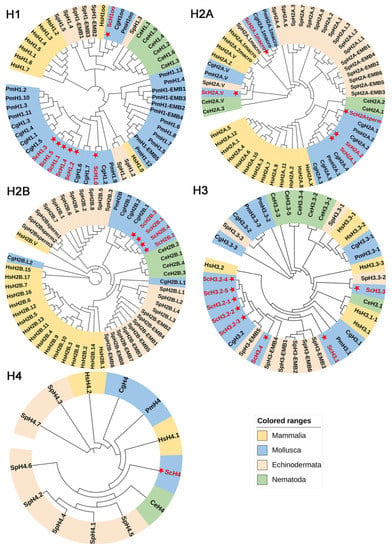
Figure 3.
Phylogenetic analysis of the histone gene family in S. constricta and other species (see also Supplemental Table S3). The histone genes of S. constricta are labeled with red font and star. Sc: S. constricta, Cg: C. gigas, Pm: P. maximus, Hs: H. sapiens, Ce: C. elegans, Sp: S. purpuratus.
3.3. Gene Structure and Conserved Motif Characteristics of S. constricta Histone Genes
The exon–intron structures of histone genes were generated based on genome sequences to clarify the structural evolution in S. constricta (Figure 4). The tree topology categorized the histone genes into five major groups, which was consistent with BI tree (Figure 4A and Supplementary Figure S5). Similar to other molluscs, the core and linker histone genes contained a single exon, whereas the histone variants had multiple exons. Specifically, the schematic structures clearly revealed that all the core and linker histones genes shared only one exon structure, except that ScH3.2-4, ScH2Asperm, ScH2A.V, ScH2A.1macro, and ScH1oo variants had three, two, five, eight, and two exons, respectively (Figure 4B). ScH2A.1macro contained the longest introns and also was the longest gene, followed by ScH2A.V, containing four introns. Further, MEME was used to detect conserved motifs in the S. constricta histone family. Obviously, members of the same group shared similar compositions of conserved motifs, but there was a distinct difference of motifs among the five groups (Supplementary Table S4, Figure 4C). In the H3 group, the motif 4, 1, 7 existed in most members, except that motif 7 was lost in ScH3.2-3. Similarly, the motif 6, 2, 9, 3, 12 was present in all members of the H2B group. Interestingly, ScH3.3 had five motifs, of which the motifs 6, 2 located in the N-terminus were consistent with H2B, and the latter three motifs 9, 3, 12 located in the C-terminus were in accord with H3. Therefore, we hypothesized that ScH3.3 might be a recombination product of H2B and H3. Moreover, groups H1 and H2A were also highly conserved. Only one motif was detected in the H4 group. In addition, the ubiquity of two characteristic sequences was discovered at the 3′-UTR of almost all core histone genes except for its variants. One was a 16-bp palindrome sequence forming a stem-loop structure, which was highly conserved and consisted of a stem with six base pairs (bp) and a 4-nt loop. Another was a purine-rich HDE sequence spaced 13 bp apart (Figure 4D and Supplementary Table S5). A total of two Lsm10, one Lsm11, and one Symplekin protein were identified in the S. constricta genome, but there was no SLBP protein (Supplemental file S1).

Figure 4.
Topology, motif, and gene structure analysis of histone genes in S. constricta. (A) The phylogenetic tree was constructed by the MEGA6.0 program using 25 full-length sequences of the different S. constricta histone genes. (B) Gene structure analysis of S. constricta histone genes. Untranslated region (UTR), exon, and intron are represented by yellow box, black box, and gray line, respectively. (C) Motif analysis of S. constricta histone genes. Different motifs are represented by different color blocks. (D) 3′UTR structure of histone genes in S. constricta. Conserved nucleotide sequences of S. constricta histones forming characteristic structural features: stem-loop and histone downstream element (HDE). The number in the gray box is the distance between the stop codon and the first nucleotide of the conserved stem-loop motif.
