The Enzyme-Modified Neutral Comet (EMNC) Assay for Complex DNA Damage Detection



Abstract
1. Introduction
2. Experimental Design
2.1. Materials
- Library efficient DH5a competent bacterial cells (Fisher Scientific, Loughborough, UK; Cat No.: 11573117)
- Rosetta2 (DE3) pLysS bacterial cells (Merck-Millipore, Watford, UK; Cat No.: 71403)
- Bacterial expression plasmid for His-tagged APE1 (e.g., Addgene, Teddington, UK; Cat No.: 70757; or available on request from authors)
- Bacterial expression plasmid for His-tagged OGG1 (available on request from authors)
- Bacterial expression plasmid for His-tagged NTH1 (available on request from authors)
- Agar granules (Fisher Scientific, Loughborough, UK; Cat No.: 10572775)
- IPTG (Sigma-Aldrich, Dorset, UK; Cat No.: I6758-5G)
- Glucose (Sigma-Aldrich, Dorset, UK; Cat. No.: 16325-1KG)
- LB Broth, Miller granulated (Fisher Scientific, Loughborough, UK; Cat No.: 11345992)
- Lysozyme from chicken egg white (Sigma-Aldrich, Dorset, UK; Cat No.: L4919-1G)
- Novex™ WedgeWell™ 10% SDS-PAGE gel (Fisher Scientific, Loughborough, UK; Cat No.: 15496794)
- Leupeptin (Fisher Scientific, Loughborough, UK; Cat No.: 11884101)
- Chemostatin (Sigma-Aldrich, Dorset, UK; Cat No.: 230790-10MG)
- Pepstatin (Fisher Scientific, Loughborough, UK; Cat No.: 10036263)
- Aprotinin (Fisher Scientific, Loughborough, UK; Cat No.: 11854101)
- PMSF (Fisher Scientific, Loughborough, UK; Cat No.: 10485015)
- Imidazole (Sigma-Aldrich, Dorset, UK; Cat. No.: I2399-500G)
- TGS, 10x (Bio-Rad Laboratories, Watford, UK; Cat No.: 161-0772)
- TG, 10x (Bio-Rad Laboratories, Watford, UK; Cat No.: 161-0771)
- Methanol (Fisher Scientific, Loughborough, UK; Cat No.: 10675112)
- Mercaptoethanol (Sigma-Aldrich, Dorset, UK; Cat. No.: M6250-250ML)
- Glycerol (Fisher Scientific, Loughborough, UK; Cat No.: 10795711)
- SDS (Sigma-Aldrich, Dorset, UK; Cat. No.: L5750-500G)
- Bromophenol blue (Fisher Scientific, Loughborough, UK; Cat No.: 10497573)
- Anti-HisTag antibody (Merck-Millipore, Watford, UK; Cat No.: 70796-3)
- Instant Blue Protein Stain (Sigma-Aldrich, Dorset, UK; Cat. No.: ISB1L)
- Agarose low melting point (Fisher Scientific, Loughborough, UK; Cat. No.: 10583355)
- Agarose normal melting point (Fisher Scientific, Loughborough, UK; Cat. No.: 10688973)
- PBS (Ca2+ and Mg2+ free) (Sigma-Aldrich, Dorset, UK; Cat. No.: D8537-500ML)
- NaCl (Sigma-Aldrich, Dorset, UK; Cat. No.: 31434-1KG-M)
- NaOH (Sigma-Aldrich, Dorset, UK; Cat. No.: 30620-1KG-M)
- EDTA disodium salt solution (Sigma-Aldrich, Dorset, UK; Cat. No.: E5134-500G)
- Tris base (Sigma-Aldrich, Dorset, UK; Cat. No.: T1503-1KG)
- Triton X-100 (Fisher Scientific, Loughborough, UK; Cat. No.: 10717503)
- N-Lauroylsarcosine (Sigma-Aldrich, Dorset, UK; Cat. No.: L5125-500G)
- Dimethyl sulphoxide (Sigma-Aldrich, Dorset, UK; Cat. No.: D5879-500ML)
- Boric acid (Sigma-Aldrich, Dorset, UK; Cat. No.: B7660-1KG)
- KCl (Sigma-Aldrich, Dorset, UK; Cat. No.: P3911-500G)
- MgCl2 (Sigma-Aldrich, Dorset, UK; Cat. No.: M8266-100G)
- DTT (Fisher Scientific, Loughborough, UK; Cat. No.: 10592945)
- BSA (Fisher Scientific, Loughborough, UK; Cat. No.: 12827172)
- KOH (Sigma-Aldrich, Dorset, UK; Cat. No.: P1767-500G)
- SYBR Gold (Fisher Scientific, Loughborough, UK; Cat. No.: 10358492)
