Chestnut Shell Extract Modulates Immune Parameters in the Rainbow Trout Oncorhynchus mykiss

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Antioxidant Activity, Total Polyphenol Content, and Composition of Chestnut Shell Extracts
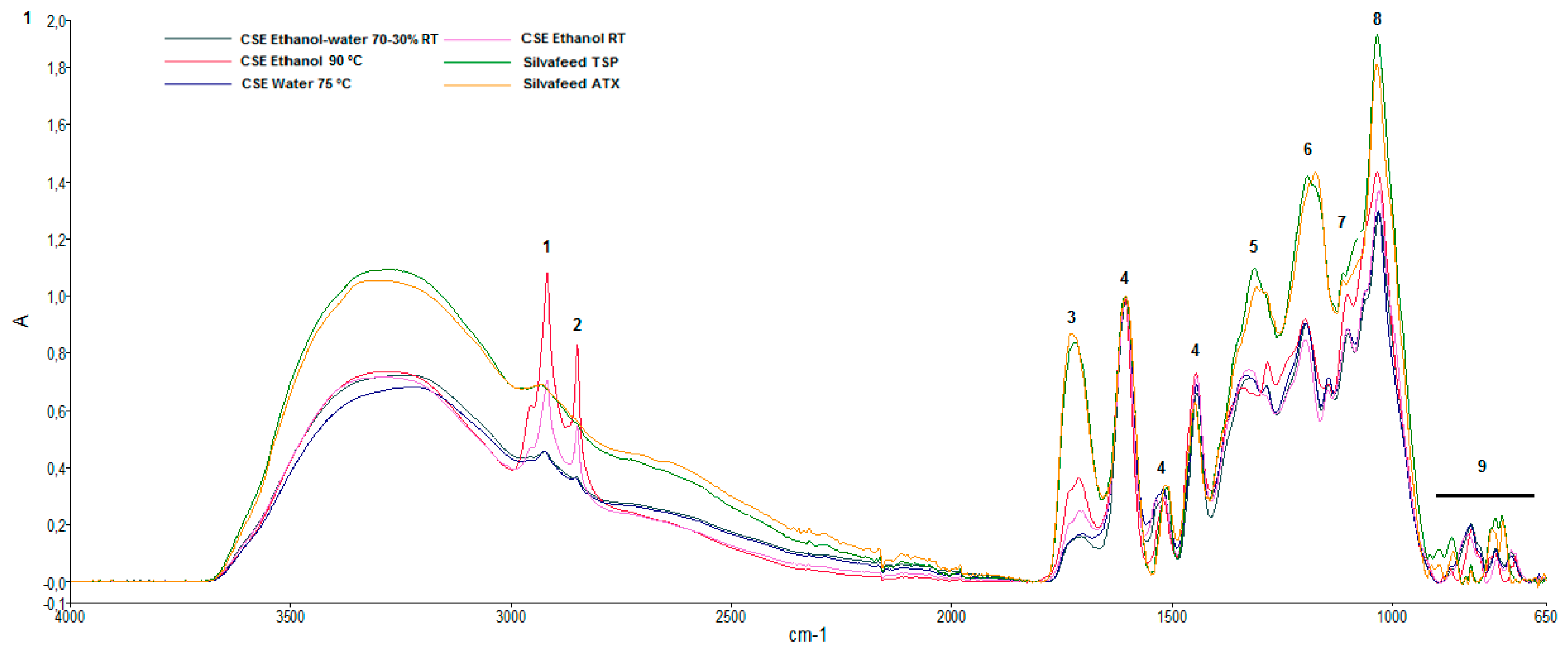
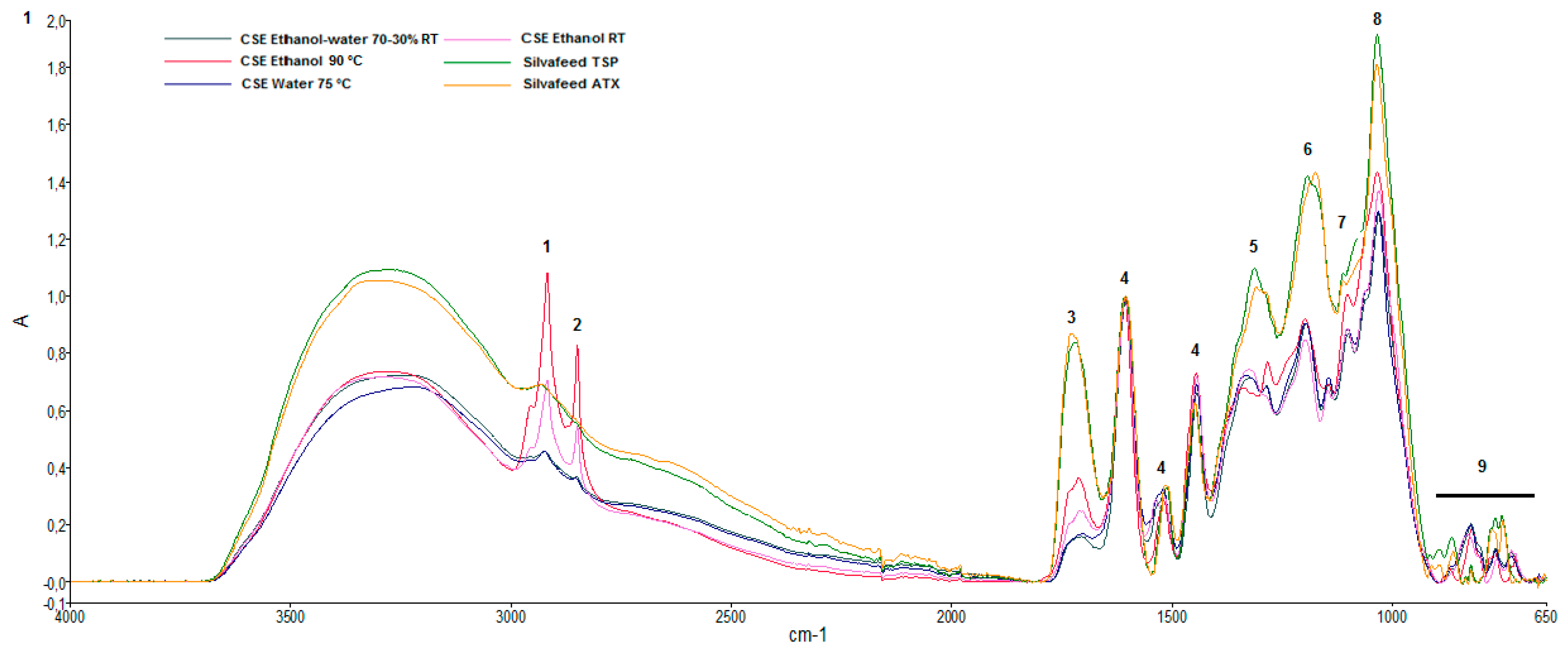
2.2. Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) Characterization of Chestnut Extracts