3.4. Gene Expression in S. constricta Assessed by Transcriptomics
The expression patterns of histone genes were investigated in seven different organs/tissues from S. constricta. As shown in Figure 5, histone types presented a tissue-dependent expression pattern, of which most histone genes were primarily expressed in the gill and hepatopancreas. Consistently high expression of ScH3.2-3, ScH3.2-4, ScH3.4, ScH2A.V, and ScH2A.1macro was found in the gill. A similar pattern was observed for ScH1oo, ScH1.2, ScH2B.1-1, ScH3.2-2, and ScH4-1 in the hepatopancreas, indicating that these genes might be involved in the same biological processes. In contrast, ScH1.1 was highly expressed in the mantle, whereas Sc3.2-1 was not expressed in any of the examined tissues. In contrast, the expression of ScH3.3 was not detected among selected organs/tissues. Considering its different motif components, we suggest that ScH3.3 might be a newly generated member of the histone gene family but does not function as histone H3. The mRNA levels of histones showed highly dynamic changes in the gill of S. constricta during V. parahaemolyticus infection (Figure 6). Specifically, the highest expression levels of ScH1.3-1, ScH2A.1-1, ScH2B.1-1, ScH3.1-1, and ScH4-1 were observed at 48 h after exposure. Meanwhile, the variants (i.e., ScH1.0, ScH2A.V, and ScH2A.1macro) showed dramatic up-regulation during 24–96 h after V. parahaemolyticus infection.
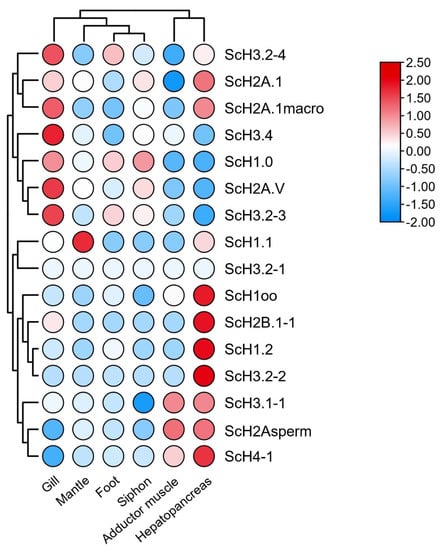
Figure 5.
Expression profiles of histone genes in different organs/tissues of adult S. constricta. Log10-transformed FPKM (fragments per kilobase of exon model per million mapped fragments) values were used to plot the heatmap. The FPKM values of the histone genes are presented in Table S6.

Figure 6.
Relative levels of expression of core histones and variants in the gill of S. constricta at different times after V. parahaemolyticus infection. The final concentration of V. parahaemolyticus infection was 1 × 108 cfu/mL. Data are represented as mean ± S.E. (n = 3). Different lowercase letters above the bars indicate significant differences among time-points (p < 0.05).
4. Discussion
It is commonly recognized that histones are an appropriate model system for the investigation of organization, structure, and expression of multigene families, which provides insight into their evolution and phylogeny. Here, we first reported the genome-wide identification of the histone gene repertoire in the genome of S. constricta. A total of 114 histone genes were identified and classified into 25 histone types. Among core histones, ScH2A.1, ScH2B.1, ScH3.1, and ScH4 showed the highest similarities with their homologous genes. Additionally, the copy numbers of core histones were high, and existed in numbers comparable to the ratio 19:22:19:23 (H2A.1:H2B.1:H3.1:H4), which were of considerable importance in sustaining the stoichiometry of all four core histone classes for new DNA strand packaging during replication [13]. Notably, the numbers of histone genes varied greatly among selected species, as observed in that the histone family expanded significantly in S. constricta compared to other molluscs. In general, the numbers of the histone H1 subtype range from four to seven in animals and plants, whereas the maximum number of the subtype in mammals is eleven [42]. In our study, the numbers of histone H1 genes in S. constricta was significantly higher than those in other invertebrates. The histone H2A had the greatest number of variants, including ScH2A.V, ScH2A.1macro, and ScH2Asperm, which was similar to the findings in mammals [43]. Additionally, the relatively high number of H4 genes may contribute to forming nucleosomes, which are coupled with H2A variants. Previous studies have concluded that the wide range of amino acid substitutions contributes to functional differentiation [44]. Consistent with that notion, these findings indicate that the histone genes may evolve into more functions in razor clam, in addition to the classical functions of DNA packaging, transcription, and recombination. Similarly, the evolved H1.3 in the model plant Arabidopsis is more responsible for adaptive responses to both low light and drought stress than H1.1 and H1.2, although they have the overall same binding properties as the main H1 variants [45]. In addition, comparative genomic analysis of the cytochrome P450 (CYP) gene family has shown that the new gained CYP genes are essential for detoxification and metabolic processes under adverse environmental stresses, particularly in the plant Fragilariopsis cylindrus with good adaptability to cold resistance [46]. In consequence, we hypothesize that the large number and varied types of histone genes may play important roles in adaptation of extreme environmental conditions in S. constricta.