- Trypsin/EDTA (for cell collection) (Sigma-Aldrich, Dorset, UK; Cat. No.: T4049-100ML)
2.2. Equipment
- QIAprep Spin Miniprep Kit (Qiagen, Southampton, UK; Cat No.: 27104)
- Syringe filter, 0.45 µm (Fisher Scientific, Loughborough, UK; Cat. No.: 15216869)
- Syringe filter, 1.1 µm (Fisher Scientific, Loughborough, UK; Cat. No.: 15372378)
- Sonicator (Sonics, Newtown, CT, USA; Cat No.: VCX 130)
- Nanodrop (Fisher Scientific, Loughborough, UK; Cat. No.: 13-400-518)
- Rotary Shaker (Kuhner, Birsfelden, Switzerland; Cat No.: SMX1700)
- HisTrap HP affinity chromatography column (GE Healthcare, Little Chalfont, UK; Cat No.: 17-5247-01)
- AKTA FPLC (GE Healthcare, Little Chalfont, UK; e.g., Cat No.: 18-1900-26)
- Oak Ridge tubes (Sigma-Aldrich, Dorset, UK; Cat. No.: T1418-10EA)
- Superloop, 10 mL (Fisher Scientific, Loughborough, UK; Cat. No.: 11330122)
- Immobilon FL membrane (Merck-Millipore, Watford, UK; Cat No.: IPFL00010)
- Superfrosted microscope slides (Fisher Scientific, UK; Cat. No.: 10149870)
- Coverslips 50 mm × 22 mm (Fisher Scientific, Loughborough, UK; Cat. No.: 12362128)
- Coverslips 22 mm × 22 mm (Fisher Scientific, Loughborough, UK; Cat. No.: 12333128)
- Coplin jars (Fisher Scientific, Loughborough, UK; Cat. No.: 10284922)
- Humidified chamber (Sigma-Aldrich, Dorset, UK; Cat. No.: Z670146-1EA)
- Comet electrophoresis tank (Appleton Woods, Birmingham, UK; Cat. No.: CS1602)
- Power supply unit (Fisher Scientific, Loughborough, UK; Cat. No.: 12613546)
- Fluorescent microscope Olympus BX61 (Olympus, Hamburg, Germany)
2.3. Software
- Komet 6.0 image analysis software (Andor Technology, Belfast, Northern Ireland)
2.4. Solutions
- LB broth media: 12.5 g of LB broth granules were added to 500 mL of dH2O (2.5% w/v) and the pH was adjusted to 7.2 with 5 M of NaOH.
- LB Agar: 7.5 g of agar granules were added to 500 mL of LB broth (1.5% w/v).
- 3× SDS-PAGE loading dye: 750 μL of 1 M of Tris-HCl (25 mM, pH = 6.8), 750 μL of 100% mercaptoethanol (2.5%), 3 mL of 10% SDS (1%), 3 mL of 100% glycerol (10%), 1.5 mL of 1 mg/mL of bromophenol blue (0.05 mg/mL), and 60 μL of 500 mM of EDTA (1 mM) were added to 940 μL of dH2O. For the working solution, 2:1 protein extract was diluted in 3× SDS dye.
- Enzyme Purification Lysis Buffer: 25 mM of Tris-HCl (12.5 mL of a 1 M solution, pH = 8.0), 500 mM of NaCl (50 mL of a 5 M solution), 5% Glycerol (25 mL of a 100% solution), and 5 mM of Imidazole (0.170 g) were added to 412.5 mL of dH2O. The complete solution was prepared prior to use by adding a mixture of protease inhibitors (30 μL of leupeptin, chemostatin, pepstatin, aprotinin (all 1 mg/mL), and 100 μL PMSF (100 mM)).
- Enzyme Purification Elution Buffer: 25 mM of Tris-HCl (12.5 mL of a 1 M solution, pH = 8.0), 500 mM of NaCl (50 mL of a 5 M solution), 5% Glycerol (25 mL of a 100% solution), and 500 mM of imidazole (17.02 g) were added to 412.5 mL of dH2O. The complete solution was prepared prior to use by adding 100 μL of PMSF (100 mM).
- Enzyme Storage Buffer: 50 mM of Tris-HCl (12.5 μL of a 1 M solution, pH = 8.0), 50 mM of KCl (12.5 μL of a 1 M solution), 1 mM of EDTA (10 μL of a 0.1 M solution), and 10% glycerol (10 μL of a 100% solution) were added to 955 μL of dH2O.