2.3. Isolation of Non-Adherent Leukocytes from Blood and Intestine
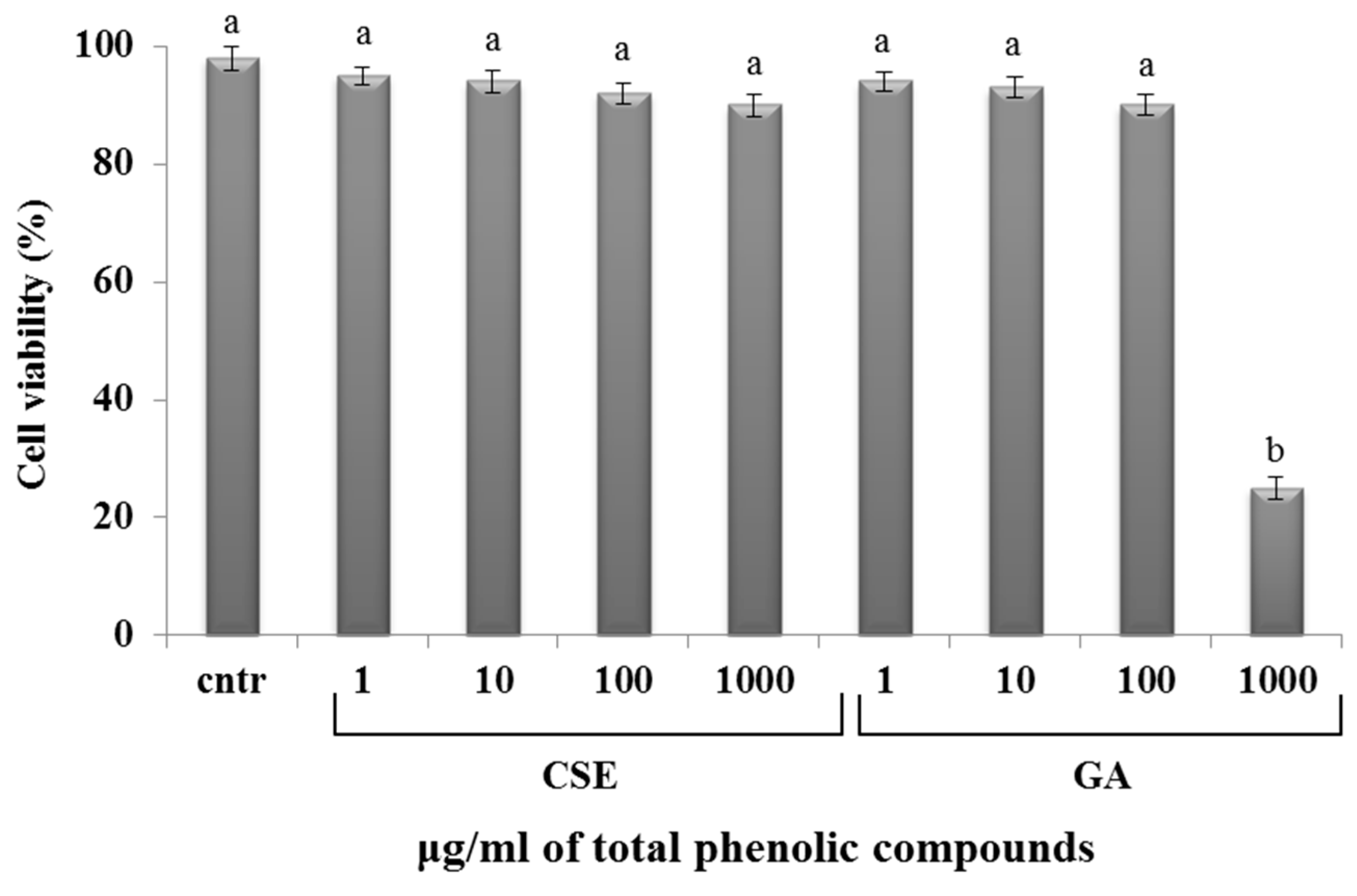
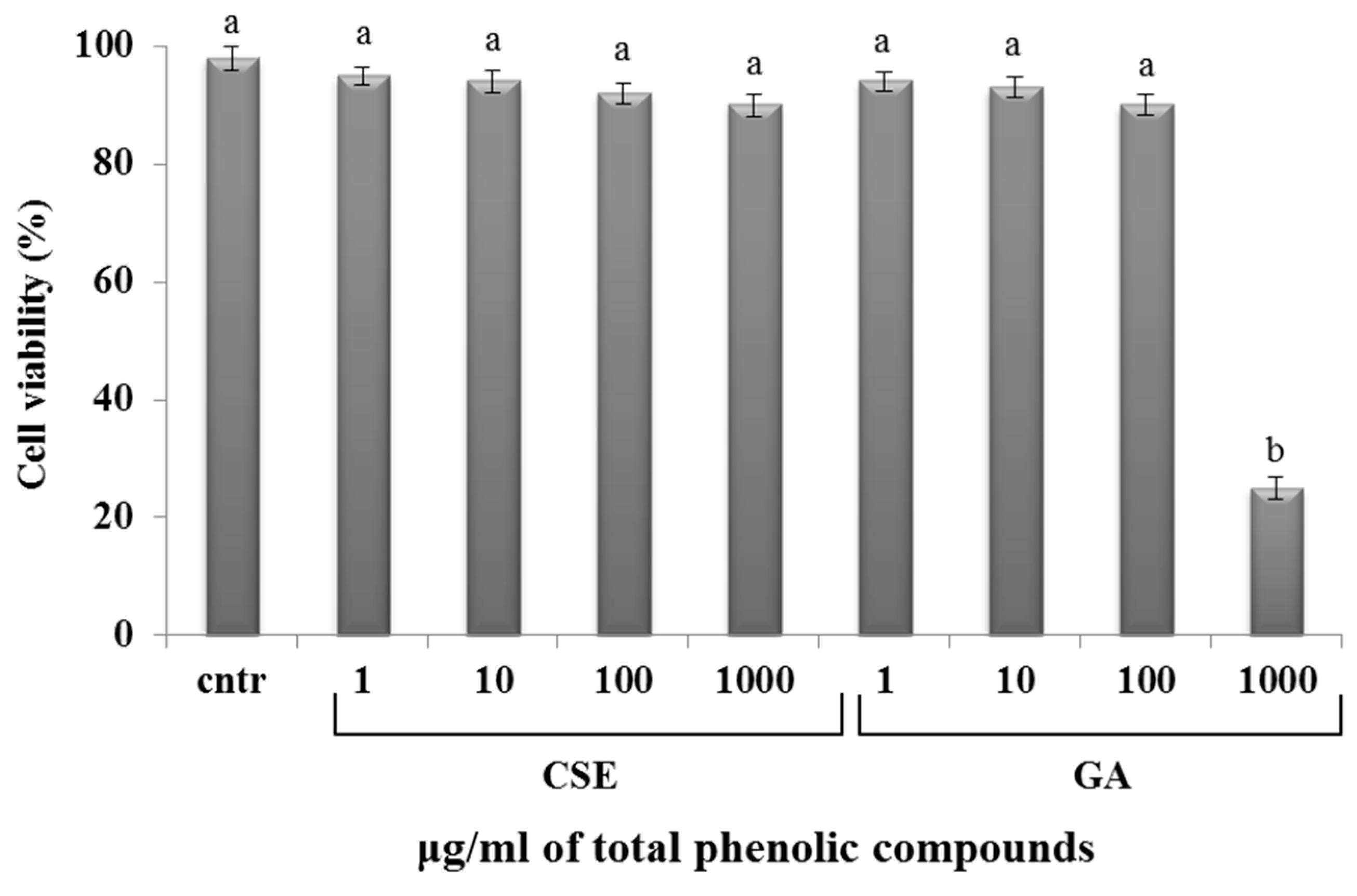
2.4. Cytotoxicity
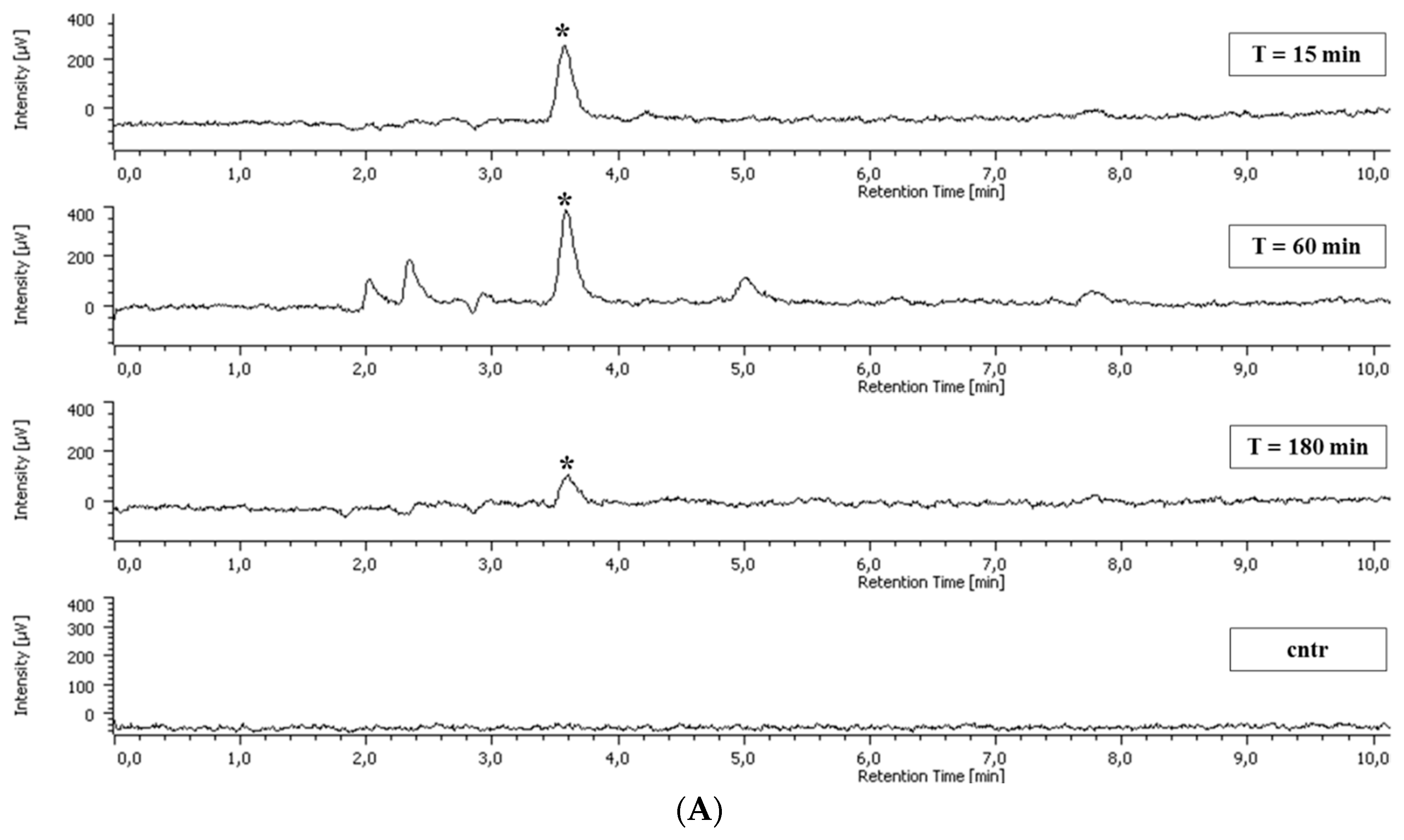
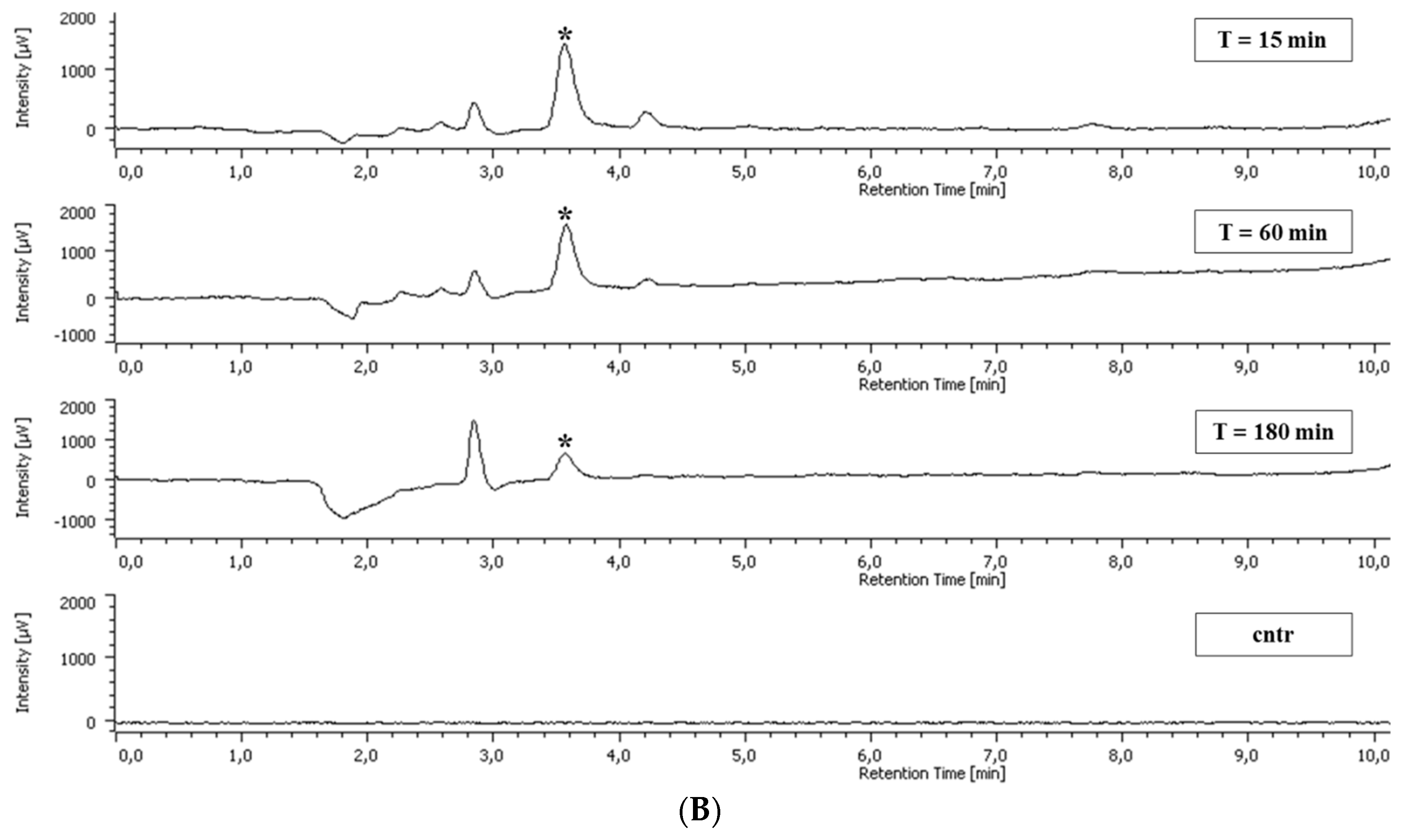
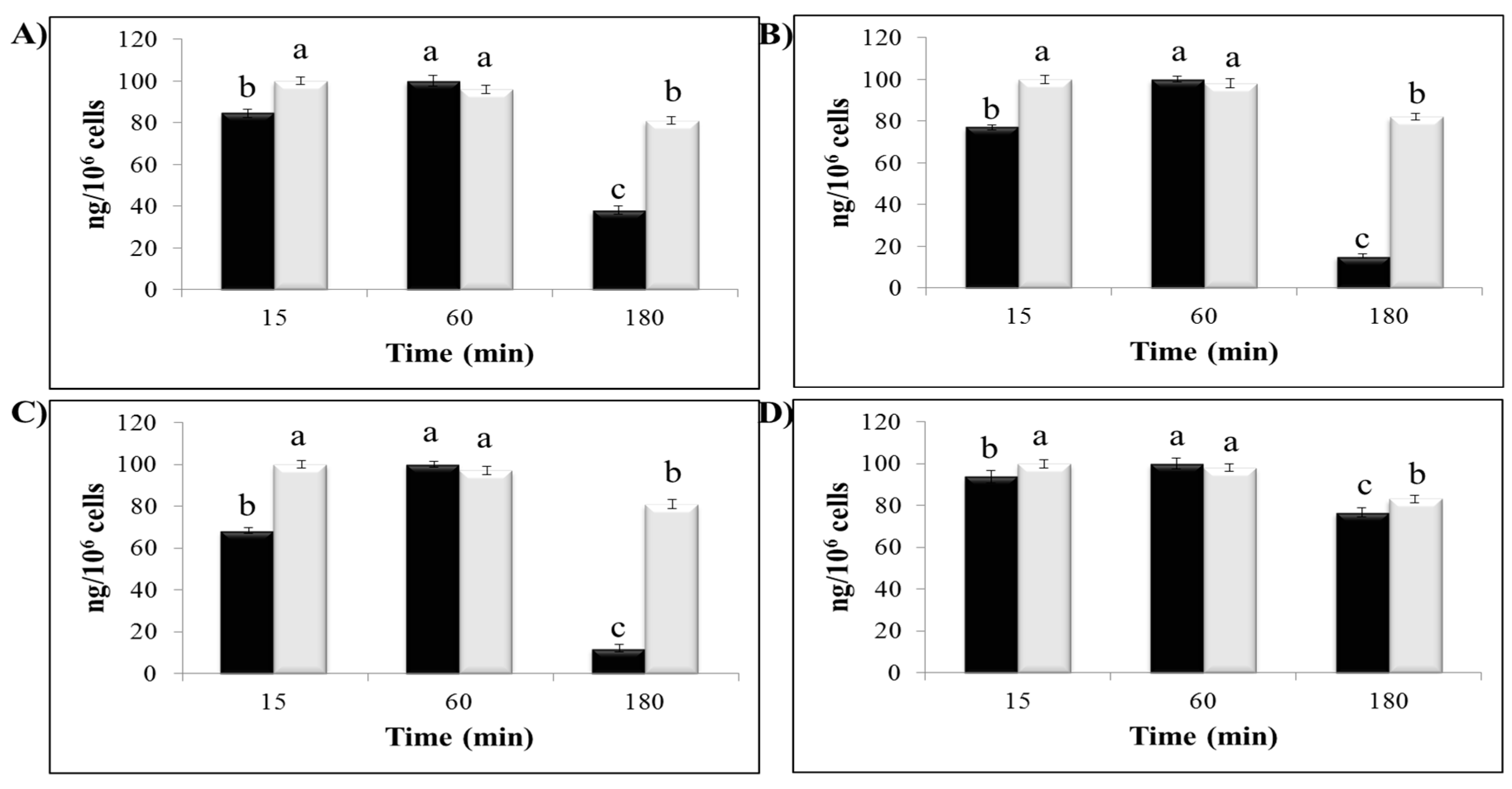
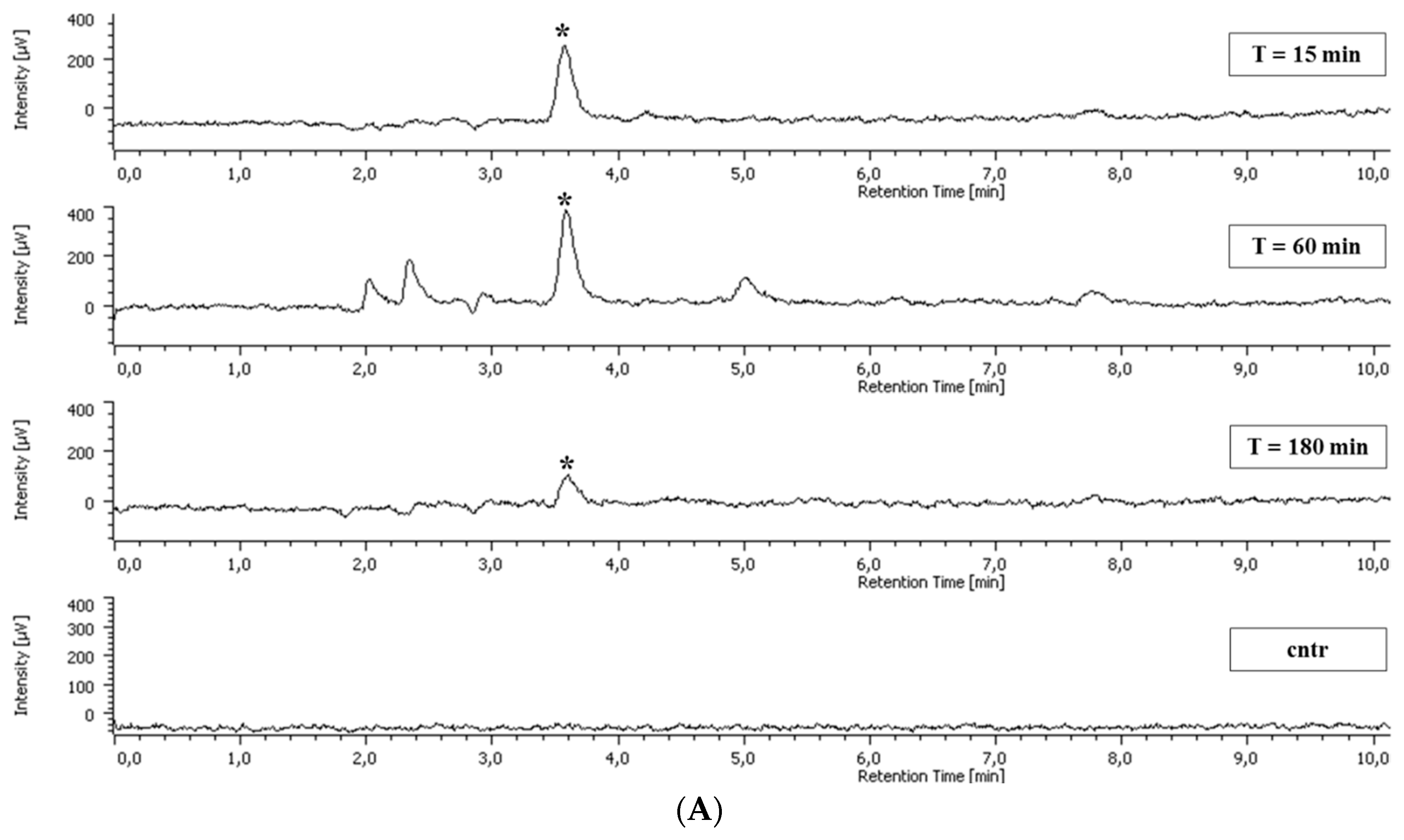
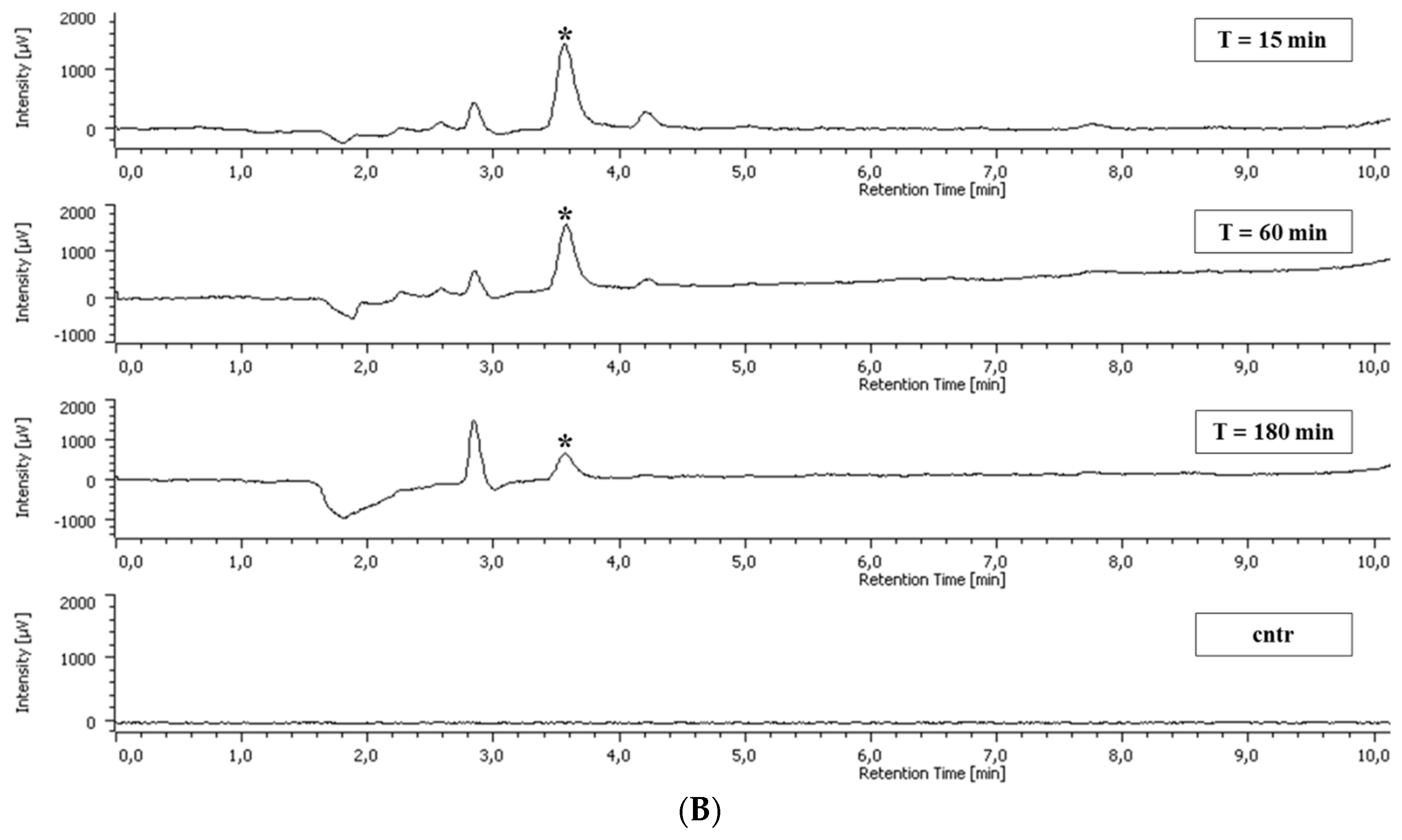
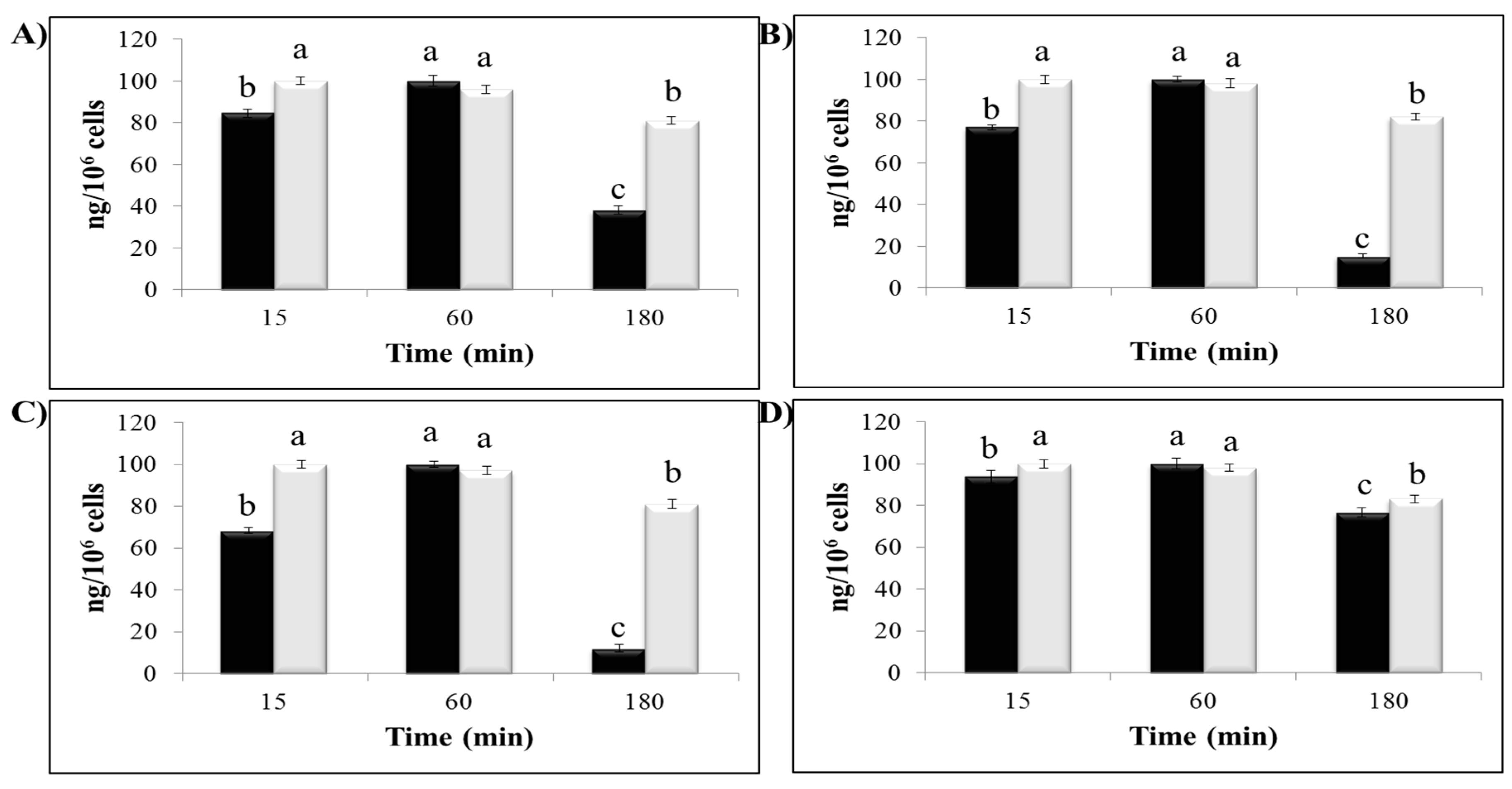
2.5. Polyphenol Cellular Content