In numerous organisms, canonical histone genes are distributed in clusters containing multi-copies of the five histone genes [47]. Studies have shown the emergence of two fundamental arrangements of histone genes in genomic distribution. In the first case, the histone genes are clustered, and their clusters have been repeated in tandem. As an example, quartets containing four core histone genes are observed in coral [48]. Moreover, similar arrangements have been reported in the nematode C. elegans, and an independently organized single-copy H1 genes was also observed [49]. Most histone genes in the echinoderm S. purpuratus are clustered in tandem repeats, and contain one copy of each of the five histone genes [47]. In another case, the histone genes are clustered but scattered throughout the whole genome, as in human, mouse, and chicken [2]. However, among metazoans, molluscs are considered to be the paradigm of arrangement diversity, which exhibit three different permutations of histone gene clusters, even within a single species. There are three types of tandem repeated units in blue mussel Mytilus edulis: five histone genes cluster, core histone gene class devoid of H1 linker histone genes and H1 cluster [50]. In this study, we found that the histone distribution in S. constricta was more similar to the first arrangement. The major clusters on Hic_asm_1, ctg4484, ctg8584, and ctg10813 contain H1, H2A-H2B, and H3-H4. It is notable that several incomplete histone clusters were observed on short scaffolds, which was likely due to the limited integrity of the S. constricta genome assembly. Furthermore, the histone H1 in S. constricta was scattered throughout the entire gene cluster. In contrast to M. edulis, H1 genes were all located in tandem and not adjacent to core histone clusters [50]. The four variants occupied solitary locations within the genome, but were not located in histone gene clusters. This finding was consistent with previous studies [2,17].
The evolutionary origin of histones can be traced back to Archaebacteria. It is noteworthy that the four core histones gradually display diversification and differentiation due to the mechanism of recurrent gene duplication, ultimately promoting DNA compaction in the time of transition to the eukaryotic cell [51]. Our investigation on evolution of histone proteins was carried out based on a Bayesian tree, which was an essential step for phylogenetic analysis. The long-term evolution of histone genes is directed by a birth-and-death process, thereby contributing to genetic diversity [51], which is consistent with the alignment results of each histone group among molluscs and human. Additionally, the phylogenetic relationships of histone genes among vertebrate and invertebrate species are coincident with their species relationships. Here, the histones in S. constricta are more closely related to those of mollusks than mammals and other selected species, even though histone gene sequences are highly conserved in eukaryotes.
The core histone genes are generally intronless, while their variants contain introns. In contrast, prior studies have reported the appearance of introns in core histones H3 and H4 in Volvox, hydra, and soybean [13,52,53]. Similarly, there were introns in core histone ScH3.2-4 here. Interestingly, motif 15 existed only in Sc3.2-4 and variant ScH2A.1macro. We conjecture that the emergence of the newly evolved motif and the different pattern of conserved motifs would facilitate the process of functional differentiation among different histone types. Moreover, the conserved stem-loop structure and purine-rich HDE element existed at 3′ UTRs of most S. constricta canonical histones, and both are involved in mRNA processing of replication-dependent genes. These findings imply that the regulatory mechanism of histone expression in S. constricta is similar to those of other eukaryotes [14]. It has been concluded that the regulatory factors, including SLBP, Lsm10, Lsm11, U7 snRNP, and Symplekin proteins, play essential roles in histone mRNA processing [54]. Among them, the 5′ terminal portion of the U7 snRNP interacts with the HDE element by forming base-pairing. The SLBP binds to the stem-loop structure and then enables it to stabilize the binding of the U7 snRNP to the HDE, while Lsm10 and Lsm11 are subunits specific to the U7 snRNP. In addition, Symplekin participates in polyadenylation. Intriguingly, with the exception of SLBP, the above-mentioned proteins could be retrieved from the S. constricta genome. Similarly, the Phytophthora, Oxytricha, Paramecium, and Trichomonas histones contain a stem-loop structure but no SLBP [12]. Based on these findings, we hypothesize that there might be another alternative protein that is similar to SLBP. However, further investigation is needed to explore the underlying cause of the absence of some functional elements in a minority of histone genes and the transcriptional regulation of these genes in S. constricta.
