Solvent-Free Synthesis of 2,5-Bis((dimethylamino)methylene)cyclopentanone


Abstract
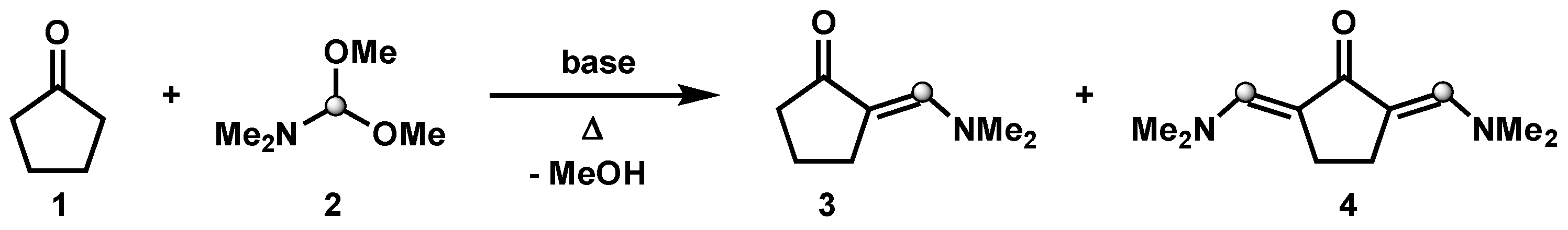
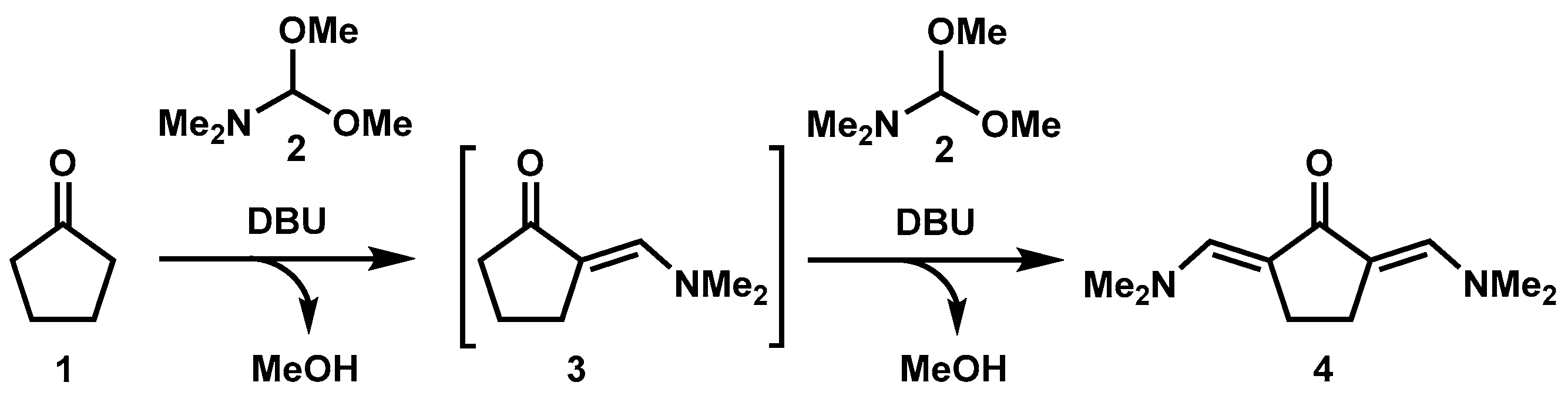
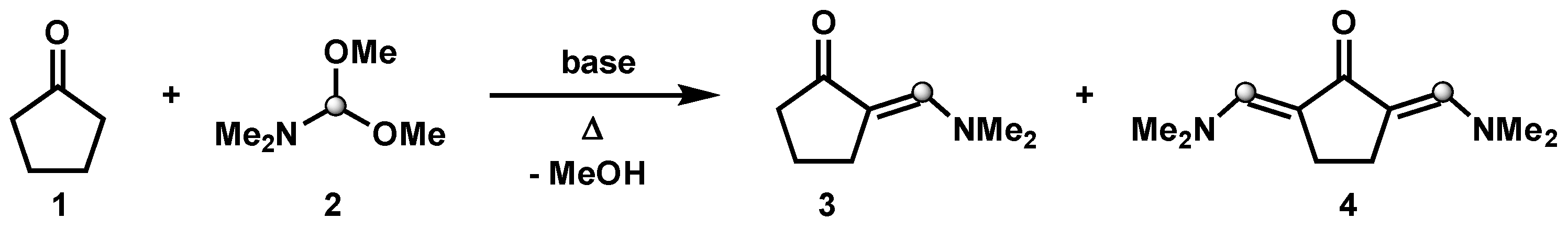
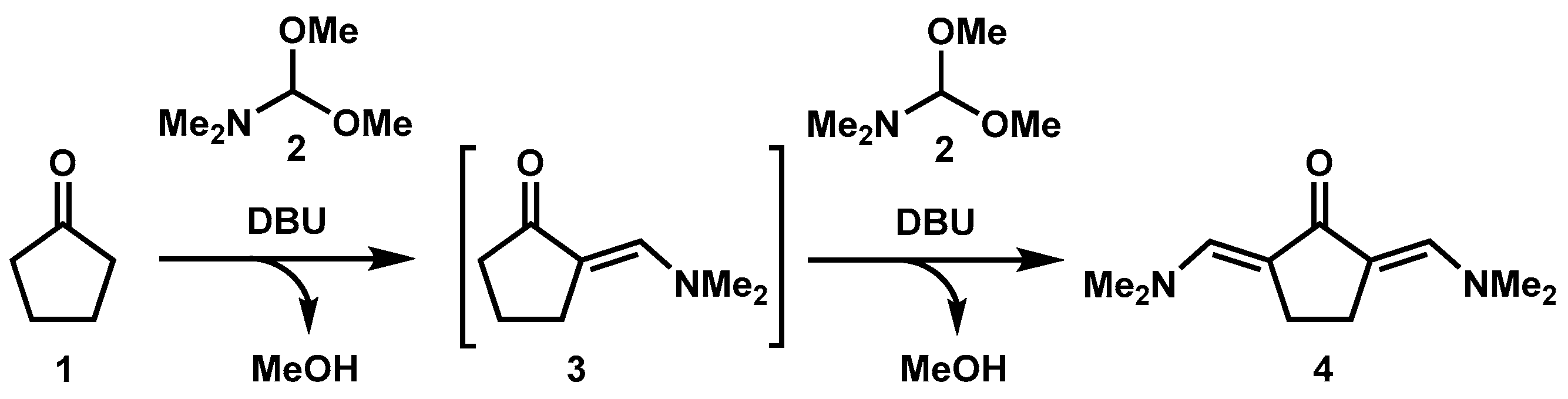
:1. Introduction
2. Experimental Design
2.1. Materials
- Cyclopentanone (Alfa Aesar)
- N,N-Dimethylformamide dimethyl acetal (Fluorochem)
- 1,8-Diazabicyclo[5.4.0]undec-7-ene, DBU (Fluorochem)
- Methyl tert-butyl ether, MTBE
2.2. Equipment
- Stirring plate (IKA)
- Water circulating system (Julabo)
- NMR (Bruker MX300 spectrometer)
3. Procedure
3.1. Synthesis of 2,5-Bis((dimethylamino)methylene)cyclopentanone (Time for Completion: 16 h)
- Add cyclopentanone (0.3 mL, 3.4 mmol), N,N-dimethylformamide dimethyl acetal (2 mL, 13.6 mmol, 4 equiv) and DBU (50 µL, 0.36 mmol, 10 mol%) to a 10 mL round-bottom flask equipped with a stir bar.
- Connect a condenser with a circulating water system at 60 °C to the flask and allow the reaction mixture to stir at 190 °C.