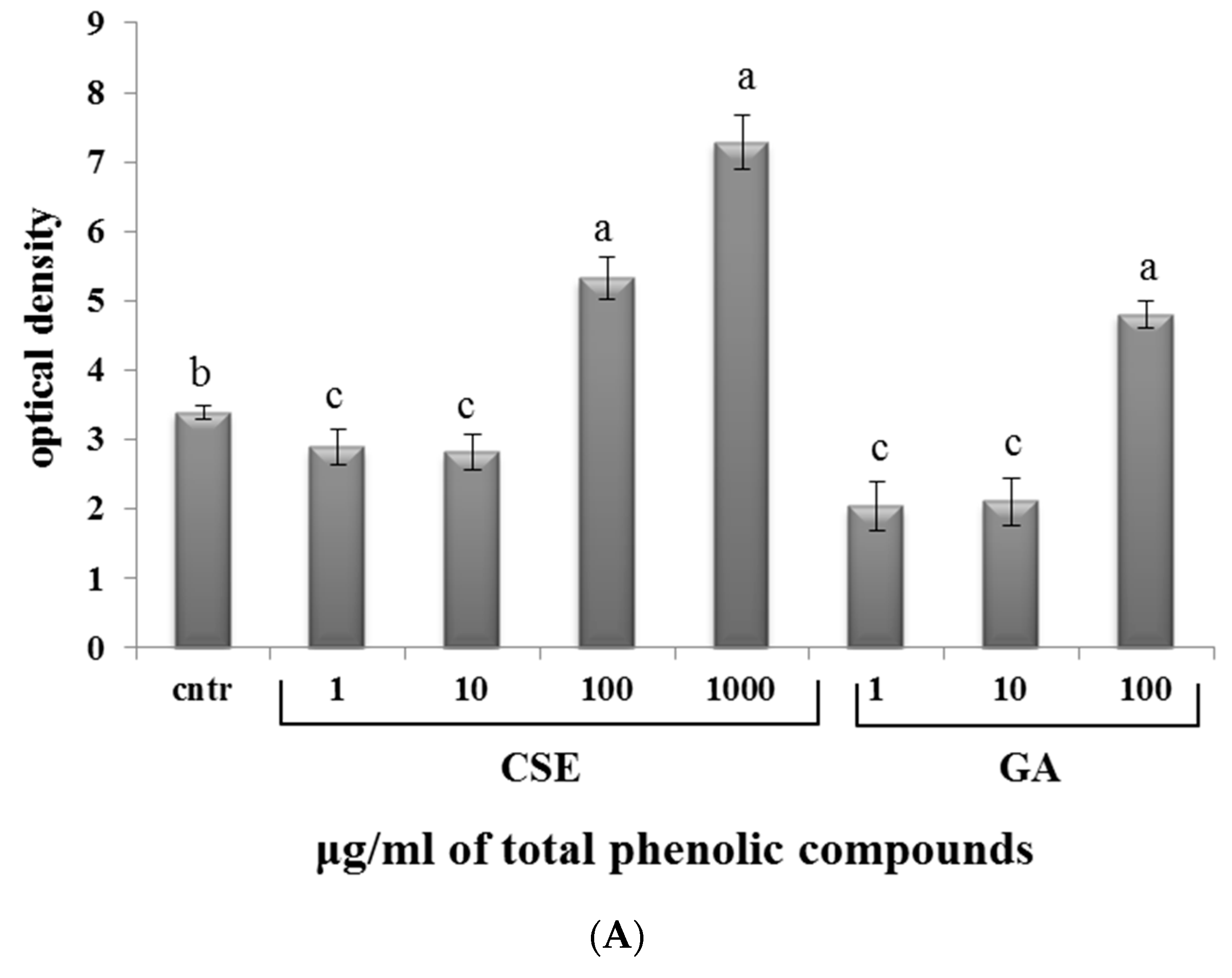
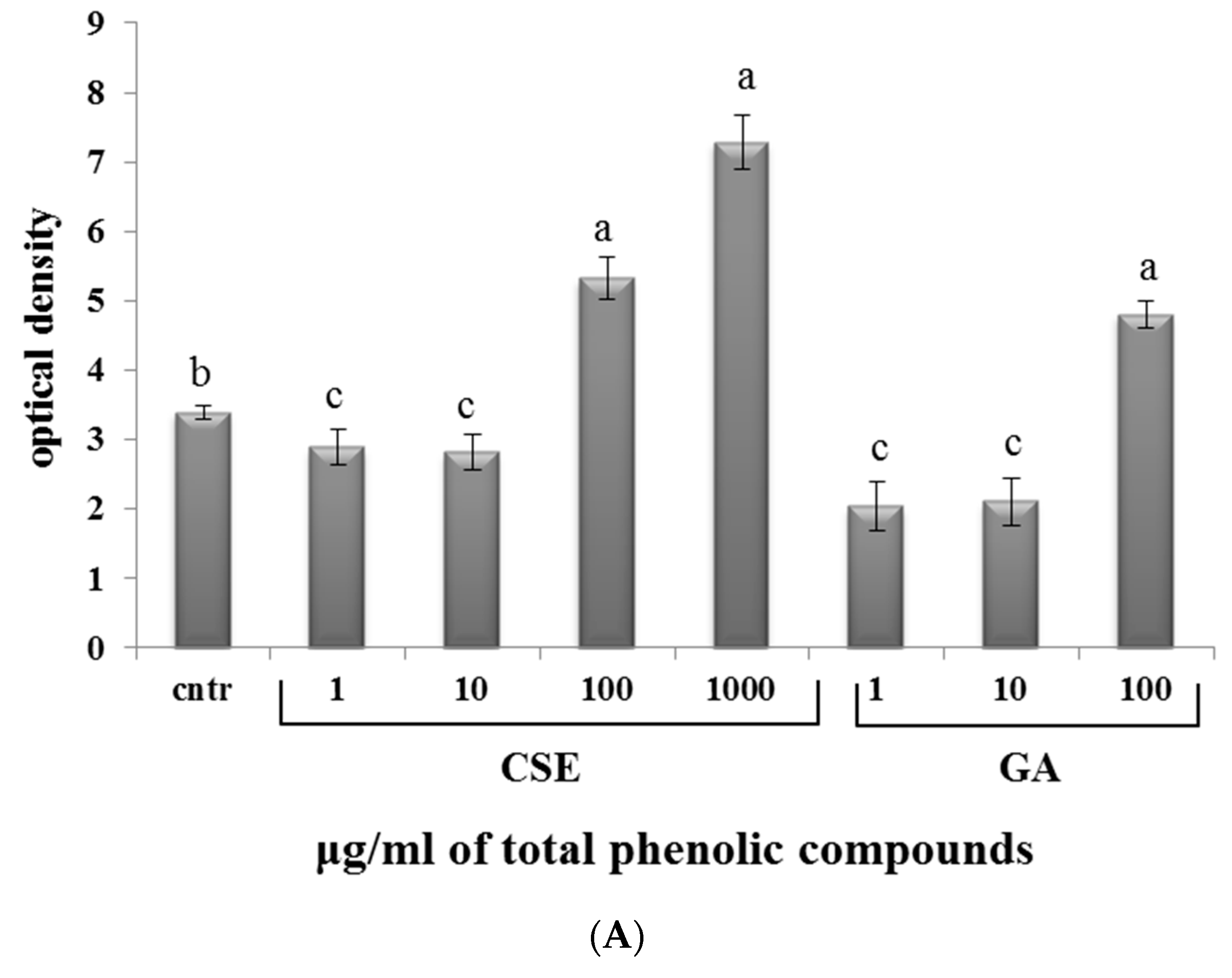
2.6. Effect of CSE on Superoxide Anion Production
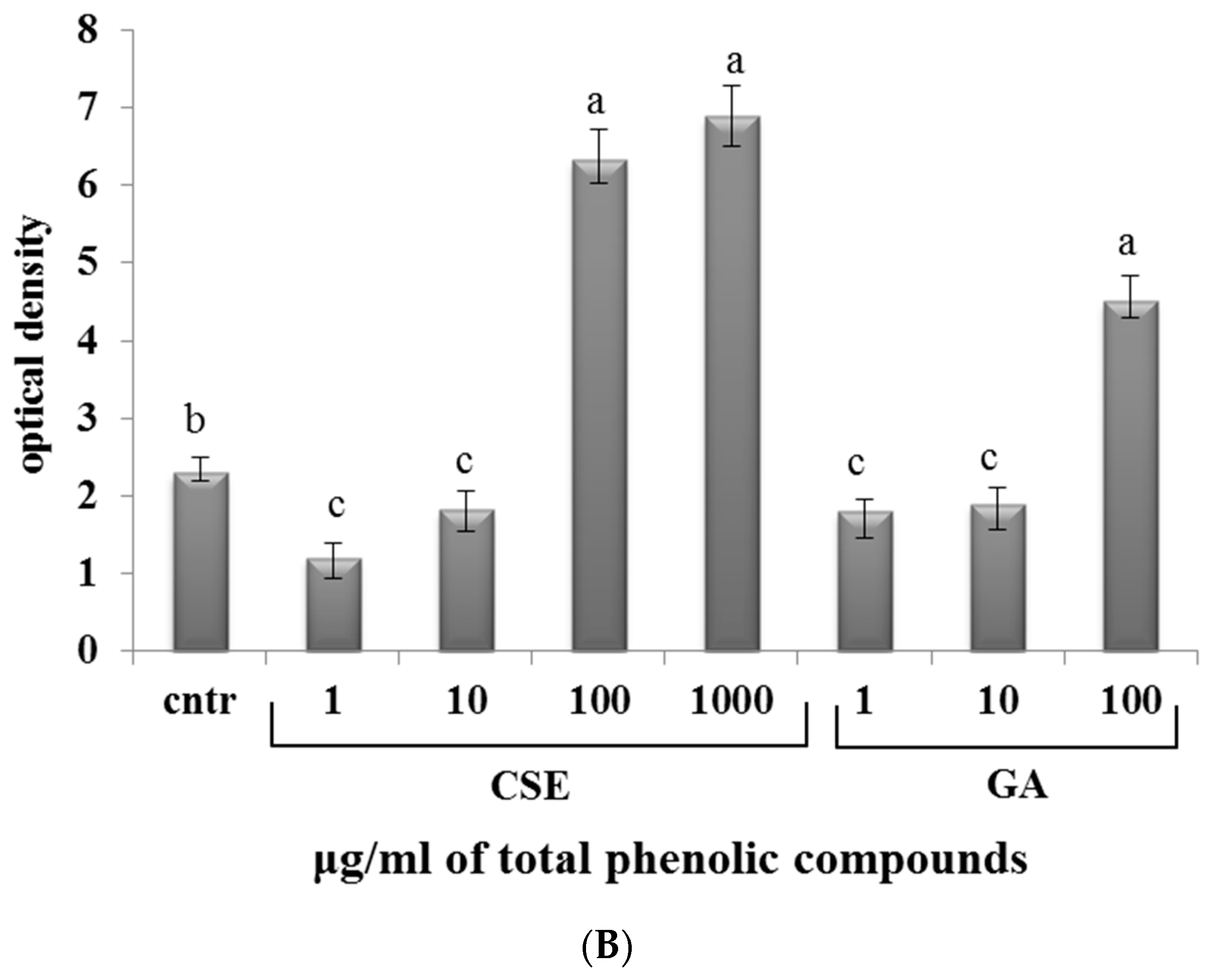
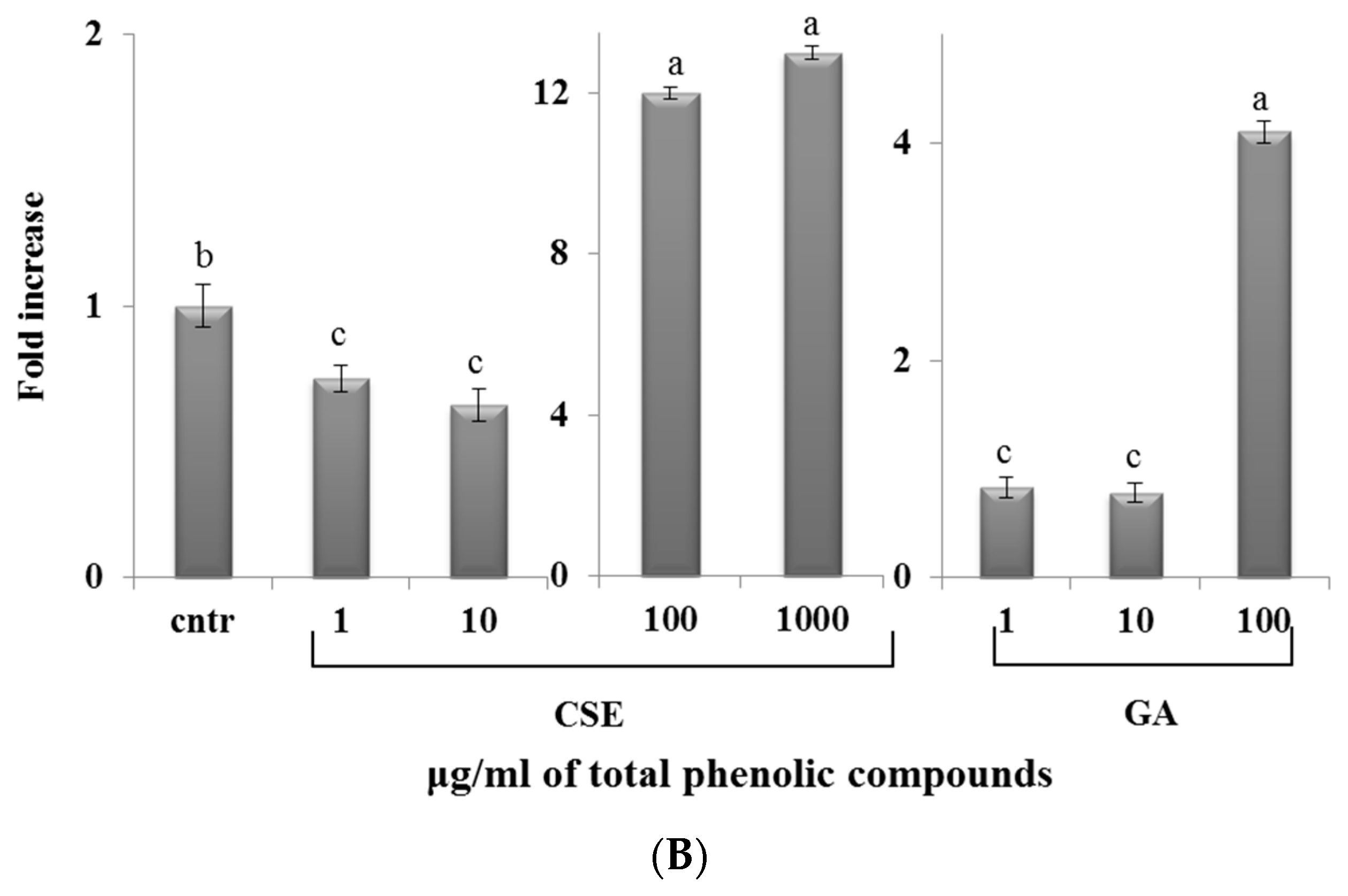
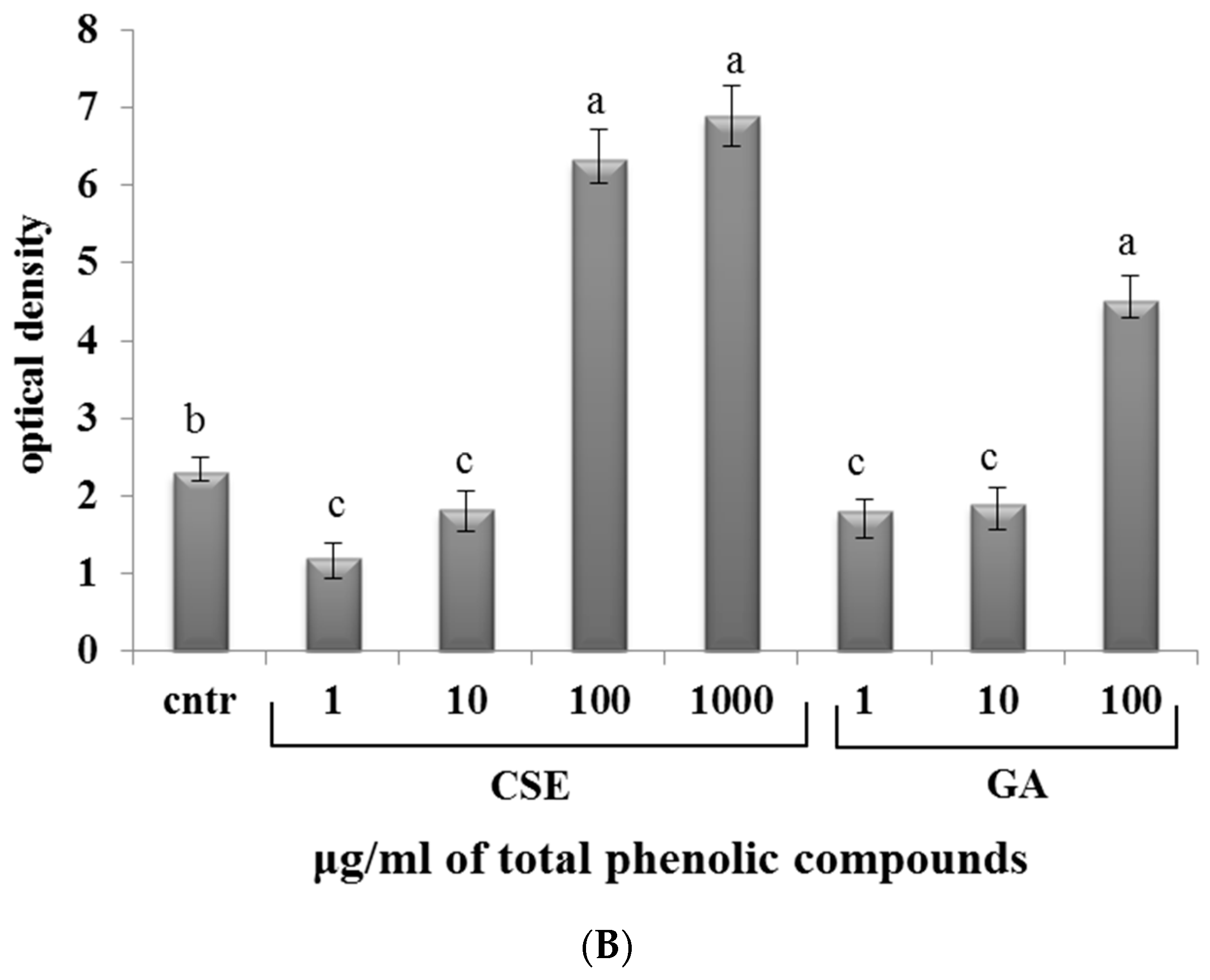
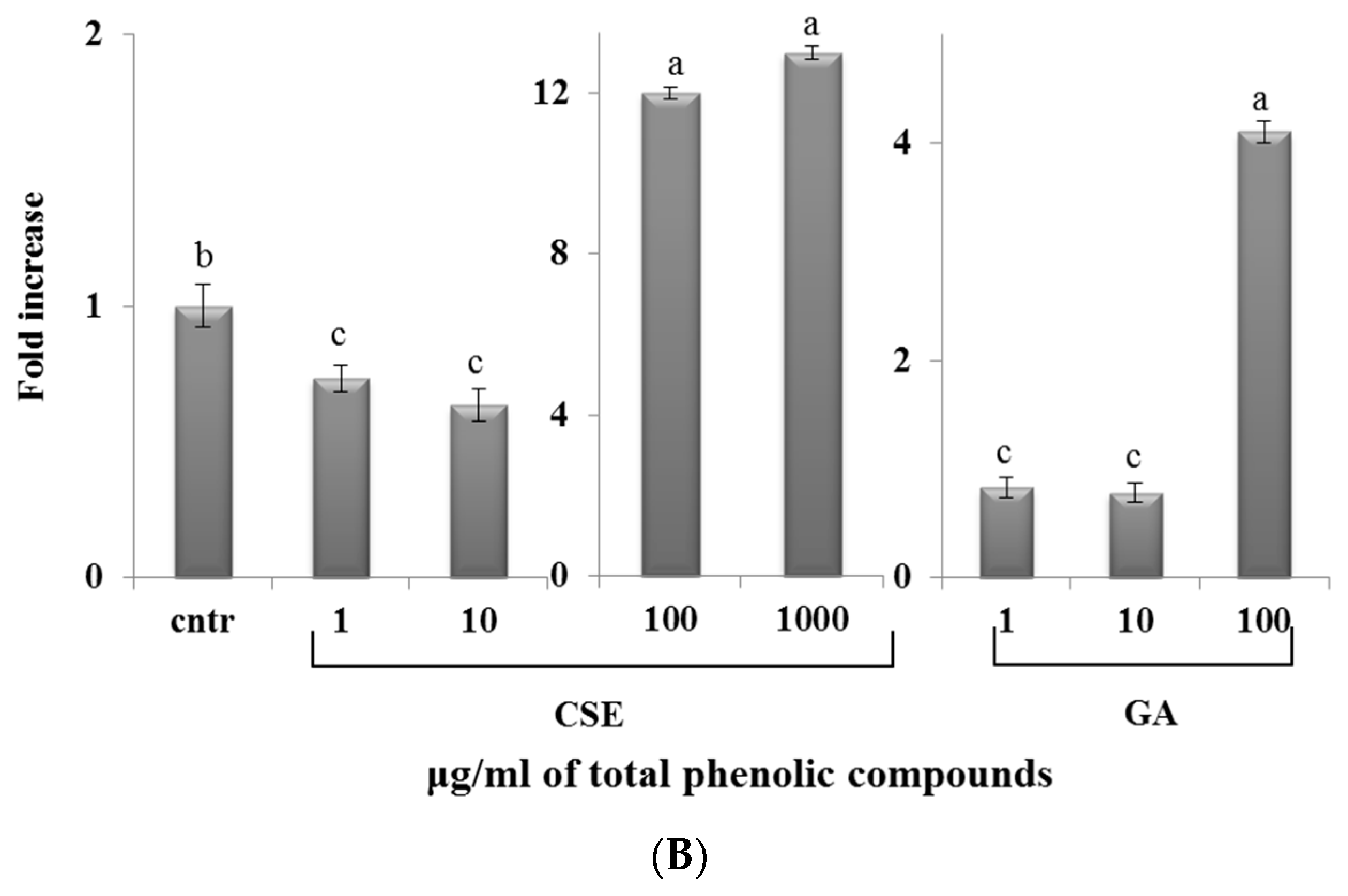
2.7. Effect of CSE on Phagocytosis
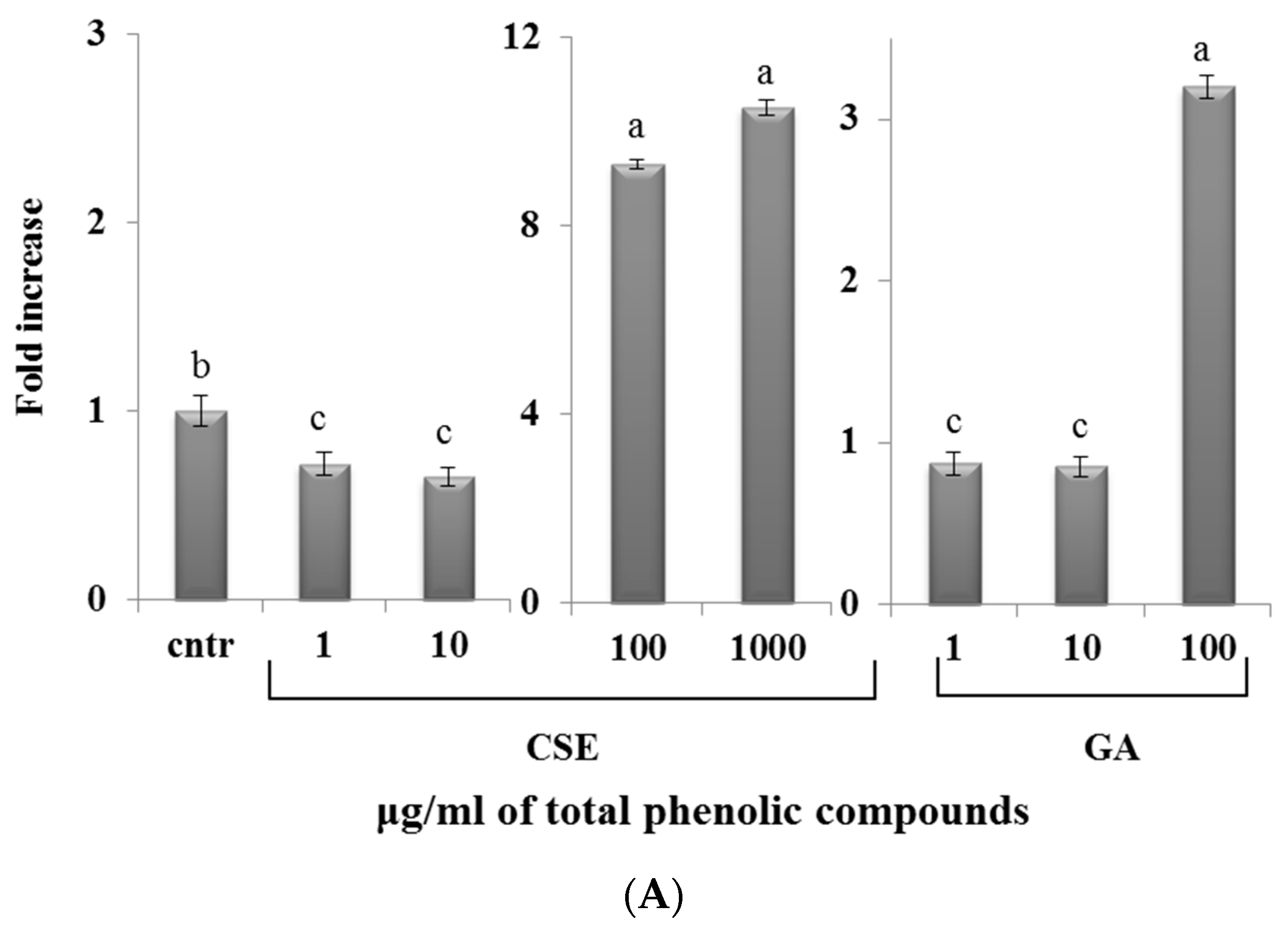
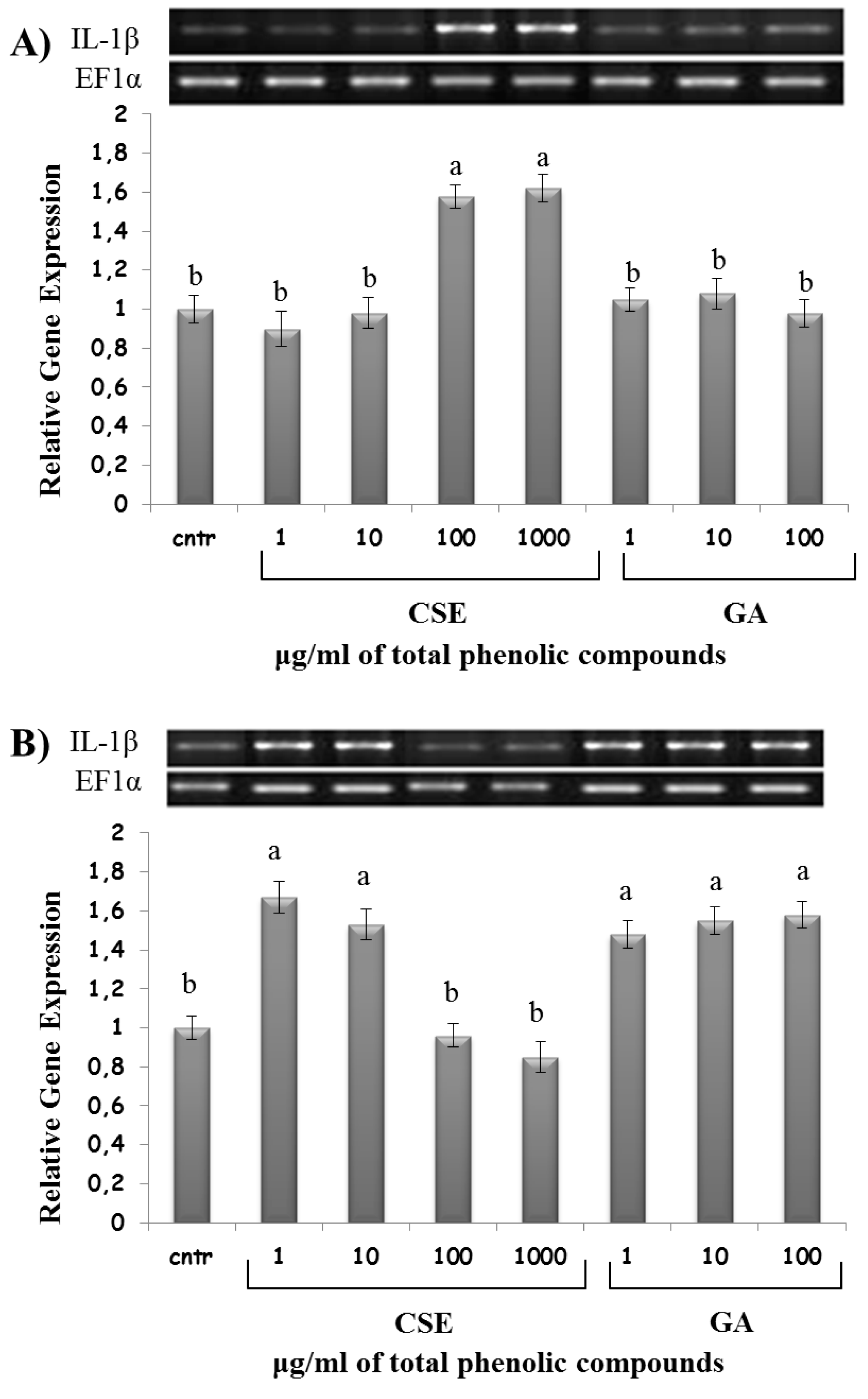
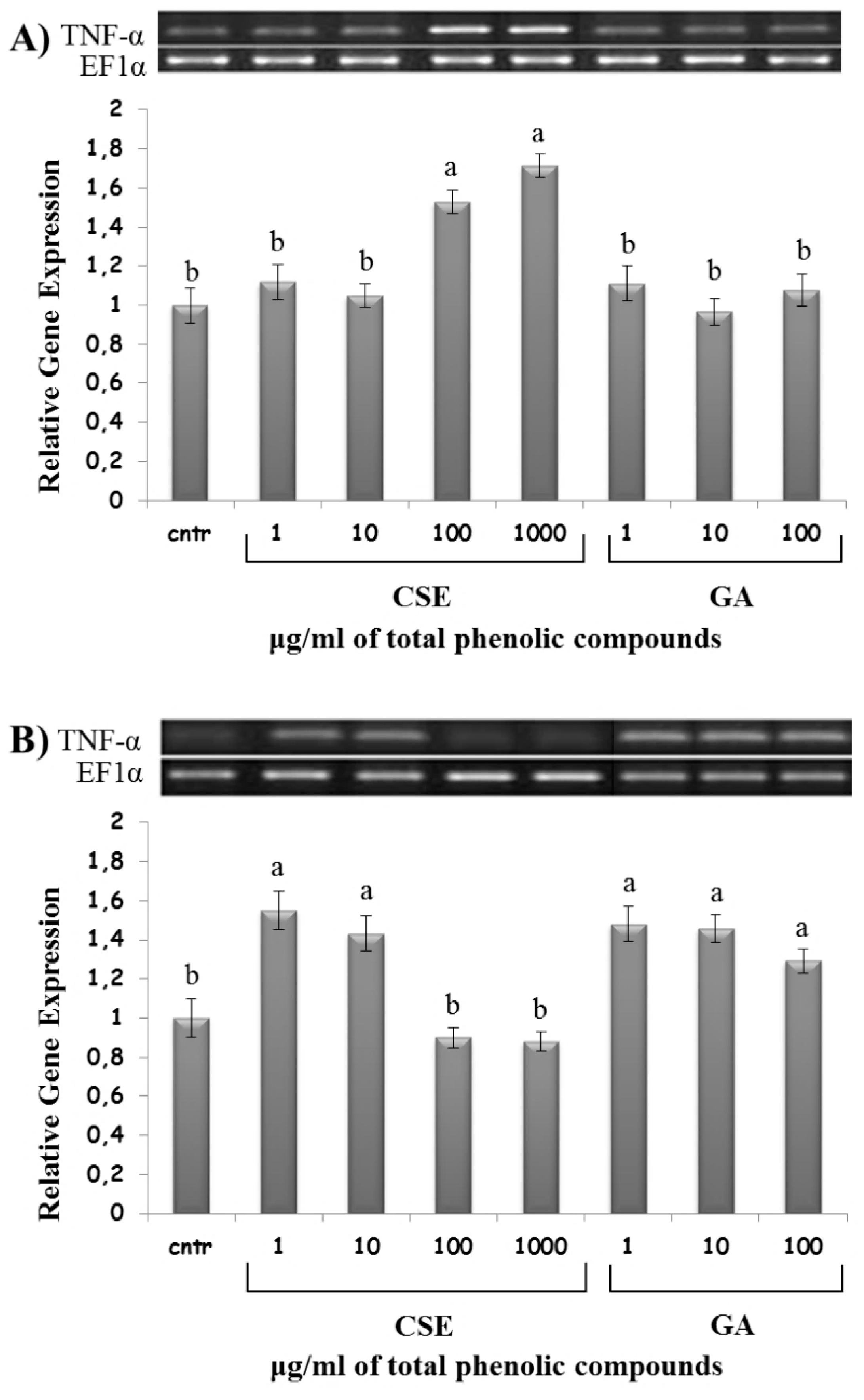