The histone genes in S. constricta showed apparent differential expression patterns. Most of the histones were expressed at low levels among all organs/tissues. In contrast, consistently high expression levels of ScH1.0, ScH2A.V and ScH3.2-3 were observed in all organs/tissues, implying that these genes may play similar roles in the same biological processes (Supplementary Table S6). Among six organs/tissues, most histone genes were predominantly expressed in the gill and hepatopancreas compared to other organs/tissues. Notably, the gill and hepatopancreas play critical roles in maintaining homeostasis and immune defense [55,56]. In this regard, the histone genes highly expressed in gill and hepatopancreas may be required not only for the packaging of DNA into chromatin, but also for cell homeostasis and immune response. H1oo and H2Asperm are oocyte- and sperm-specific variants of H1 and H2A, which exist in oocytes and zygotes and are important to meiotic maturation of the sex cells [57,58]. Curiously, ScH1oo and ScH2Asperm both showed high expression in the hepatopancreas. One possible reason for this anomaly is that the specific anatomical structure in that gonads are encapsulated together with hepatopancreas in razor clam, so the tissue of hepatopancreas is easily mixed with gonadal debris when sampling. Studies have demonstrated that the H2A.V protein has multiple functions, such as active transcription [59,60], telomeric stabilization [61], DNA damage repair [1], and maintenance of genomic integrity [1,60,61]. In this regard, the high expression of H2A variants (ScH2A.1macro and ScH2Asperm) might contribute to the response to gill microstructural damage in stressful situations.
Long before the integration of histones as chromatin structural elements in eukaryotes, it has been shown that histones play an important role in the ancient host defense system against pathogenic microorganisms [62]. In recent years, its antibacterial effect has also been widely demonstrated. It has been reported that the expression level of histone genes exhibited sharp increases in epithelial cells of macaque Macuca mulatta kidney after monkeypox virus infection [63]. Similarly, the linker histone H1 has been observed as having a significant up-regulation in the skin of zebrafish Danio rerio when infected with Citrobacter freundii [64]. Besides this, relevant studies on C. virginica have revealed that the histone H2B has strong antimicrobial activity against two Gram-negative bacteria, V. parahaemolyticus and V. vulnificus [24], while similar function has been confirmed in the core histones isolated from hemolymph of Pacific white shrimp L. vannamei [21]. The razor clam lives in mudflats, a habitat that is full of various microbes. Thus, its innate immune system is vulnerable to attack. Recent studies have shown that the expression of partial histones can decrease the load of Vibrio in some marine filter feeders [65]. Consistently, the core histones and variants were significantly upregulated in the gill of S. constricta after V. parahaemolyticus infection in this study, suggesting their potential roles in immune defense. Collectively, these findings indicated that the expanded histone genes might be beneficial for razor clam to protect itself against microbes. However, further work is needed to investigate the differences of antibacterial ability among different types of histone proteins.