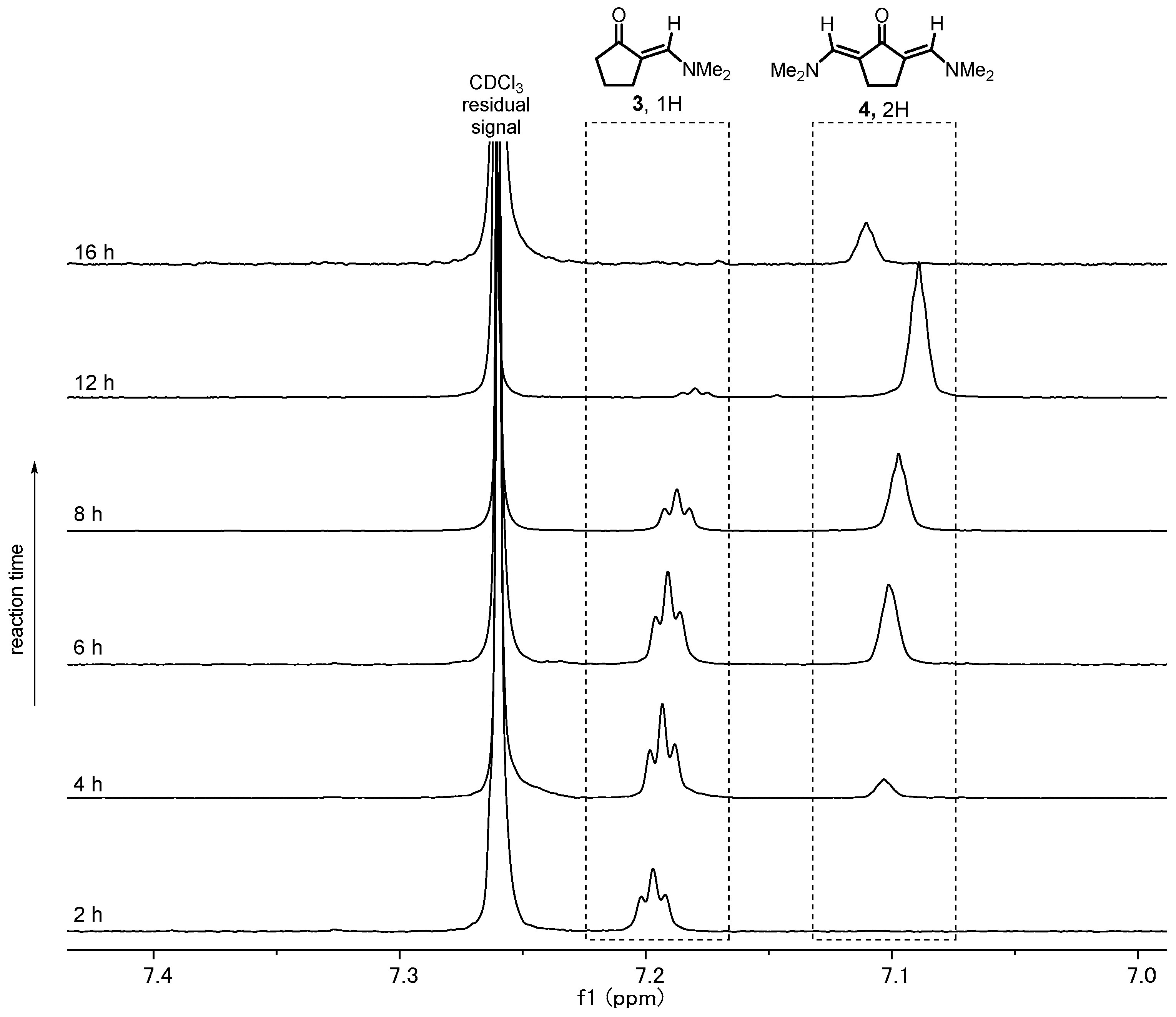
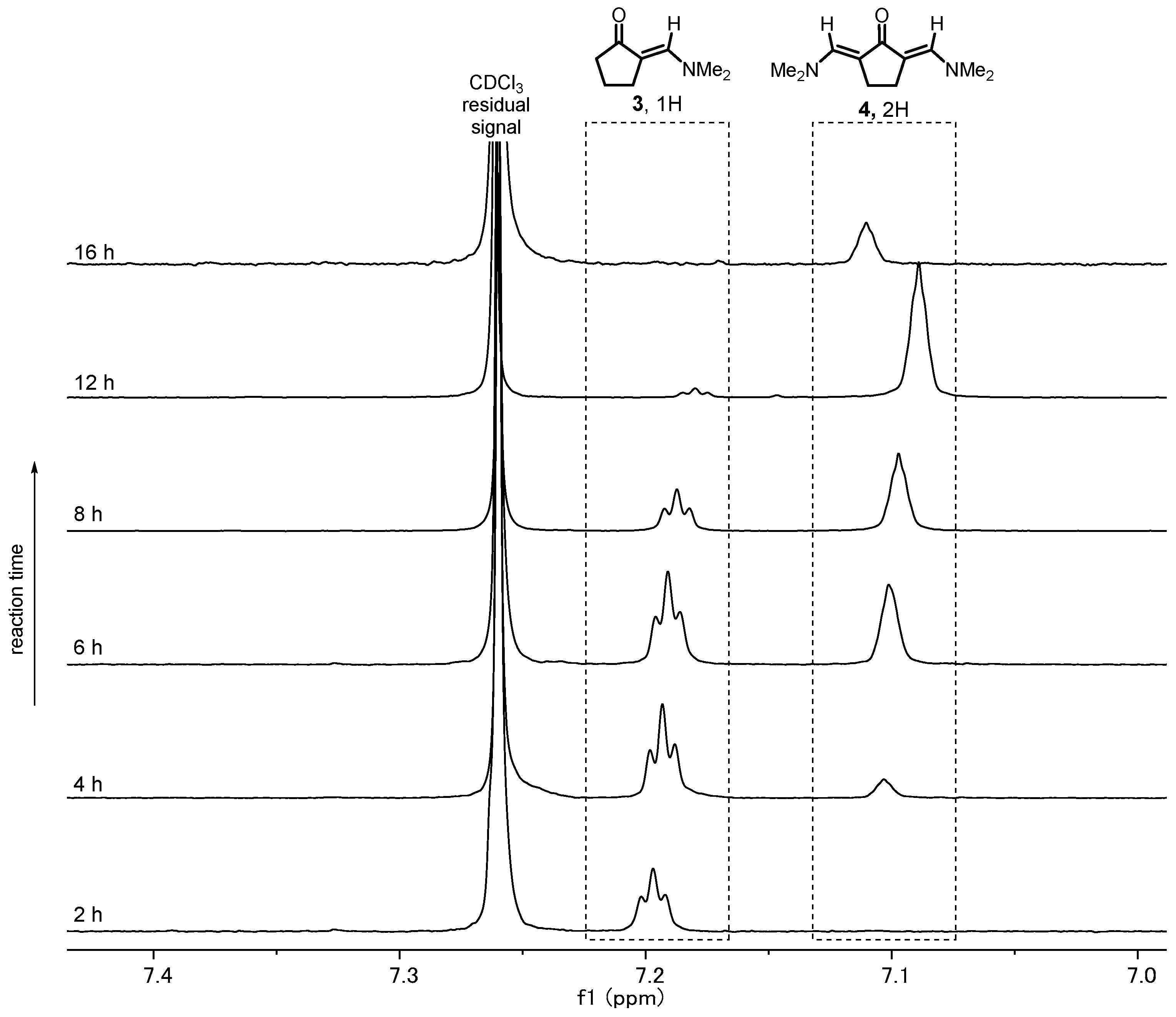
![Mps 02 00069 i001]() CRITICAL STEP The circulating water temperature of the condenser allows the methanol to evaporate from the reaction mixture while condensing the other components (see Appendix A).OPTIONAL STEP Follow the reaction by 1H NMR analysis of crude reaction mixture.
CRITICAL STEP The circulating water temperature of the condenser allows the methanol to evaporate from the reaction mixture while condensing the other components (see Appendix A).OPTIONAL STEP Follow the reaction by 1H NMR analysis of crude reaction mixture.![Mps 02 00069 i002]() PAUSE STEP The heating can be stopped overnight for safety reasons and restarted the next day.
PAUSE STEP The heating can be stopped overnight for safety reasons and restarted the next day. - After completion (usually 16 h), allow the reaction mixture to cool down to room temperature and observe the crystallization of product (see Appendix B).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.2. Isolation of 2,5-Bis((dimethylamino)methylene)cyclopentanone (Time for Completion: 30 min)
- Wash the crystals of product with MTBE (3 × 15 mL).
- Dry the product under vacuum.OPTIONAL STEP Recrystallize from hot acetone.