- Comet Lysis Buffer: NaCl (146.1 g, 2.5 M), EDTA disodium salt (37.2 g, 100 mM), Tris base (1.2 g, 10 mM), and 1 % N-lauroylsarcosine (10 g) were added to 800 mL of dH2O. The solution was heated to ~45 °C if necessary. The pH was set to 9.5 by the addition of NaOH (8 g) and 5 M of NaOH dropwise, then adjusted to 1 L and stored at 4 °C. The complete solution was prepared prior to use by adding a mixture of 1 ml of DMSO and 1 mL of Triton X-100 to 98 mL of cold lysis buffer.
- Comet Electrophoresis Buffer: A 5× TBE solution was prepared by adding 54 g of Tris base, 27.5 g of boric acid, and 20 mL of 0.5 M EDTA (pH = 8.0) to ~800 mL of dH2O. The pH was adjusted to 8.3, with volume of 1 L made up and stored at room temperature (RT). For working electrophoresis buffer (1× TBE), 300 mL of 5× TBE with 1200 mL of cold dH2O were mixed just before use.
- Enzyme Activity Buffer: A 10× enzyme buffer solution was prepared by adding Tris base (605.7 mg, 50 mM), KCl (745.5 mg, 100 mM), MgCl2 (47.6 mg, 5 mM), EDTA (26.2 mg, 1 mM), DTT (15.4 mg, 1 mM), and BSA (10 mg) to 10 mL of dH2O. The pH was set to 8 by the addition of 5 M of KOH dropwise; 1 mL aliquots were stored at −20 °C. For the working enzyme buffer (1×), 1 mL of 10× solution with 9 mL of dH2O were mixed just before use.
- Enzyme Solution: concentrations of enzymes to use depended on the level of purity and activity (see Section 3.5). As a guide, we routinely added 0.6 pmol of APE1, 6.0 pmol of NTH1, and 5.2 pmol of OGG1 diluted in 1x enzyme buffer per treatment.
- Staining Solution: SYBR Gold 1 was diluted in 20,000 in dH2O, pH = 8.0.
3. Procedure
3.1. Purification and Expression of Recombinant Enzymes
3.1.1. Overexpression of Recombinant His-Tagged APE1, OGG1, and NTH1
- Thaw Rossetta2 (DE3) pLysS cells on ice and add 20 µL of cells into the appropriate number of 1.5 mL tubes.
- Add 1 µL of each bacterial expression plasmid (5 ng/µL) expressing either APE1, OGG1, or NTH1 to each of three tubes, as necessary, and mix carefully by flicking the bottom of the tube.
- Incubate cells plus plasmid on ice for 5 min, heat shock for exactly 30 s at 42 °C and return to the ice for at least 2 min.
- Add 500 µL of LB media pre-warmed to 37 °C to each of the tubes containing the cells and incubate on a rotary shaker for 1 h at 37 °C.
- Plate 50 µL of each mix onto separate pre-prepared 10 cm LB agar plates (containing the appropriate antibiotics) using bacterial spreaders. The remainder of the mix can be stored at 4 °C, in case further required.
- Invert the plates and incubate overnight in a static incubator set at 37 °C.
- The following day, select a single bacterial colony, add to 5 mL of LB containing the appropriate antibiotics, and incubate at 37 °C with shaking at 225 rpm overnight. We would advise selecting 2–3 different colonies and incubating in separate tubes to ensure at least one efficiently grown culture.
- Add 300 µL of overnight culture to feed a 30 mL culture (i.e. 1:100) containing the appropriate antibiotics and 60 µL of glucose (20%), and grow at 37 °C with shaking at 225 rpm until an OD600nm of 0.6–0.8 is achieved (~3 h).
- Add 30 mL of culture to feed a 300 mL culture (i.e., 1:10) containing the appropriate antibiotics and 600 µL of glucose (20 %), and grow at 37 °C with shaking at 225 rpm until an OD600 nm of 0.6–0.8 is achieved (~1.5 h). Any culture from the original 30 mL culture can be used to create a glycerol stock by removing 150 µL and adding to 50 µL of 50% glycerol, which can then be stored at −80 °C.
- Induce the 300 mL culture with 1 mM of IPTG (330 µL from 1 M stock) and grow for a further 3 h at 30 °C with shaking.
- Centrifuge the culture at 8000 rpm for 20 min, remove the supernatant and freeze the bacterial cell pellets at −80 °C.