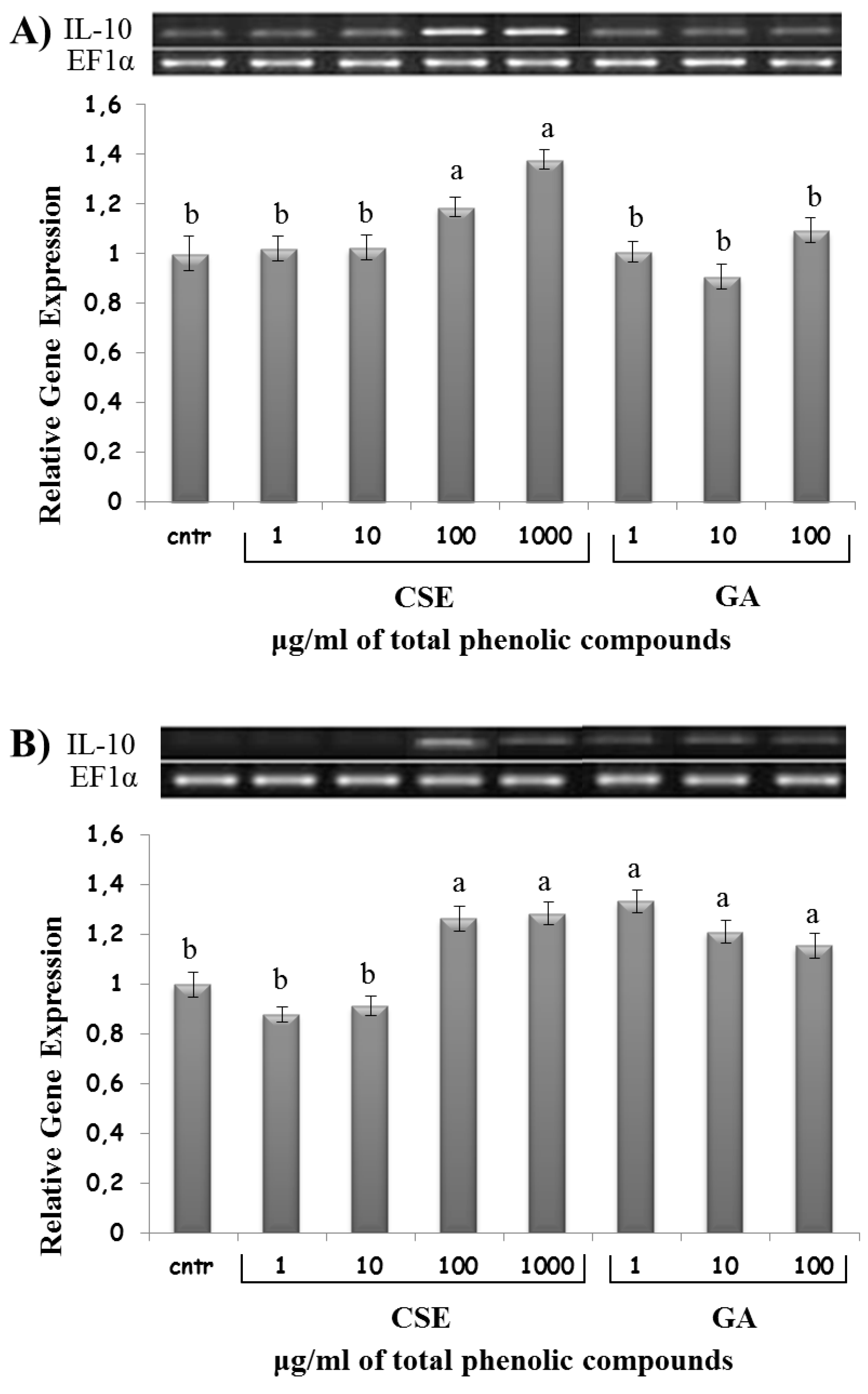
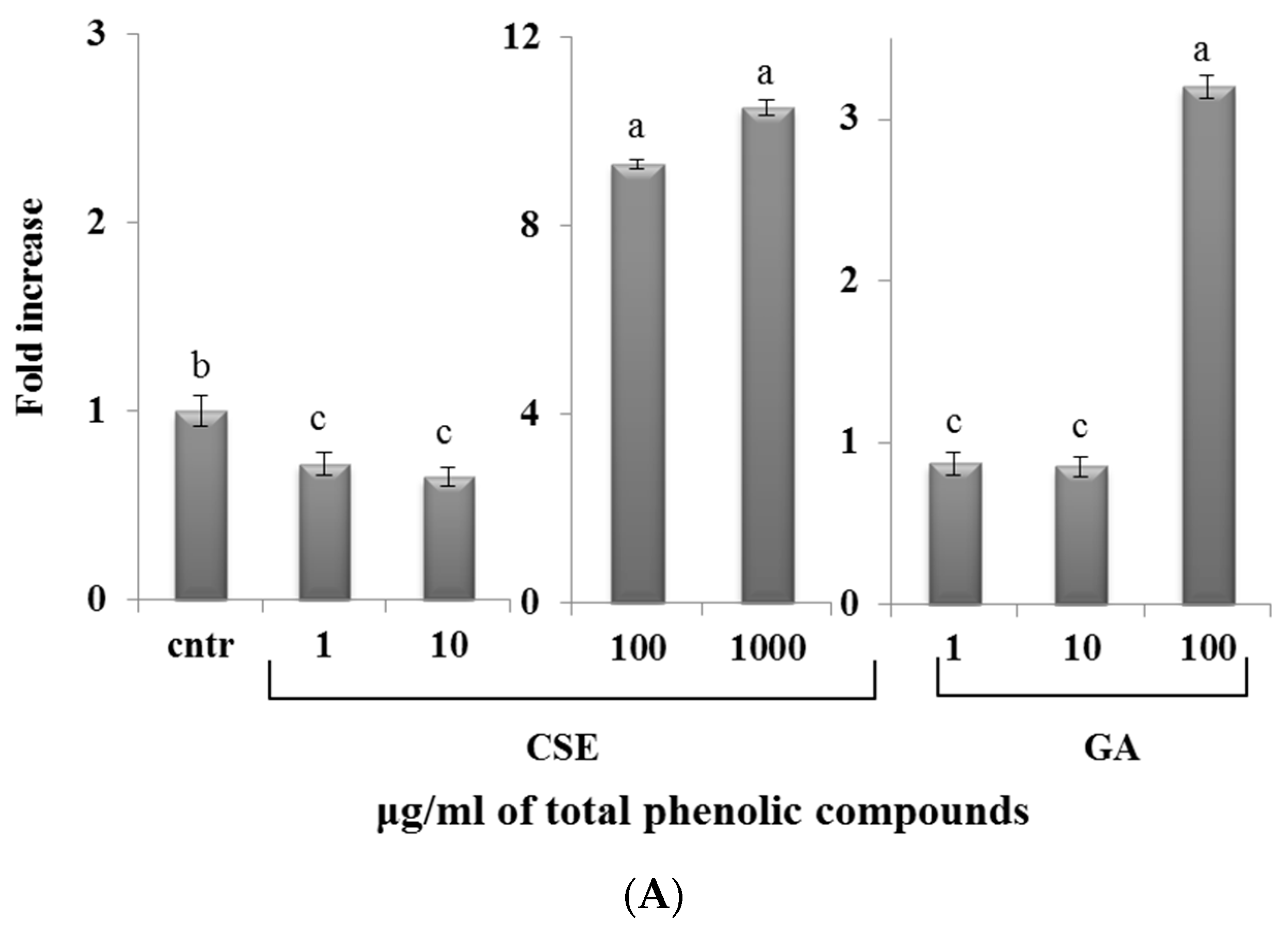
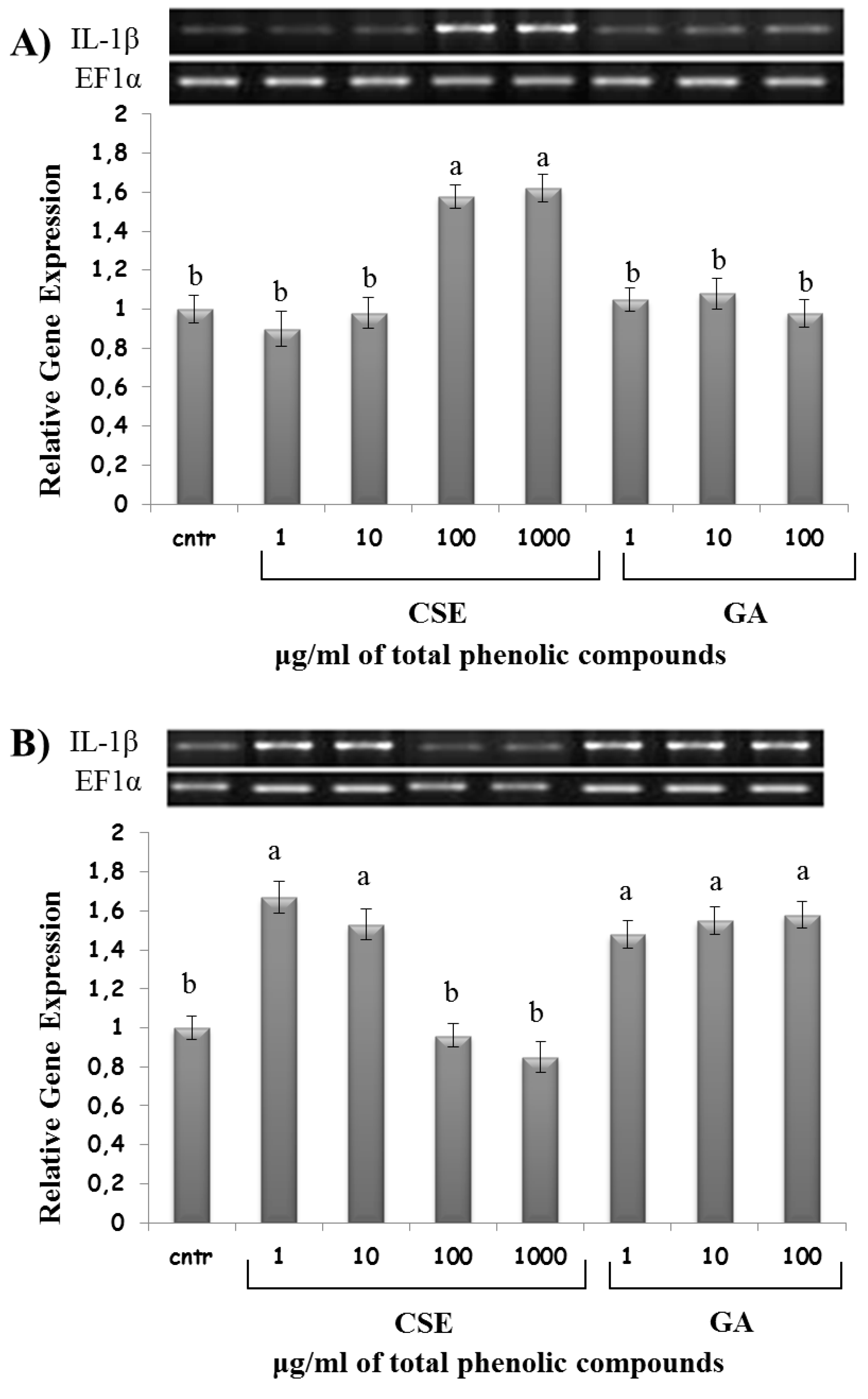
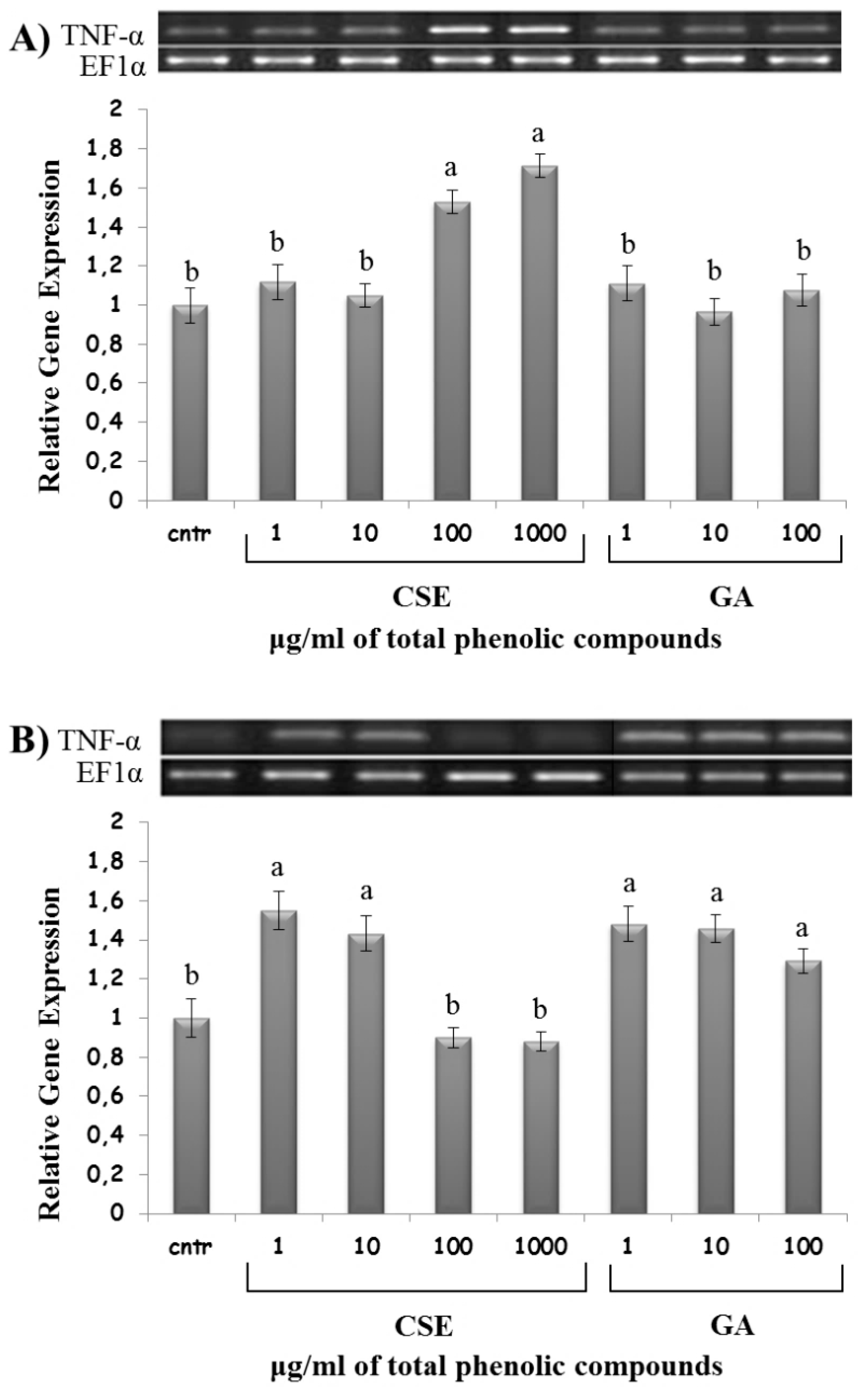
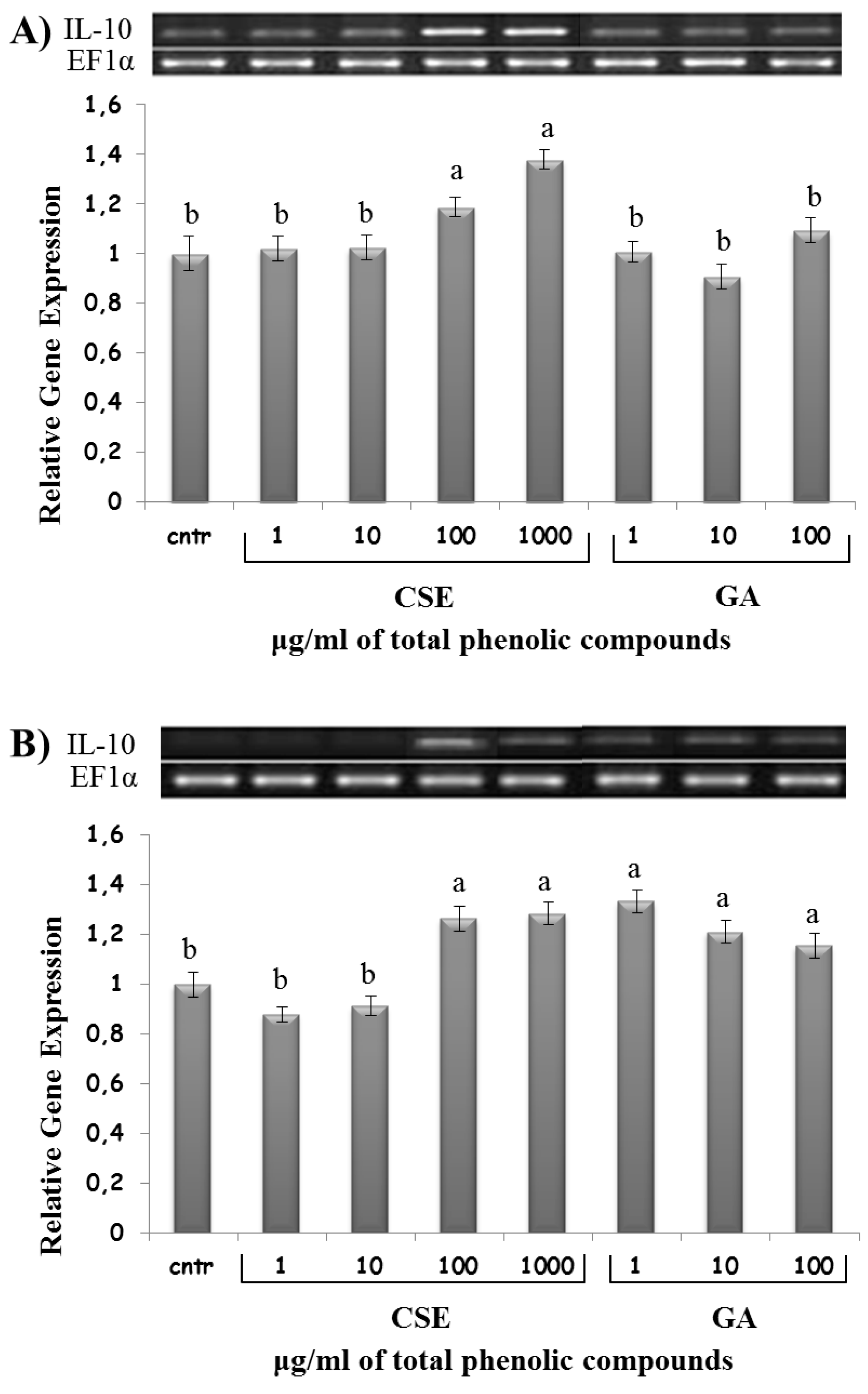
2.8. Effect of CSE on Cytokine Expression
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Chestnut Shell Extract (CSE) Preparation
4.3. Total Polyphenol Content and Antioxidant Activity of CSE
4.4. High-Performance Liquid Chromatography (HPLC) of CSE
4.5. Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) Analysis of CSE
4.6. Experimental Fish
4.7. Cell Culture
4.8. Cytotoxicity Assay
4.9. Cellular Content of Polyphenols
4.10. Superoxide Anion Production Assay
4.11. Phagocytosis Assay
4.12. qPCR
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reverter, M.; Bontemps, N.; Lecchini, D.; Banaigs, B.; Sasal, P. Use of plant extracts in fish aquaculture as an alternative to chemotherapy: Current status and future perspectives. Aquaculture 2014, 433, 50–61. [Google Scholar] [CrossRef]
- Landers, T.F.; Cohen, B.; Wittum, T.E.; Larson, E.L. Review of Antibiotic Use in Food Animals: Perspective, Policy, and Potential. Public Health Rep. 2012, 127, 4–22. [Google Scholar] [CrossRef] [PubMed]
- FAO Report. 2018. Available online: http://www.fao.org/documents/card/en/c/I9540EN/ (accessed on 10 June 2018).