5. Conclusions
To our knowledge, this is the first study to provide comprehensive and genome-wide analysis of the histone repertoire in razor clam S. constricta. A total of 114 histone genes were identified and divided into 25 types. The summary of histone genes provided insight into their genomic organization, genic structures, and evolutionary relationships. The expression profilings of histone genes in adult organs/tissues and following V. parahaemolyticus infection imply their potential functions in bivalves, such as homeostasis maintenance and immune defense. Taken together, our findings facilitate the understanding of important functions and evolution of histone genes in bivalve molluscs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/fishes7010005/s1, Supplementary Figure S1. Multiple sequence alignment of H1 and H1oo amino acid sequences from selected members of molluscs and human. Supplementary Figure S2. Multiple sequence alignment of H2A variants’ amino acid sequences from selected members of molluscs and human. Supplementary Figure S3. Multiple sequence alignment of H3 amino acid sequences from selected members of molluscs and human. Supplementary Figure S4. Multiple sequence alignment of H4 amino acid sequences from selected members of molluscs and human. Supplementary Figure S5. Phylogenetic analysis of histone gene family-related 390-amino acid sequences across 11 animals. Supplementary Table S1. Detailed information of razor clam histone genes sequences. Supplementary Table S2. Comparison of the genomic sequences between duplicated genes. Supplementary Table S3. All the histone amino acid sequences used for phylogenetic analysis. Supplementary Table S4. Sequences of the 15 motifs detected by MEME. Supplementary Table S5. Alignment of stem-loop motifs of all genes in S. constricta. Supplementary Table S6. FPKM values of the adult tissues. Largest values among tissues are colored orange. Supplementary file S1: Related protein factors of histones mRNA 3′ terminal processing.
Author Contributions
Conceptualization, Y.D., Z.L. and L.L.; Formal analysis, J.L. and L.L.; Funding acquisition, Z.L., Y.D. and L.L.; Investigation, J.L. and L.L.; Supervision, Z.L. and Y.D.; Visualization, J.L., L.L. and Y.D.; Writing—original draft preparation, J.L. and L.L.; Writing—review and editing, L.L., Y.D., J.L., H.Y. and Z.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2018YFD0901405), Zhejiang Major Program of Science and Technology (2021C02069-7), Zhejiang Provincial Top Discipline of Biological Engineering (Level A) (KF2020010), Ningbo Major Project of Science and Technology (2019B10005), National Marine Genetic Resource Center Program.
Institutional Review Board Statement
All experimental procedures in this study were approved by the Institutional Animal Care and Use Committee (IACUC)of Zhejiang Wanli University, China (Approval code: 20211222001).
Data Availability Statement
Data is contained within the article and supplementary materials. Supplementary materials contain figures and tables necessary for interpreting the results.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gonzalez-Romero, R.; Rivera-Casas, C.; Frehlick, L.J.; Mendez, J.; Ausio, J.; Eirín-López, J.M. Histone H2A (H2A.X and H2A.Z) variants in molluscs: Molecular characterization and potential implications for chromatin dynamics. PLoS ONE 2012, 7, e30006. [Google Scholar]
- Li, C.; Song, L.; Zhao, J.; Zou, H.; Su, J.; Zhang, H. Genomic organization, nucleotide sequence analysis of the core histone genes cluster in Chlamys farreri and molecular evolution assessment of the H2A and H2B. DNA Seq. 2006, 17, 440–451. [Google Scholar] [CrossRef]
- Wolffe, A.P.; Kurumizaka, H. The nucleosome: A powerful regulator of transcription. Prog. Nucleic Acid Res. Mol. Biol. 1998, 61, 379–422. [Google Scholar]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Zlatanova, J.; Bishop, T.C.; Victor, J.-M.; Jackson, V.; van Holde, K. The nucleosome family: Dynamic and growing. Structure 2009, 17, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Allan, J.; Hartman, P.; Crane-Robinson, C.; Aviles, F. The structure of histone H1 and its location in chromatin. Nature 1980, 288, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Reinberg, D. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 2011, 21, 175–186. [Google Scholar] [CrossRef]