4. Results
4.1. Reaction Optimization
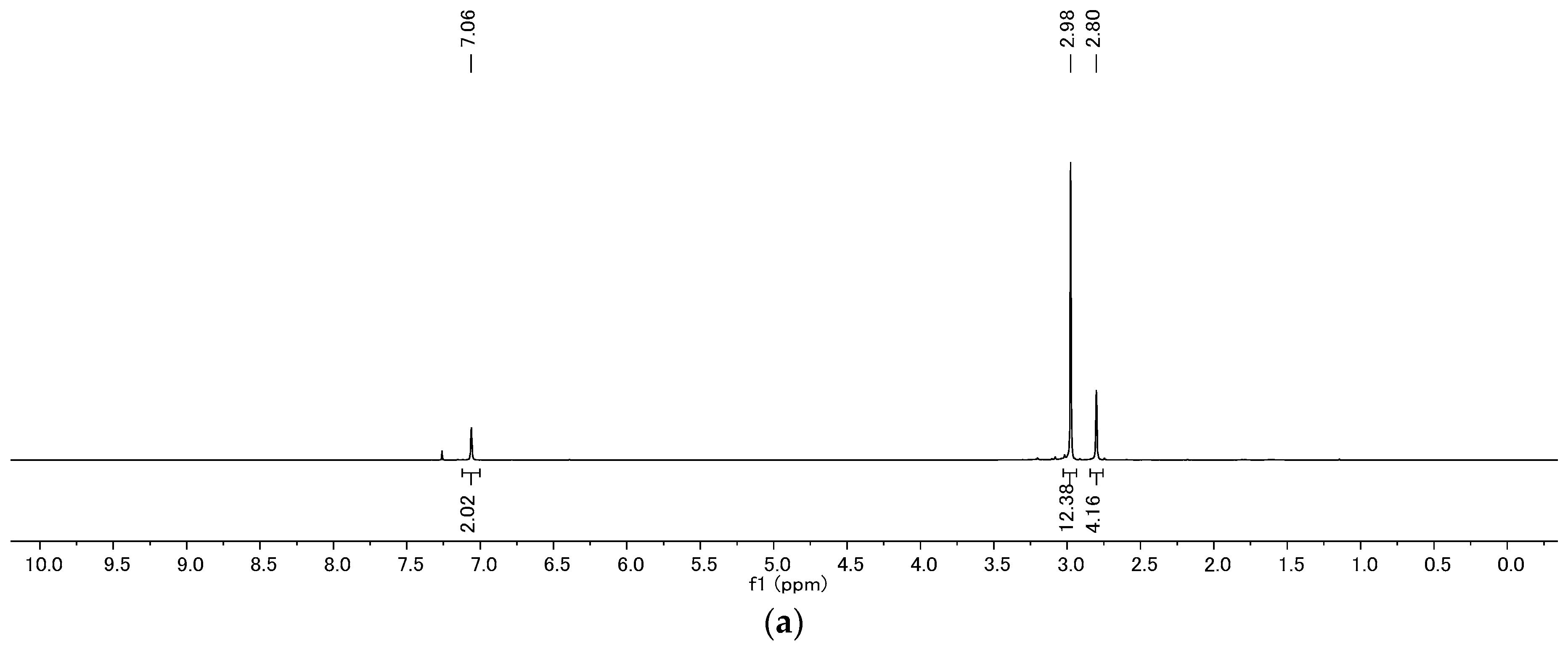
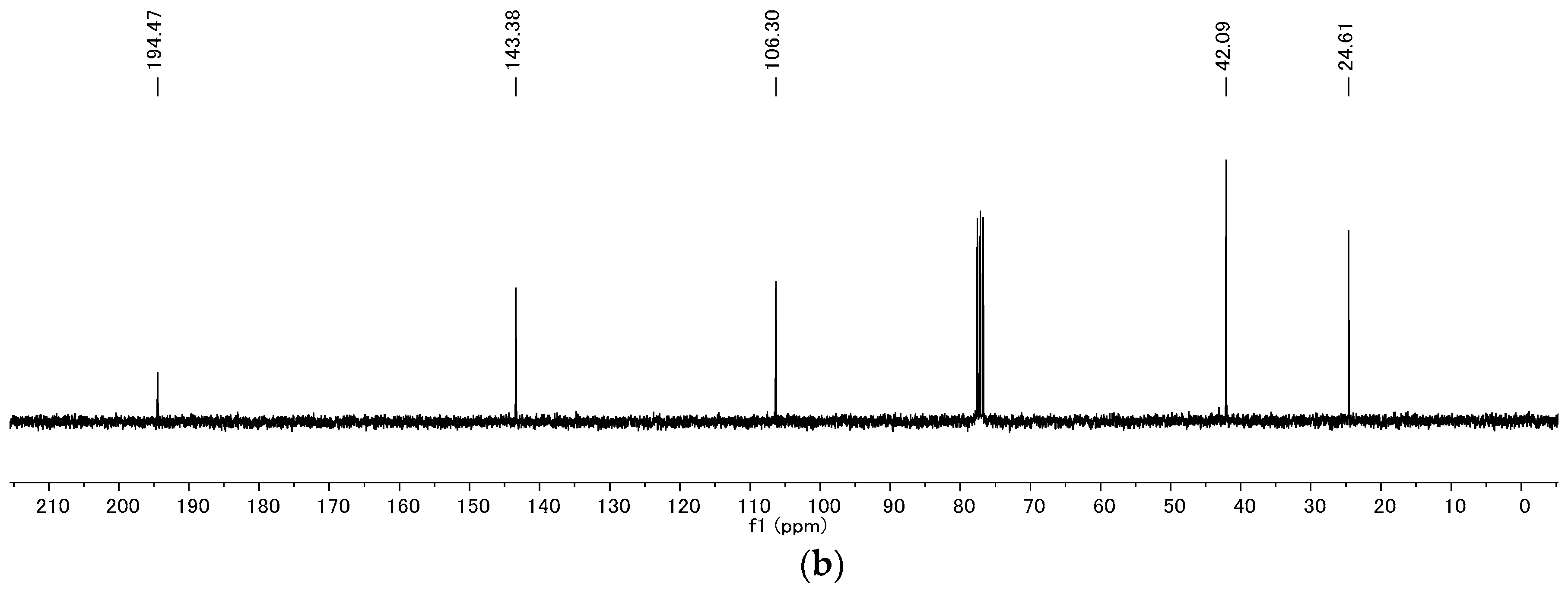
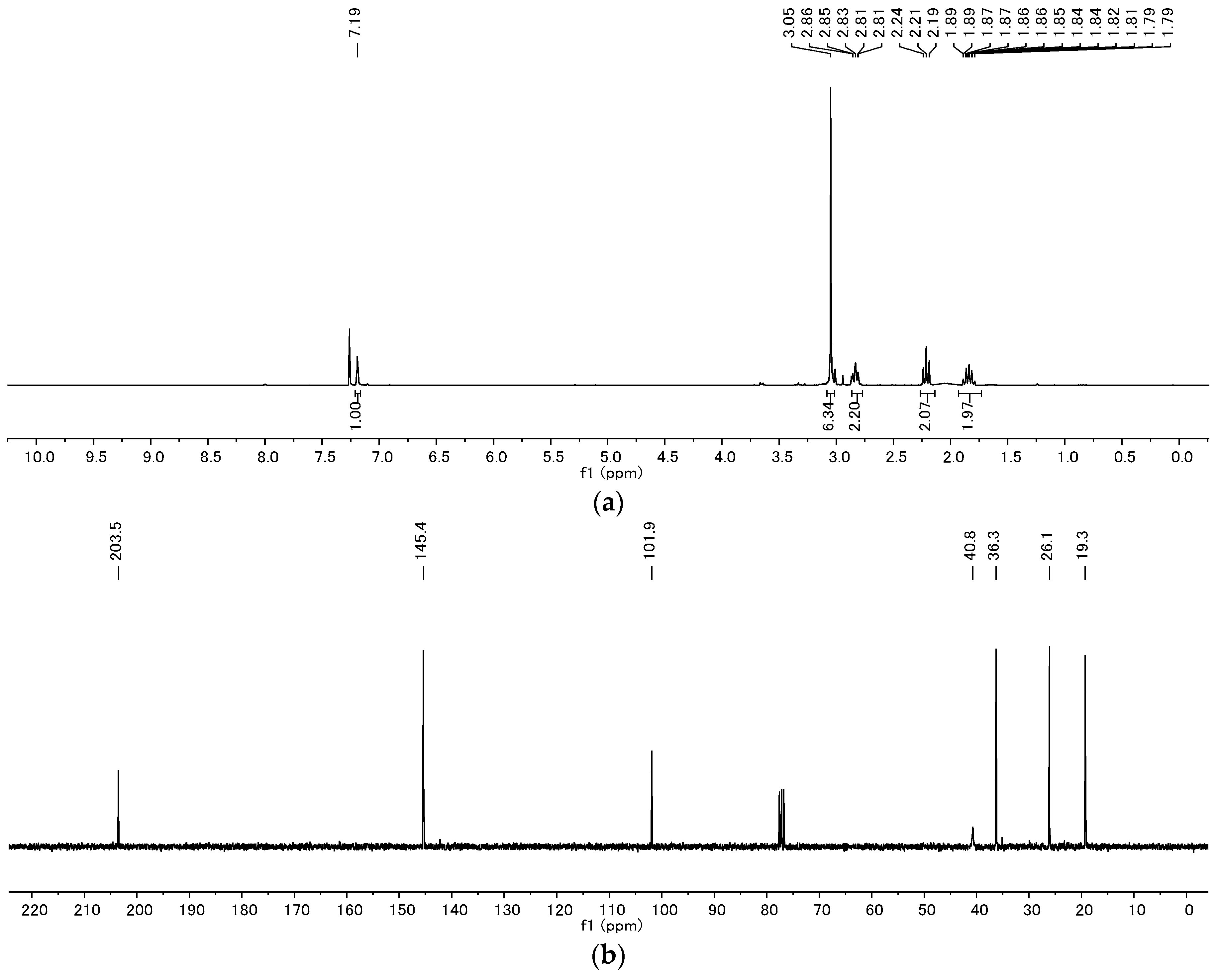
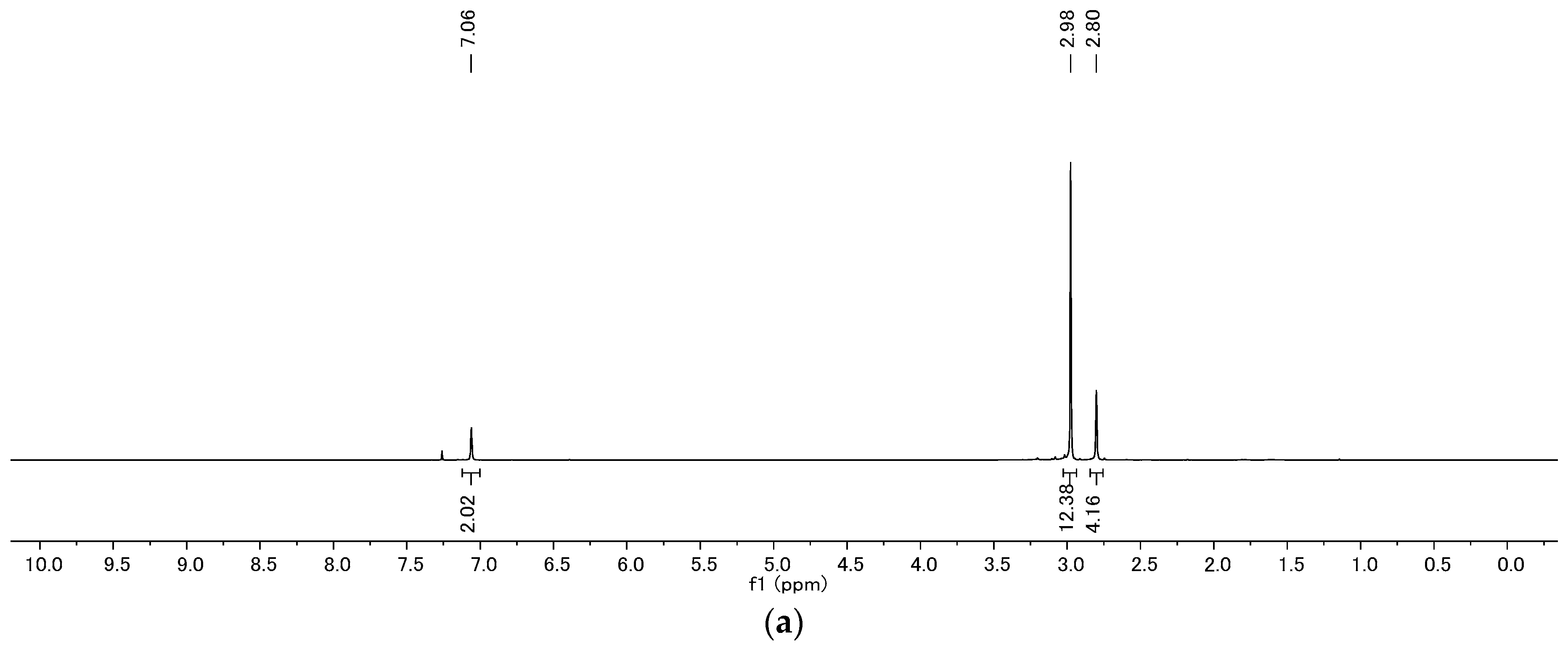
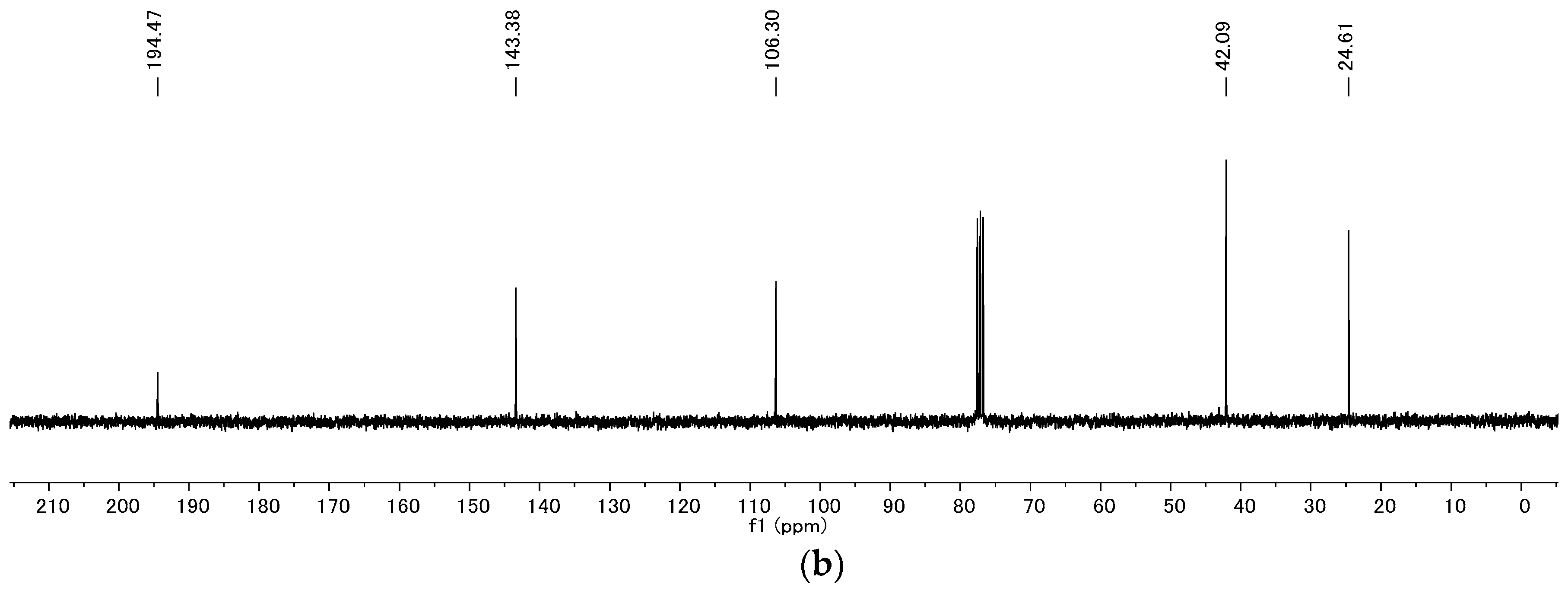
4.2. Product Characterization
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

Appendix B

References
- Law, K.Y. Organic photoconductive materials: Recent trends and developments. Chem. Rev. 1993, 93, 449–486. [Google Scholar] [CrossRef]
- Langhals, H. Color Chemistry: Syntheses, Properties, and Applications of Organic Dyes