- Murray, A.G.; Peeler, E.J. A framework for understanding the potential for emerging diseases in aquaculture. Prev. Vet. Med. 2005, 67, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.K.; Manna, S.K.; Thomas Allnutt, F.C. Viral vaccines for farmed finfish. Virus Dis. 2014, 25, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Muktar, Y.; Tesfaye, S.; Tesfaye, B. Present status and future prospects of fish vaccination: A review. J. Vet. Sci. Technol. 2016, 7, 2. [Google Scholar] [CrossRef]
- Harikrishnan, R.; Balasundaram, C.; Heo, M.-S. Impact of plant products on innate and adaptive immune system of cultured finfish and shellfish. Aquaculture 2011, 317, 1–15. [Google Scholar] [CrossRef]
- FAOSTAT Food and Agriculture Organization of the United States. 2017. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 15 May 2017).
- Vazquez, G.; Fontenla, E.; Santos, J.; Freire, M.S.; Gonzalez-Alvarez, J.; Antorrena, G. Antioxidant activity and phenolic content of chestnut (Castanea sativa) shell and eucalyptus (Eucalyptus globulus) bark extracts. Ind. Crops Prod. 2008, 28, 279–285. [Google Scholar] [CrossRef]
- Vázquez, G.; González-Alvarez, J.; Santos, J.; Freire, M.S.; Antorrena, G. Evaluation of potential applications for chestnut (Castanea sativa) shell and eucalyptus (Eucalyptus globulus) bark Extracts. Ind. Crops Prod. 2009, 29, 364–370. [Google Scholar] [CrossRef]
- Pokorny, J.; Korczak, J. Preparation of natural antioxidants. In Antioxidants in Food. Practical Applications; Pokorny, J., Yanishlieva, N., Gordon, M., Eds.; CRC Press: Boca Raton, FL, USA, 2001; pp. 311–330. [Google Scholar]
- Aires, A.; Carvalho, R.; Saavedra, M.J. Valorization of solid wastes from chestnut industry processing: Extraction and optimization of polyphenols, tannins and ellagitannins and its potential for adhesives, cosmetic and pharmaceutical industry. Waste Manag. 2016, 48, 457–464. [Google Scholar] [CrossRef]
- De Vasconcelos, M.C.; Bennett, R.N.; Rosa, E.A.; Ferrera-Cardoso, J.V. Composition of European chestnut (Castanea sativa Mill.) and association with health effects: Fresh and processed products. J. Sci. Food Agric. 2010, 90, 1578–1589. [Google Scholar] [CrossRef]
- Hao, J.J.; Liu, H.; Donis-Gonzalez, I.R.; Lu, X.H.; Jones, A.D.; Fulbright, D.W. Antimicrobial activity of chestnut extracts for poten-tial use in managing soilborne plant pathogens. Plant Dis. 2012, 96, 354–360. [Google Scholar] [CrossRef]
- Lee, N.K.; Jung, B.S.; Na, D.S.; Yua, H.H.; Kim, J.S.; Paik, H.D. The impact of antimicrobial effect of chestnut inner shell extracts against Campylobacter jejuni in chicken meat. LWT—Food Sci. Technol. 2016, 65, 746–750. [Google Scholar] [CrossRef]
- Ramudu, K.R.; Dash, G. A Review on Herbal Drugs Against Harmfull Pathogens in Aquaculture. Am. J. Drug Discovery Dev. 2013, 3, 209–219. [Google Scholar] [CrossRef]
- Pagliarulo, C.; Sateriale, D.; Scioscia, E.; De Tommasi, N.; Colicchio, R.; Pagliuca, C.; Scaglione, E.; Jussila, J.; Makkonen, J.; Salvatore, P.; et al. Growth, Survival and Spore Formation of the Pathogenic Aquatic Oomycete Aphanomyces astaci and Fungus Fusarium avenaceum Are Inhibited by Zanthoxylum rhoifolium Bark Extracts In Vitro. Fishes 2018, 3, 12. [Google Scholar] [CrossRef]
- Sateriale, D.; Scioscia, E.; Colicchio, R.; Pagliuca, C.; Salvatore, P.; Varricchio, E.; Volpe, M.G.; Paolucci, M.; Pagliarulo, C. Italian acacia honey exhibits lytic effects against the crayfish pathogens Aphanomyces astaci and Fusarium avenaceum. Lett. Appl. Microbiol. 2018. [Google Scholar] [CrossRef]
- Niedzwiecki, A.; Roomi, M.W.; Kalinovsky, T.; Rath, M. Anticancer Efficacy of Polyphenols and Their Combinations. Nutrients 2016, 8, 552. [Google Scholar] [CrossRef]
- Mirabella, N.; Castellani, V.; Sala, S. Beyond the throwaway society: A life cycle-based assessment of the environmental benefit of reuse. Integr. Environ. Assess. Manag. 2015, 11, 373–382. [Google Scholar]
- Yang, C.S.; Sang, S.; Lambert, J.D.; Lee, M.-J. Bioavailability issues in studying the health effects of plant polyphenolic compounds. Mol. Nutr. Food Res. 2008, 52, S139–S151. [Google Scholar] [CrossRef]
- Sorice, A.; Siano, F.; Capone, F.; Guerriero, E.; Picariello, G.; Budillon, A.; Ciliberto, G.; Paolucci, M.; Costantini, S.; Volpe, M.G. Potential anticancer effects of polyphenols from chestnut shell extracts: Modulation of cell growth, and cytokinomic and metabolomic profiles. Molecules 2016, 21, 1411. [Google Scholar] [CrossRef]
- Ricci, A.; Olejar, K.J.; Parpinello, G.P.; Kilmartin, P.A.; Versari, A. Application of Fourier transform infrared (FTIR) spectroscopy in the characterization of tannins. Appl. Spectrosc. Rev. 2015, 50, 407–442. [Google Scholar] [CrossRef]
- Ricci, A.; Lagel, M.-C.; Parpinello, G.P.; Pizzi, A.; Kilmartin, P.A.; Versari, A. Spectroscopy analysis of phenolic and sugar patterns in a food grade chestnut tannin. Food Chem. 2016, 203, 425–429. [Google Scholar] [CrossRef]
- Dos Santos Grasel, F.; Flôres Ferrão, M.; Wolf, C.R. Development of methodology for identification the nature of the polyphenolic extracts by FTIR associated with multivariate analysis. Spectrochim. Acta A. Mol. Biomol. Spectrosc. 2016, 153, 94–101. [Google Scholar] [CrossRef]
- Sun-Waterhouse, D. The development of fruit-based functional foods targeting the health and wellness market: A review. Int. J. Food Sci. Technol. 2011, 46, 899–920. [Google Scholar] [CrossRef]
- Noh, J.-R.; Gang, G.-T.; Kim, Y.-H.; Yang, K.-J.; Hwang, J.-H.; Lee, H.-S.; Oh, W.-K.; Song, K.-S.; Lee, C.-H. Antioxidant effects of the chestnut (Castanea crenata) inner shell extract in t-BHP-treated HepG2 cells, and CCl4- and high-fat diet-treated mice. Food Chem. Toxicol. 2010, 48, 3177–3183. [Google Scholar] [CrossRef]
- Danneh, D.; Josuttis, M. Cultivar and Production Effects on Bioactive Polyphenols. In Plants Isolation, Purification and Extract Preparation; Watson, R.R., Ed.; Academic Press: New York, NY, USA, 2014; pp. 3–13. [Google Scholar]
- Turkmen, N.; Sari, F.; Velioglu, Y.S. Effects of extraction solvents on concentration and antioxidant activity of black and black mate tea polyphenols determined by ferrous tartrate and Folin–Ciocalteu methods. Food Chem. 2006, 99, 835–841. [Google Scholar] [CrossRef]
- Vella, F.M.; Laratta, B.; La Cara, F.; Morana, A. Recovery of bioactive molecules from chestnut (Castanea sativa Mill.) by-products through extraction by different solvents. Nat. Prod. Res. 2018, 32, 1022–1032. [Google Scholar] [CrossRef]
- Shi, J.; Nawaz, H.; Pohorly, J.; Mittal, G.; Kakuda, Y.; Jiang, Y. Extraction of polyphenolics from plant material for functional foods—Engineering and technology. Food Rev. Int. 2005, 21, 139–166. [Google Scholar] [CrossRef]
- Biesaga, M. Influence of extraction methods on stability of flavonoids. J. Chromatogr. 2011, 1218, 2505–2512. [Google Scholar] [CrossRef]
- Laghi, L.; Parpinello, G.P.; Del Rio, D.; Calani, L.; Mattioli, A.U.; Versari, A. Fingerprint of enological tannins by multiple techniques approach. Food Chem. 2010, 121, 783–788. [Google Scholar] [CrossRef]
- Vázquez, G.; Pizzi, A.; Freire, M.S.; Santos, J.; Antorrena, G.; González-Alvarez, J. MALDI-TOF, HPLC-ESI-TOF and 13C-NMR characterization of chestnut (Castanea sativa) shell tannins for wood adhesives. Wood Sci. Technol. 2013, 47, 523–535. [Google Scholar] [CrossRef]
- Vàzquez, G.R.; Guerrero, G.A. Characterization of blood cells and hematological parameters in Cichlasoma dimerus (Teleostei, Perciformes). Tissue Cell. 2017, 39, 151–160. [Google Scholar] [CrossRef]
- Chernyavskikh, S.D.; Kuet, D.H.; Trikula, L.N.; Buslovskaya, L.K.; Kovtunenko, A.Y.; Makarova, Y.A. Hematologic profile for Cyprinus carpio. Indo Am. J. Pharm. Sci. 2017, 4, 3155–3161. [Google Scholar]
- Bernard, D.; Six, A.; Rigottier-Gois, L.; Messiaen, S.; Chilmonczyk, S.; Quillet, E.; Boudinot, P.; Benmansour, A. Phenotypic and Functional Similarity of Gut Intraepithelial and Systemic T Cells in a Teleost Fish. J. Immunol. 2006, 176, 3942–3949. [Google Scholar] [CrossRef]
- Lançon, A.; Delma, D.; Osman, H.; Thénot, J.-P.; Latruffe, B.J.N.; Latruffe, N. Human hepatic cell uptake of resveratrol: Involvement of both passive diffusion and carrier-mediated process. Biochem. Biophys. Res. Commun. 2004, 316, 1132–1137. [Google Scholar] [CrossRef]
- Salucci, M.; Stivala, L.A.; Maiani, G.; Bugianesi, R.; Vannini, V. Flavonoids uptake and their effect on cell cycle of human colon adenocarcinoma cells (Caco2). Br. J. Cancer 2002, 86, 1645–1651. [Google Scholar] [CrossRef]
- Jannin, B.; Menzel, M.; Berlot, J.-P.; Delmas, D.; Lançon, A.; Latruffe, N. Transport of resveratrol, a cancer chemopreventive agent, to cellular targets: Plasmatic protein binding and cell uptake. Biochem. Pharmacol. 2004, 68, 1113–1118. [Google Scholar] [CrossRef]
- Cvorovi, J.; Ziberna, L.; Fornasaro, S.; Tramer, F.; Passamonti, S. Bioavailability of Flavonoids: The Role of Cell Membrane Transporters. In Polyphenols: Mechanisms of Action in Human Health and Disease, 2nd ed.; Watson, R.R., Preedy, V.R., Zibadi, S., Eds.; Elsevier, Inc.: London, UK, 2018; pp. 295–320. [Google Scholar]
- Carocho, M.; Ferreira, I.C.F.R. A review on antioxidants, prooxidants and related controversy: Natural and synthetic compounds, screening and analysis methodologies and future perspectives. Food Chem. Toxicol. 2013, 51, 15–25. [Google Scholar] [CrossRef]
- Villanueva, C.; Kross, R.D. Antioxidant-induced stress. Int. J. Mol. Sci. 2012, 13, 2091–2109. [Google Scholar] [CrossRef]
- Gomes, C.A.; Cruz, T.G.; Andrade, J.L.; Milhazes, N.; Borges, F.; Marques, M.P.M. Anticancer activity of phenolic acids of natural or synthetic origin: A structure–activity study. J. Med. Chem. 2003, 46, 5395–5401. [Google Scholar] [CrossRef]
- Galati, G.; O’Brien, P.J. Potential toxicity of flavonoids and other dietary phenolics: Significance for their chemopreventive and anticancer properties. Free Radic. Biol. Med. 2004, 37, 287–303. [Google Scholar] [CrossRef]
- Halliwell, B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch. Biochem. Biophys. 2008, 476, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Procházková, D.; Boušová, I.; Wilhelmová, N. Antioxidant and prooxidant properties of flavonoids. Fitoterapia 2011, 82, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Yordi, E.G.; Pérez, E.M.; Matos, M.J.; Villares, E.U. Structural alerts for predicting clastogenic activity of pro-oxidant flavonoid compounds. J. Biomol. Screen. 2012, 17, 216–224. [Google Scholar] [CrossRef]