- PAUSE STEP: Pellets can be stored indefinitely at −80 °C.

3.1.2. Purification of Recombinant His-Tagged Proteins
- Resuspend the bacterial cell pellet thoroughly in 30 mL of complete Enzyme Purification Lysis Buffer.
- Add lysozyme to 0.1 mg/mL (3 mg) and incubate on ice for 15 min.
- Lyse the cells by sonication using 3 × 15 s bursts with 30 s intervals on ice.
- Centrifuge the cell lysate in Oak Ridge tubes at 25,000 rpm for 20 min at 4 °C.
- Collect the supernatant and filter through 1.1 µm syringe pre-filters, and then through 0.45 µm syringe filters.
- Prepare 150 mL of the Enzyme Purification Lysis Buffer and 150 mL of the Enzyme Purification Elution Buffer, each containing 0.1 mM PMSF.
- Wash the 1 mL HisTrap column (usually stored in ethanol) with three volumes of water, followed by three volumes of Enzyme Purification Lysis Buffer containing 0.1 mM PMSF using an FPLC in a cold cabinet/room (4 °C).
- Add the filtered supernatant to the washed column using a 10 or 50 mL superloop.
- Wash column with lysis buffer containing 0.1 mM of PMSF until no more protein elutes (~3–5 column volumes).
- Gradient elute using 20 column volumes (20 mL) of Enzyme Purification Elution Buffer containing 0.1 mM PMSF, collecting 0.5 mL fractions.
- Remove 5 µL of protein-containing fractions, add 5 µL water and 5 µL 3× SDS PAGE loading dye, and analyze by 10 % SDS-PAGE in 1× TGS Buffer.
- Transfer proteins to nitrocellulose or PVDF membrane in 1× TG Buffer containing 20% methanol and immunoblot using anti-HisTag antibodies (diluted 1:1000).
- Use the gel following protein transfer, and stain with Instant blue for > 15 min to identify fractions containing high purity enzyme(s) relative to bacterial contaminants
- Store protein fractions at −80 °C until required. Once APE1, OGG1, or NTH1-containing relatively pure (> 90%) protein fractions have been identified, combine these for the individual proteins and buffer exchange or dialyze into the Enzyme Storage Buffer. If proteins are not of sufficient purity, proceed with further purification (e.g., ion exchange chromatography).
- It is recommended that the activity of the enzymes are checked using oligonucleotide substrates containing the appropriate site-specific DNA damage (e.g., AP site for APE1, 8-oxoguanine for OGG1, and thymine glycol for NTH1; [20]).
3.2. Agarose Preparation (10 min)
3.2.1. For Slides Coating
- Prepare 1% normal melting point agarose by mixing powdered agarose with distilled water in a glass beaker or bottle.
- Place bottle in the microwave at low power for short intervals (~30 s), avoiding vigorous boiling of the agarose and ensuring that all of the agarose is dissolved.
- Agarose solution can be used immediately or stored at RT.
3.2.2. For Cell Embedding
- Prepare 1% low melting point agarose by mixing powdered agarose with PBS in a glass beaker or bottle.
- Place bottle in the microwave at low power for short intervals (~30 s), avoiding vigorous boiling of the agarose and ensuring that all of the agarose is dissolved.
- Agarose solution can be used immediately (once cooled to 37 °C) or stored at RT.
3.3. Slide Coating (10 min)
- Prepare slides by adding 800 µL of molten normal melting point agarose to a microscope slide, add a 22 × 50 mm coverslip, and leave agarose to set (~2–5 min) on a flat surface. Remove coverslip, carefully sliding sideward, and air dry slides overnight.