- Jiang, D.; Borg, M.; Lorkovi, Z.J.; Montgomery, S.A.; Osakabe, A.; Yelagandula, R.; Axelsson, E.; Berger, F. The evolution and functional divergence of the histone H2B family in plants. PLoS Genet. 2020, 16, e1008964. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. Histone variants on the move: Substrates for chromatin dynamics. Nat. Rev. Mol. Cell Biol. 2017, 18, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Gamble, M.J.; Kraus, W.L. Multiple facets of the unique histone variant macroH2A: From genomics to cell biology. Cell Cycle 2010, 9, 2568–2574. [Google Scholar] [CrossRef]
- Bönisch, C.; Hake, S.B. Histone H2A variants in nucleosomes and chromatin: More or less stable? Nucleic Acids Res. 2012, 40, 10719–10741. [Google Scholar] [CrossRef] [PubMed]
- López, M.D.; Samuelsson, T. Early evolution of histone mRNA 3′ end processing. RNA 2008, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.C.; Ubhe, S.; Sirwani, N.; Lohokare, R.; Galande, S. Rapid divergence of histones in Hydrozoa (Cnidaria) and evolution of a novel histone involved in DNA damage response in hydra. Zoology 2017, 123, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Knowles, B.; Solter, D.; Messerschmidt, D. Epigenetic control of early mouse development. Curr. Top. Dev. Biol. 2016, 120, 311–360. [Google Scholar]
- Rooney, A.P.; Piontkivska, H.; Nei, M. Molecular evolution of the nontandemly repeated genes of the histone 3 multigene family. Mol. Biol. Evol. 2002, 19, 68–75. [Google Scholar] [CrossRef]
- Wu, X.M.; Cao, L.; Nie, P.; Chang, M.X. Histone H2A cooperates with RIP2 to induce the expression of antibacterial genes and MHC related genes. Dev. Comp. Immunol. 2019, 101, 103455. [Google Scholar] [CrossRef]
- Richards, R.C.; O’Neil, D.B.; Thibault, P.; Ewart, K.V. Histone H1: An antimicrobial protein of Atlantic salmon (Salmo salar). Biochem. Biophys. Res. Commun. 2001, 284, 549–555. [Google Scholar] [CrossRef]
- Arockiaraj, J.; Gnanam, A.J.; Kumaresan, V.; Palanisamy, R.; Bhatt, P.; Thirumalai, M.K.; Roy, A.; Pasupuleti, M.; Kasi, M. An unconventional antimicrobial protein histone from freshwater prawn Macrobrachium rosenbergii: Analysis of immune properties. Fish Shellfish Immunol. 2013, 35, 1511–1522. [Google Scholar] [CrossRef]
- Patat, S.A.; Carnegie, R.B.; Kingsbury, C.; Gross, P.S.; Chapman, R.; Schey, K.L. Antimicrobial activity of histones from hemocytes of the Pacific white shrimp. Eur. J. Biochem. 2004, 271, 4825–4833. [Google Scholar] [CrossRef] [PubMed]
- De Zoysa, M.; Nikapitiya, C.; Whang, I.; Lee, J.-S.; Lee, J. Abhisin: A potential antimicrobial peptide derived from histone H2A of disk abalone (Haliotis discus discus). Fish Shellfish Immunol. 2009, 27, 639–646. [Google Scholar] [CrossRef]
- Li, C.; Song, L.; Zhao, J.; Zhu, L.; Zou, H.; Zhang, H.; Wang, H.; Cai, Z. Preliminary study on a potential antibacterial peptide derived from histone H2A in hemocytes of scallop Chlamys farreri. Fish Shellfish Immunol. 2007, 22, 663–672. [Google Scholar] [CrossRef]
- Seo, J.-K.; Stephenson, J.; Crawford, J.M.; Stone, K.L.; Noga, E.J. American oyster, Crassostrea virginica, expresses a potent antibacterial histone H2B protein. Mar. Biotechnol. 2010, 12, 543–551. [Google Scholar] [CrossRef]
- Li, A.; Eirín-López, J.M.; Ausió, J. H2AX: Tailoring histone H2A for chromatin-dependent genomic integrity. Biochem. Cell Biol. 2005, 83, 505–515. [Google Scholar] [CrossRef] [PubMed]
- González-Romero, R.; Ausió, J.; Méndez, J.; Eirín-López, J.M. Histone genes of the razor clam Solen marginatus unveil new aspects of linker histone evolution in protostomes. Genome 2009, 52, 597–607. [Google Scholar] [CrossRef]
- Shen, H.; Qi, Z.; Wei, H. Molecular evolution of the histone gene family in Schistosoma japonicum. Int. J. Med. Parasit. Dis. 2015, 42, 67–73. [Google Scholar]