- Esteban, M.; Cuesta, A.; Chaves-Pozo, E.; Meseguer, J. Phagocytosis in Teleosts. Implications of the new cells involved. Biology 2015, 4, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Sohi, K.K.; Mittal, N.; Hundal, M.K.; Khanduja, K.L. Gallic acid, an antioxidant, exhibits antiapoptotic potential in normal human lymphocytes: A Bcl-2 independent mechanism. J. Nutr. Sci. Vitaminol. 2003, 49, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Tallarida, R.J. Drug synergism: Its detection and applications. J. Pharmacol. Exp. Ther. 2001, 298, 865–872. [Google Scholar] [PubMed]
- Liu, R.H. Health benefits of fruit and vegetables are from additive and synergistic combinations of phytochemicals. Am. J. Clin. Nutr. 2003, 78, 517S–520S. [Google Scholar] [CrossRef]
- Santangelo, C.; Varì, R.; Scazzocchio, B.; Di Benedetto, R.; Filesi, C.; Masella, R. Polyphenols, intracellular signalling and inflammation. Ann. Ist. Super. Sanita 2007, 43, 394–405. [Google Scholar]
- Leyva-López, N.; Gutierrez-Grijalva, E.; Ambriz-Perez, D.; Heredia, J. Flavonoids as cytokine modulators: A possible therapy for inflammation-related diseases. Int. J. Mol. Sci. 2016, 17, 921. [Google Scholar] [CrossRef]
- Blonska, M.; Czuba, Z.P.; Krol, W. Effect of flavone derivatives on interleukin-1beta (IL-1beta) mRNA expression and IL-1beta protein synthesis in stimulated RAW 264.7 macrophages. Scand. J. Immunol. 2003, 57, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Comalada, M.; Ballester, I.; Bailón, E.; Sierra, S.; Xaus, J.; Gálvez, J.; de Medina, F.S.; Zarzuelo, A. Inhibition of pro-inflammatory markers in primary bone marrow-derived mouse macrophages by naturally occurring flavonoids: Analysis of the structure–activity relationship. Biochem. Pharmacol. 2006, 72, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.-D.; Choi, C.-H.; Bark, H.; Son, H.-Y.; Park, H.-H.; Lee, S.; Park, J.-W.; Park, E.-K.; Shin, H.-I.; Kim, S.-H. Quercetin inhibits expression of inflammatory cytokines through attenuation of NF-κB and p38 MAPK in HMC-1 human mast cell line. Inflamm. Res. 2007, 56, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.S.; Catravas, J.D.; Odoms, K.; Denenberg, A.; Malhotra, V.; Wong, H.R. Epigallocatechin-3-gallate, a green tea-derived polyphenol, inhibits IL-1 beta-dependent proinflammatory signal transduction in cultured respiratory epithelial cells. J. Nutr. 2004, 134, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Secombes, C.J. The function of fish cytokines. Biology 2016, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.R.; Xu, Q.; Secombes, C.J.; Wang, T. Two types of TNF-α exist in teleost fish: Phylogeny, expression, and bioactivity analysis of type-II TNF-α- in rainbow trout Oncorhynchus mykiss. J. Immunol. 2013, 191, 5959–5972. [Google Scholar] [CrossRef]
- Zhang, A.; Chen, D.; Wei, H.; Du, L.; Zhao, T.; Wang, X.; Zhou, H. Functional characterization of TNF-α in grass carp head kidney leukocytes: Induction and involvement in the regulation of NF kB signaling. Fish Shellfish Immunol. 2012, 33, 1123–1132. [Google Scholar] [CrossRef]
- Raida, M.K.; Buchmann, K. Development of adaptive immunity in rainbow trout, Oncorhynchus mykiss (Walbaum) surviving an infection with Yersinia ruckeri. Fish Shellfish Immunol. 2008, 25, 533–541. [Google Scholar] [CrossRef]
- Uribe, C.; Folch, H.; Enriquez, R.; Moran, G. Innate and adaptive immunity in teleost fish: A review. Vet. Med. 2011, 56, 486–503. [Google Scholar] [CrossRef]
- Zhang, Z.; Swain, T.; Bøgwald, J.; Dalmo, R.A.; Kumari, J. Bath immunostimulation of rainbow trout (Oncorhynchus mykiss) fry induces enhancement of inflammatory cytokine transcripts, while repeated bath induce no changes. Fish Shellfish Immunol. 2009, 26, 677–684. [Google Scholar] [CrossRef]
- Bakhiet, M.; Al-Salloom, F.S.; Qareiballa, A.; Bindayna, K.; Farid, I.; Botta, G.A. Induction of α and β chemokines by intestinal epithelial cells stimulated with Campylobacter jejuni. J. Infect. 2004, 48, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, G.; Hernell, O.; Hammarstrom, S.; Hammarstrom, M.-L. Concomitant increase of IL-10 and pro-inflammatory cytokines in intraepithelial lymphocyte subsets in celiac disease. Int. Immunol. 2007, 19, 993–1001. [Google Scholar] [CrossRef]
- De Santis, S.; Cavalcanti, E.; Mastronardi, M.; Jirillo, E.; Chieppa, M. Nutritional keys for intestinal barrier modulation. Front. Immunol. 2015, 6, 612. [Google Scholar] [CrossRef]
- Dinarello, C.A. Historical Review of Cytokines. EUR. J. Immunol. 2007, 37, S34–S45. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Aziz, M.; Jacob, A.; Yang, W.L.; Matsuda, A.; Wang, P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J. Leukoc. Biol. 2013, 93, 329–342. [Google Scholar] [CrossRef]
- Chuang, C.-C.; Martinez, K.; Xie, G.; Kennedy, A.; Bumrungpert, A.; Overman, A.; Jia, W.; McIntosh, M.K. Quercetin is equally or more effective than resveratrol in attenuating tumor necrosis factor-α mediated inflammation and insulin resistance in primary human adipocytes. Am. J. Clin. Nutr. 2010, 92, 1511–1521. [Google Scholar] [CrossRef]
- Singleton, V.L.; Rossi, J.A. Colourimetry of total phenolics with phosphomolybdic–phospholungistic acid reagents. Am. J. Enol. Viticult. 1965, 16, 144–158. [Google Scholar]
- Mariano, G.; Stilo, R.; Terrazzano, G.; Coccia, E.; Vito, P.; Varricchio, E.; Paolucci, M. Effects of recombinant trout leptin in superoxide production and NF-κB/MAPK phosphorylation in blood leukocytes. Peptides 2013, 48, 59–69. [Google Scholar] [CrossRef]
- McMillan, D.N.; Secombes, C.J. Isolation of rainbow trout (Oncorhynchus mykiss) intestinal intraepithelial lymphocytes (IEL) and measurement of their cytotoxic activity. Fish Shellfish Immunol. 1997, 7, 527–541. [Google Scholar] [CrossRef]
- Komatsu, K.; Tsutsui, S.; Hino, K.; Araki, K.; Yoshiura, Y.; Yamamoto, A.; Nakamura, O.; Watanabe, T. Expression profiles of cytokines released in intestinal epithelial cells of the rainbow trout, Oncorhynchus mykiss, in response to bacterial infection. Dev. Comp. Immunol. 2009, 33, 499–506. [Google Scholar] [CrossRef]
- Ferrari, M.; Fornasiero, M.C.; Isetta, A.M. MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J. Immunol. Methods 1990, 131, 165–172. [Google Scholar] [CrossRef]
- Reyes-Becerril, M.; Ascencio-Valle, F.; Tovar-Ramírez, D.; Meseguer, J.; Esteban, M.Á. Effects of polyamines on cellular innate immune response and the expression of immune-relevant genes in gilthead seabream leucocytes. Fish Shellfish Immunol. 2011, 30, 248–254. [Google Scholar] [CrossRef]
- Park, K.I.; Kang, S.R.; Park, H.S.; Lee, D.H.; Nagappan, A.; Kim, J.A.; Shin, S.C.; Kim, E.H.; Lee, W.S.; Chung, H.J.; et al. Regulation of Proinflammatory Mediators via NF-κB and p38 MAPK-Dependent Mechanisms in RAW 264.7 Macrophages by Polyphenol Components Isolated from Korea Lonicera japonica THUNB. Evid. Based Complement. Altern. Med. 2012, 828521. [Google Scholar] [CrossRef]
- Mazur-Bialy, A.I.; Pocheć, E. HMGB1 inhibition during zymosan-induced inflammation: The potential therapeutic action of riboflavin. Arch. Immunol. Ther. Exp. 2016, 64, 171–176. [Google Scholar] [CrossRef]
- Castro, R.; Zou, J.; Secombes, C.J.; Martin, S.A.M. Cortisol modulates the induction of inflammatory gene expression in a rainbow trout macrophage cell line. Fish Shellfish Immunol. 2011, 30, 215–223. [Google Scholar] [CrossRef]
- Morash, A.J.; Kajimura, M.; McClelland, G.B. Intertissue regulation of carnitine palmitoyl transferase I (CPTI): Mitochondrial membrane properties and gene expression in rainbow trout (Oncorhynchus mykiss). Biochim. Biophys. Acta 2008, 1778, 1382–1389. [Google Scholar] [CrossRef]
- Magrone, T.; Fontana, S.; Laforgia, F.; Dragone, T.; Jirillo, E.; Passantino, L. Administration of a Polyphenol-Enriched Feed to Farmed Sea Bass (Dicentrarchus labrax L.) Modulates Intestinal and Spleen Immune Responses. Oxid. Med. Cell. Longev. 2016, 2016, 2827567. [Google Scholar] [CrossRef]
- Kroes, B.; van den Berg, A.; Quarles van Ufford, H.; van Dijk, H.; Labadie, R. Anti-inflammatory activity of gallic acid. Planta Med. 1992, 58, 499–504. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extracts | Gallic Acid | Syringic Acid | Ellagic Acid | Rutin | Quercetin | Catechin |
|---|---|---|---|---|---|---|
| Ethanol 100% (90 °C) | 3.39 ± 0.25 | 0.79 ± 0.05 | 1.91 ± 0.20 | 0.099 ± 0.005 | 0.200 ± 0.015 | 0.61 ± 0.05 |
| Ethanol 100% RT | 1.82 ± 0.09 | 0.52 ± 0.09 | 0.75 ± 0.12 | 0.049 ± 0.003 | 0.089 ± 0.008 | 0.34 ± 0.08 |
| Ethanol 70%/water 30% RT | 2.12 ± 0.15 | 0.50 ± 0.10 | 1.05 ± 0.18 | 0.059 ± 0.007 | 0.081 ± 0.010 | 0.40 ± 0.09 |
| Water 100% (75 °C) | 3.74 ± 0.21 | 0.20 ± 0.08 | 1.65 ± 0.08 | 0.024 ± 0.009 | 0.033 ± 0.005 | 0.25 ± 0.05 |
| Extract | % Inhibition | Total Polyphenols |
|---|---|---|
| Ethanol 100% (90 °C) | 78.9 ± 0.8 | 610.7 ± 2.3 |
| Ethanol 100% RT | 79.8 ± 1.2 | 635.3 ± 1.5 |
| Ethanol 70%/water 30% RT | 78.5 ± 1.1 | 590.2 ± 1.7 |
| Water 100% (75 °C) | 74.3 ± 1.0 | 487.9 ± 1.4 |
| Gallic acid | 82.6 ± 0.9 | - |
| Main Vibrational Bands of Chestnut Shell Extracts (Wavenumbers, cm−1, Intensity) | |||||
|---|---|---|---|---|---|
| Ethanol 100% at 90 °C | Ethanol 100% at RT | Ethanol/Water 70%/30% at RT | Water 100% at 75 °C | Silvafeed® TSP and ATX | Peak Number and Assignment |
| 2918 | 2918 |
| |||
| 2850 | 2850 |
| |||
| 1711 | 1708 | 1705 | 1700 | 1727 |
|
| 1607 | 1606 | 1604 | 1605 | 1605 |
|
| 1536 | |||||
| 1519 | 1516 | 1519 | 1520 | 1515 | |
| 1445 | 1444 | 1443 | 1444 | 1447 | |
| 1342 | 1323 | 1319 | 1334 | 1308 |
|
| 1283 | 1284 | 1285 | |||
| 1198 | 1198 | 1195 | 1197 |
| |
| 1174 | |||||
| 1145 | 1146 | 1144 | 1144 | ||
| 1101 | 1101 | 1100 | 1112 |
| |
| 1034 | 1030 | 1029 | 1032 | 1035 |
|
| 911 |
| ||||
| 892 | |||||
| 869 | 861 | ||||
| 834 | |||||
| 819 | 830 | 820 | 822 | 820 | |
| 766 | 763 | 765 | 765 | 773 | |
| 749 | |||||
| 719 | 729 | 728 | 729 | 719 | |
| 703 | |||||
| 666 | 666 | 666 | 666 | 676 | |
| 654 | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coccia, E.; Siano, F.; Volpe, M.G.; Varricchio, E.; Eroldogan, O.T.; Paolucci, M. Chestnut Shell Extract Modulates Immune Parameters in the Rainbow Trout Oncorhynchus mykiss. Fishes 2019, 4, 18. https://doi.org/10.3390/fishes4010018
Coccia E, Siano F, Volpe MG, Varricchio E, Eroldogan OT, Paolucci M. Chestnut Shell Extract Modulates Immune Parameters in the Rainbow Trout Oncorhynchus mykiss. Fishes. 2019; 4(1):18. https://doi.org/10.3390/fishes4010018
Chicago/Turabian StyleCoccia, Elena, Francesco Siano, Maria Grazia Volpe, Ettore Varricchio, Orhan Tufan Eroldogan, and Marina Paolucci. 2019. "Chestnut Shell Extract Modulates Immune Parameters in the Rainbow Trout Oncorhynchus mykiss" Fishes 4, no. 1: 18. https://doi.org/10.3390/fishes4010018
APA StyleCoccia, E., Siano, F., Volpe, M. G., Varricchio, E., Eroldogan, O. T., & Paolucci, M. (2019). Chestnut Shell Extract Modulates Immune Parameters in the Rainbow Trout Oncorhynchus mykiss. Fishes, 4(1), 18. https://doi.org/10.3390/fishes4010018