3.4. Cell Embedding and Lysis (30 min)
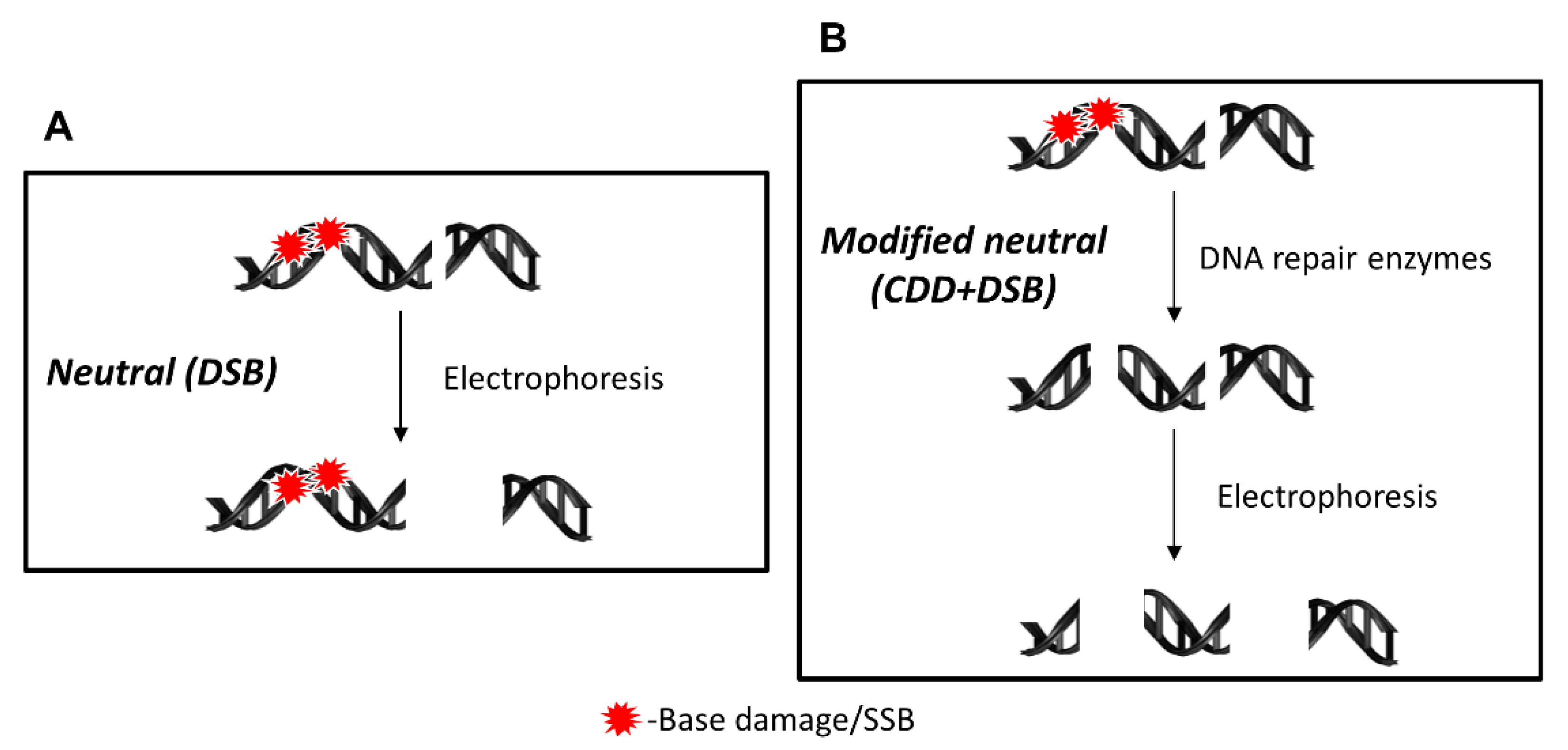
- Trypsinise cells and dilute to ~1 × 105 cells/mL. Add 250 µL of cell suspension per well of a 24-well plate on ice to prevent repair and adhesion. Induce DNA damage by chemical or physical stress within plate. Note that chemicals with a long half-life will continue to induce DNA damage in cells in suspension, so these are not recommended to be used using this method. Alternatively, cells can be grown as monolayer and exposed separately to stress according to the experiment design (compound concentrations, time exposure) and then trypsinized. The amount of DNA damaging agent should be determined empirically, but should induce a DNA damage level of <35% tail DNA to ensure this is not too extensive for the cell to repair, if analyzing DNA repair efficiency.
- Add 500 µL of low melting point agarose (previously melted and maintained at 37 °C) to cells, mix gently, and add 70 µL of this cell suspension to two areas on a normal melting point agarose-coated slide (equivalent to duplicate treatments). Add two 22 × 22 mm coverslips and place on the ice tray for ~2 min for the agarose to set. Two slides per treatment should be prepared (one for the buffer treatment only detecting DSBs, and one for the enzyme treatment detecting both DSBs and CDD).
- A negative control (no DNA damage treatment) should be prepared. Ideally, the experiment would also include a positive control with a treatment known to induce CDD (e.g., high-LET radiation).
- If analyzing DNA repair efficiency, place slides in a humidified chamber at 37 °C to allow repair according to experiment time-point (e.g., up to 6 h).
- After incubation, carefully remove coverslips by sliding sideward and place slides in coplin jars containing fresh cold lysis buffer. Lyse cells for at least 1 h at 4 °C.