- Montes-Rodríguez, I.M.; Rodríguez-Pou, Y.; González-Méndez, R.R.; Lopez-Garriga, J.; Ropelewski, A.J.; Cadilla, C.L. Characterization of histone genes from the bivalve Lucina pectinata. Int. J. Environ. Res. Public Health 2018, 15, 2170. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.-P.; Qian, Y.-H.; Zeng, Q.-T. Phylogenetic relationships of Drosophila melanogaster species group deduced from spacer regions of histone gene H2A-H2B. Mol. Phylogenetics Evol. 2004, 30, 336–343. [Google Scholar] [CrossRef]
- Dong, Y.; Zeng, Q.; Ren, J.; Yao, H.; Lv, L.; He, L.; Ruan, W.; Xue, Q.; Bao, Z.; Wang, S.; et al. The chromosome-level genome assembly and comprehensive transcriptomes of the razor clam (Sinonovacula constricta). Front. Genet. 2020, 11, 664. [Google Scholar] [CrossRef]
- Morton, B. The functional morphology of Sinonovacula constricta with a discussion on the taxonomic status of the Novaculininae (Bivalvia). J. Zool. 1984, 202, 299–325. [Google Scholar] [CrossRef]
- Wang, W.X.; Xu, Z.Z. Larval swimming and postlarval drifting behavior in the infaunal bivalve Sinonovacula constricta. Mar. Ecol. Prog. Ser. 1997, 148, 71–81. [Google Scholar] [CrossRef]
- Sousa, H.; Hinzmann, M. Review: Antibacterial components of the bivalve’s immune system and the potential of freshwater bivalves as a source of new antibacterial compounds. Fish Shellfish Immunol. 2020, 98, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.-Y.; Liang, X.-F.; He, S. Genome-wide identification and characterization of olfactory receptor genes in Chinese perch, Siniperca chuatsi. Genes 2019, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Gerard, T.; Jose, C. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Ponte, I.; Romero, D.; Yero, D.; Suau, P.; Roque, A. Complex evolutionary history of the mammalian histone H1.1-H1.5 gene family. Mol. Biol. Evol. 2017, 34, 545–558. [Google Scholar] [CrossRef]
- Eirin-Lopez, J.M.; Ishibashi, T.; Ausio, J. H2A.Bbd: A quickly evolving hypervariable mammalian histone that destabilizes nucleosomes in an acetylation-independent way. FASEB J. 2008, 22, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, A. Nuclear and nucleolar activity of linker histone variant H1.0. Cell. Mol. Biol. Lett. 2016, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Rutowicz, K.; Puzio, M.; Halibart-Puzio, J.; Lirski, M.; Kotliński, M.; Kroteń, M.A.; Knizewski, L.; Lange, B.; Muszewska, A.; Śniegowska-Świerk, K.; et al. A specialized histone H1 variant is required for adaptive responses to complex abiotic stress and related DNA methylation in Arabidopsis. Plant Physiol. 2015, 169, 2080–2101. [Google Scholar] [CrossRef]
- Teng, L.; Fan, X.; Nelson, D.R.; Han, W.; Zhang, X.; Xu, D.; Renault, H.; Markov, G.V.; Ye, N. Diversity and evolution of cytochromes P450 in stramenopiles. Planta 2019, 249, 647–661. [Google Scholar] [CrossRef]
- Hentschel, C.C.; Birnstiel, M.L. The organization and expression of histone gene families. Cell 1981, 25, 301–313. [Google Scholar] [CrossRef]
- Miller, D.; Harrison, P.; Mahony, T.; McMillan, J.; Miles, A.; Odorico, D.; Ten Lohuis, M. Feature of histone gene structure and organization are common to diploblastic and tripoblastic metazoans. J. Mol. Evol. 1993, 37, 245–253. [Google Scholar] [CrossRef]
- Sanicola, M.; Ward, S.; Childs, G.; Emmons, S.W. Identification of a Caenorhabditis elegans histone H1 gene family: Characterization of a family member containing an intron and encoding a poly (A)+ mRNA. J. Mol. Biol. 1990, 212, 259–268. [Google Scholar] [CrossRef]
- Eirín-López, J.M.; Ruiz, M.F.; Gonzalez-Tizon, A.M.; Martínez, A.; Sanchez, L.; Mendez, J. Molecular evolutionary characterization of the mussel Mytilus histone multigene family: First record of a tandemly repeated unit of five histone genes containing an H1 subtype with “orphon” features. J. Mol. Evol. 2004, 58, 131–144. [Google Scholar] [CrossRef]
- Eirín-López, J.M.; González-Romero, R.; Dryhurst, D.; Méndez, J.; Ausió, J. Long-Term Evolution of Histone Families: Old Notions and New Insights into Their Mechanisms of Diversification across Eukaryotes. 2009. Available online: https://link.springer.com/chapter/10.1007/978-3-642-00952-5_8 (accessed on 12 October 2020).