- CRITICAL STEP: duration of cell lysis may vary among different cell lines and must be empirically determined.

- PAUSE STEP: slides can be kept in lysis buffer overnight at 4 °C.

3.5. Enzyme Treatment (100 min)
- Wash slides three times, for 5 min each, in PBS in coplin jars, at 4 °C (to remove lysis buffer).
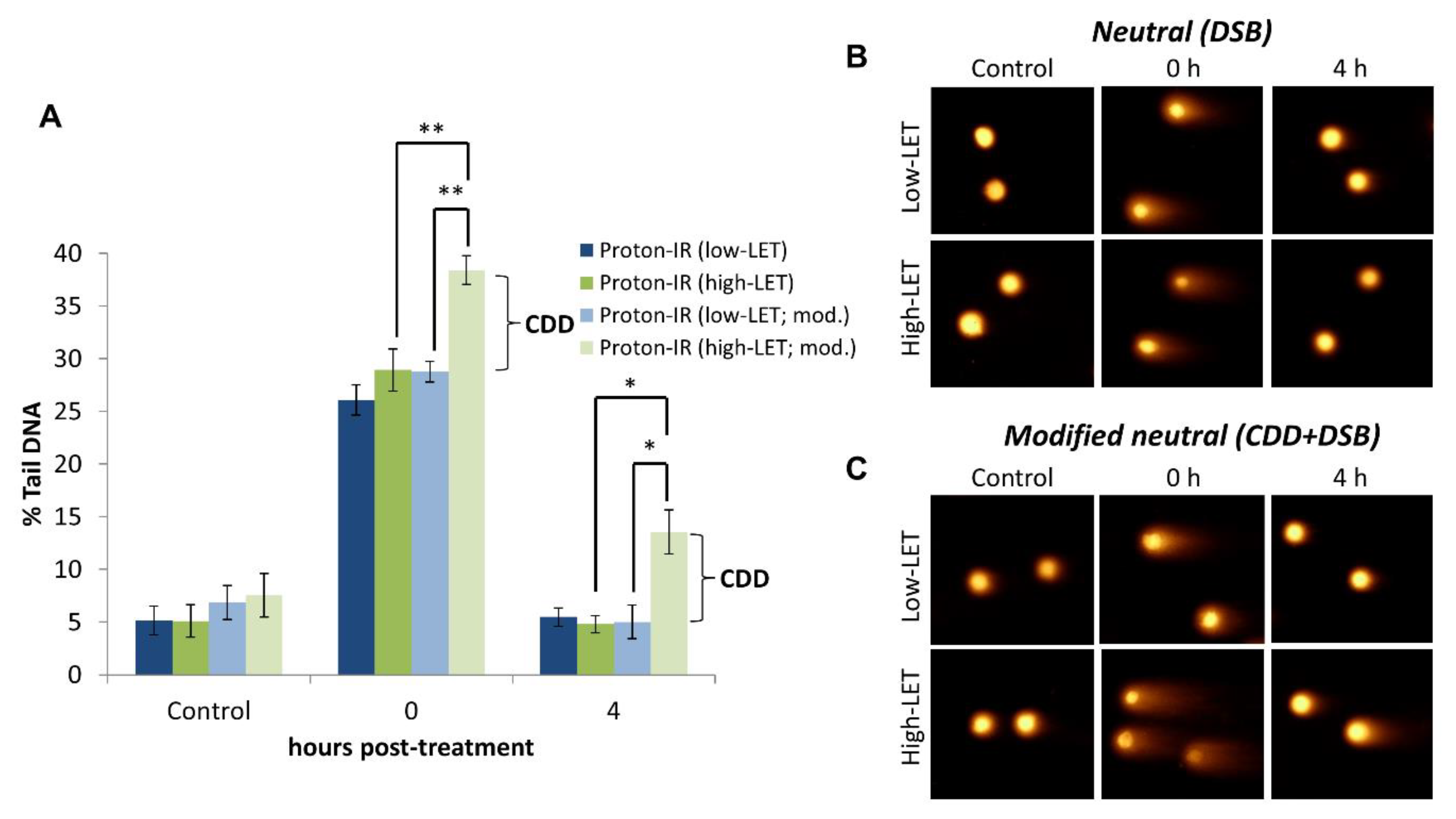
- Lay slides on a flat surface and place 50 µL of enzyme solution per agarose/cell area, or buffer alone for the untreated slides, and cover with a 22 × 22 mm coverslip. Note that the amount of enzyme to use should be predetermined beforehand, ideally by titrating each enzyme (NTH1, OGG1, and APE1) against a positive control (e.g., high-LET radiation) versus a negative control (e.g., low-LET radiation) to ensure that CDD is only revealed largely under the former conditions.
- Place slides in a humidified chamber and incubate at 37 °C for 1 h to allow enzyme processing.
3.6. Electrophoresis (105 min + Overnight Dry)
- Carefully remove coverslips by sliding sideward and wash slides three times for 5 min each in coplin jars containing 1× PBS.
- Transfer slides to an electrophoresis tank (can be placed at 4 °C if ambient temperature is relatively high), organize them into two separate columns, and cover with ~1.2 L of the fresh cold Comet Electrophoresis Buffer.
- Incubate slides for 30 min.
- Electrophorese slides at 25 V (1 V/cm, ~20 mA; note that volume of buffer may need to be removed/added to adjust to the correct current) for 25 min.
- Carefully remove slides from the electrophoresis tank to minimize movement of the slides (and potential loss of agarose/cells) and lay on a flat surface covered with paper roll. Cover agarose/gel areas with cold 1× PBS buffer (~500 µL per slide) for 5 min. Repeat twice.
- Pour off excess PBS, lay slides flat, and allow to dry overnight.
3.7. Rehydration and DNA Staining (75 min + Overnight Dry)
- Place dried slides in coplin jars containing dH2O (pH = 8.0) for 30 min to rehydrate agarose.
- Lay slides on a flat surface covered with paper roll and add enough SYBR Gold (diluted 1:20,000) to cover each slide (~500 µL) for DNA staining. Cover the slides to protect from light and incubate for 30 min.
- Remove excess stain from the slides, lay flat, and allow slides to dry overnight (while protecting from light) prior to analysis or storage in a sealed box.
3.8. Analysis
- Capture images of stained DNA from the dried slides using a fluorescent microscope equipped with a 10× objective. There is no standard procedure for measuring the intensity of light and correcting the level of DNA migration accordingly, therefore, this should be optimized empirically.
- Images can be analyzed live or offline using a validated image analysis software and by scoring 50 cells per agarose/cell square across multiple images, which are present in duplicate on each slide. In our experience, we recommend the Komet 6.0 image analysis software, although other commercial and free software is available (e.g., Comet Assay IV, OpenComet, and CometScore).
- When analyzing images, it is important to recognize the presence of apoptotic cells (also called “hedgehogs”) that have extremely high DNA damage levels (>90% of DNA in the comet tail), and that these should be quantified separately from cells containing levels of DNA damage (0–40 %) that are amenable to cellular repair.
4. Expected Results
4.1. Principles of the EMNC Assay
4.2. Detection of CDD Following Proton Irradiation
5. Conclusions
6. Troubleshooting
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cook, P.R.; A Brazell, I.; Jost, E. Characterization of nuclear structures containing superhelical DNA. J. Cell Sci. 1976, 22, 303–324. [Google Scholar] [PubMed]
- Ostling, O.; Johanson, K. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem. Biophys. Res. Commun. 1984, 123, 291–298. [Google Scholar] [CrossRef]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P.; Durand, R.E. Heterogeneity in Radiation-Induced DNA Damage and Repair in Tumor and Normal Cells Measured Using the "Comet" Assay. Radiat. Res. 1990, 122, 86. [Google Scholar] [CrossRef]
- Saha, D.T.; Davidson, B.J.; Wang, A.; Pollock, A.J.; Orden, R.A.; Goldman, R. Quantification of DNA repair capacity in whole blood of patients with head and neck cancer and healthy donors by comet assay. Mutat. Res. Toxicol. Environ. Mutagen. 2008, 650, 55–62. [Google Scholar] [CrossRef]
- Collins, A.R. The Comet Assay for DNA Damage and Repair: Principles, Applications, and Limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Nickson, C.M.; Parsons, J.L. Monitoring regulation of DNA repair activities of cultured cells in-gel using the comet assay. Front. Genet. 2014, 5, 232. [Google Scholar] [CrossRef]
- Grundy, G.J.; Parsons, J.L. Base excision repair and its implications to cancer therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef]