- Müller, K.; Schmitt, R. Histone genes of Volvox carteri: DNA sequence and organization of two H3-H4 gene loci. Nucleic Acids Res. 1988, 16, 4121–4136. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Kanazin, V.; Shoemaker, R.C. Phylogenetic utility of histone H3 intron sequences in the perennial relatives of soybean (Glycine: Leguminosae). Mol. Phylogenet. Evol. 1996, 6, 438–447. [Google Scholar] [CrossRef]
- Dominski, Z.; Marzluff, W.F. Formation of the 3′ end of histone mRNA: Getting closer to the end. Gene 2007, 396, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Cong, M.; Wu, H.; Cao, T.; Ji, C.; Lv, J. Effects of ammonia nitrogen on gill mitochondria in clam Ruditapes philippinarum. Environ. Toxicol. Pharmacol. 2018, 65, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, B.; Koner, D.; Bhuyan, G.; Saha, N. Differential expression of multiple glutamine synthetase genes in air-breathing magur catfish, Clarias magur and their induction under hyper-ammonia stress. Gene 2018, 671, 85–95. [Google Scholar] [CrossRef]
- Tanaka, M.; Hennebold, J.D.; Macfarlane, J.; Adashi, E.Y. A mammalian oocyte-specific linker histone gene H1oo: Homology with the genes for the oocyte-specific cleavage stage histone (cs-H1) of sea urchin and the B4/H1M histone of the frog. Development 2001, 128, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Tanaka, M.; Teranishi, T.; Matsumoto, K.; Hosoi, Y.; Saeki, K.; Ishimoto, H.; Minegishi, K.; Iritani, A.; Yoshimura, Y. H1foo is indispensable for meiotic maturation of the mouse oocyte. J. Reprod. Dev. 2007, 53, 895–902. [Google Scholar] [CrossRef][Green Version]
- Eirin-Lopez, J.M.; Ausio, J. H2A. Z-mediated genome-wide chromatin specialization. Curr. Genom. 2007, 8, 59–66. [Google Scholar] [CrossRef]
- Subramanian, V.; Fields, P.A.; Boyer, L.A. H2A. Z: A molecular rheostat for transcriptional control. F1000Prime Rep. 2015. Available online: https://pubmed.ncbi.nlm.nih.gov/25705384/ (accessed on 22 October 2020).
- Mattiroli, F.; D’Arcy, S.; Luger, K. The right place at the right time: Chaperoning core histone variants. EMBO Rep. 2015, 16, 1454–1466. [Google Scholar] [CrossRef]
- Kawasaki, H.; Iwamuro, S. Potential roles of histones in host defense as antimicrobial agents. Infect. Disord. Drug Targets 2008, 8, 195–205. [Google Scholar] [CrossRef]
- Alkhalil, A.; Hammamieh, R.; Hardick, J.; Ichou, M.A.; Jett, M.; Ibrahim, S. Gene expression profiling of monkeypox virus-infected cells reveals novel interfaces for host-virus interactions. Virol. J. 2010, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lü, A.; Hu, X.; Xue, J.; Zhu, J.; Wang, Y.; Zhou, G. Gene expression profiling in the skin of zebrafish infected with Citrobacter freundii. Fish Shellfish Immunol. 2012, 32, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Dorrington, T.; Villamil, L.; Gómez-chiarri, M. Upregulation in response to infection and antibacterial activity of oyster histone H4. Fish Shellfish Immunol. 2011, 30, 94–101. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).