- Azqueta, A.; Collins, A.R. The essential comet assay: A comprehensive guide to measuring DNA damage and repair. Arch. Toxicol. 2013, 87, 949–968. [Google Scholar] [CrossRef]
- Collins, A.R.; Duthie, S.J.; Dobson, V.L. Direct enzymic detection of endogenous oxidative base damage in human lymphocyte DNA. Carcinog. 1993, 14, 1733–1735. [Google Scholar] [CrossRef] [PubMed]
- Dušinská, M.; Collins, A. Detection of Oxidised Purines and UV-induced Photoproducts in DNA of Single Cells, by Inclusion of Lesion-specific Enzymes in the Comet Assay. Altern. Lab. Anim. 1996, 24, 405–411. [Google Scholar] [CrossRef]
- Collins, A.R.; Mitchell, D.L.; Zunino, A.; de Wit, J.; Busch, D. UV-sensitive rodent mutant cell lines of complementation groups 6 and 8 differ phenotypically from their human counterparts. Environ. Mol. Mutagen 1997, 29, 152–160. [Google Scholar] [CrossRef]
- Collins, A.R.; Dusinská, M.; Horská, A. Detection of alkylation damage in human lymphocyte DNA with the comet assay. Acta Biochim. Pol. 2001, 48, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Vitti, E.T.; Parsons, J.L. The Radiobiological Effects of Proton Beam Therapy: Impact on DNA Damage and Repair. Cancers 2019, 11, 946. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef]
- Eccles, L.J.; O’Neill, P.; Lomax, M.E. Delayed repair of radiation induced clustered DNA damage: Friend or foe? Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 134–141. [Google Scholar] [CrossRef]
- De Lapuente, J.; Lourenã§o, J.; Mendo, S.A.; S, M.B.; Martins, M.G.; Costa, P.M.; Pacheco, M. The Comet Assay and its applications in the field of ecotoxicology: A mature tool that continues to expand its perspectives. Front. Genet. 2015, 6, 180. [Google Scholar] [CrossRef]
- Muruzabal, D.; Collins, A.; Azqueta, A. The enzyme-modified comet assay: Past, present and future. Food Chem. Toxicol. 2021, 147, 111865. [Google Scholar] [CrossRef]
- Holt, S.M.; Georgakilas, A.G. Detection of Complex DNA Damage in γ-Irradiated Acute Lymphoblastic Leukemia Pre-B NALM-6 Cells. Radiat. Res. 2007, 168, 527–534. [Google Scholar] [CrossRef]
- Parsons, J.L.; Zharkov, D.O.; Dianov, G.L. NEIL1 excises 3’ end proximal oxidative DNA lesions resistant to cleavage by NTH1 and OGG1. Nucleic Acids Res. 2005, 33, 4849–4856. [Google Scholar] [CrossRef]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Complex DNA Damage Induced by High Linear Energy Transfer Alpha-Particles and Protons Triggers a Specific Cellular DNA Damage Response. Int. J. Radiat. Oncol. 2018, 100, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Characterisation of Deubiquitylating Enzymes in the Cellular Response to High-LET Ionizing Radiation and Complex DNA Damage. Int. J. Radiat. Oncol. 2019, 104, 656–665. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Problem | Possible Causes | Remedies |
|---|---|---|
| Agarose does not remain attached to the glass slide. |
|
|
| Cells do not show comets |
|
|
| Cells do not show more extensive comets after enzyme treatment |
|
|
| Cells in the untreated (negative) control have large comet tails |
|
|
| Cells in the treated samples (particularly positive controls) have very small comet tails |
|
|
| Comets are unevenly distributed across the slides |
|
|
| No, or poorly visible, comets are observed |
|
|
| Quality of the comet images is poor |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabbrizi, M.R.; Hughes, J.R.; Parsons, J.L. The Enzyme-Modified Neutral Comet (EMNC) Assay for Complex DNA Damage Detection. Methods Protoc. 2021, 4, 14. https://doi.org/10.3390/mps4010014
Fabbrizi MR, Hughes JR, Parsons JL. The Enzyme-Modified Neutral Comet (EMNC) Assay for Complex DNA Damage Detection. Methods and Protocols. 2021; 4(1):14. https://doi.org/10.3390/mps4010014
Chicago/Turabian StyleFabbrizi, Maria Rita, Jonathan R. Hughes, and Jason L. Parsons. 2021. "The Enzyme-Modified Neutral Comet (EMNC) Assay for Complex DNA Damage Detection" Methods and Protocols 4, no. 1: 14. https://doi.org/10.3390/mps4010014
APA StyleFabbrizi, M. R., Hughes, J. R., & Parsons, J. L. (2021). The Enzyme-Modified Neutral Comet (EMNC) Assay for Complex DNA Damage Detection. Methods and Protocols, 4(1), 14. https://doi.org/10.3390/mps4010014