and Pigments, 3rd ed.; Zollinger, H., Ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Strekowski, L. Heterocyclic Polymethine Dyes; Springer-Verlag: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Mishra, A.; Behera, R.K.; Behera, P.K.; Mishra, B.K.; Behera, G.B. Cyanines during the 1990s: A Review. Chem. Rev. 2000, 100, 1973–2012. [Google Scholar] [CrossRef] [PubMed]
- Fabian, J.; Nakazumi, H.; Matsuoka, M. Near-infrared absorbing dyes. Chem. Rev. 1992, 92, 1197–1226. [Google Scholar] [CrossRef]
- Pascal, S.; Denis-Quanquin, S.; Appaix, F.; Duperray, A.; Grichine, A.; Le Guennic, B.; Jacquemin, D.; Cuny, J.; Chi, S.; Perry, J.W.; et al. Keto-polymethines: A versatile class of dyes with outstanding spectroscopic properties for in cellulo and in vivo two-photon microscopy imaging. Chem. Sci. 2017, 8, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Daehne, S.; Resch-Genger, U.; Wolfbeis, O.S. (Eds.) Near-Infrared Dyes for High Technology Applications; Springer-Science+Business Media B.V.: Dordrecht, The Netherlands, 1997. [Google Scholar]
- Puyol, M.; Encinas, C.; Rivera, L.; Miltsov, S.; Alonso, J. Synthesis of new ketocyanine dyes for the development of optical sensors. Sens. Actuators B Chem. 2006, 115, 287–296. [Google Scholar] [CrossRef]
- Miltsov, S.; Encinas, C.; Alonso, J. Novel synthesis of ketocyanine dyes. Tetrahedron Lett. 2001, 42, 6129–6131. [Google Scholar] [CrossRef]
- Slominskii, Y.L.; Radchenko, I.D.; Popov, S.V.; Tolmachev, A.L. Polymethine dyes with hydrocarbon bridges. Enamine ketones in the chemistry of cyanine dyes. Zh. Org. Khim. 1983, 19, 2134–2142. [Google Scholar]
- Callant, P.; Louwet, J. Intermediate Compounds for the Preparation of Meso-Substituted Cyanine, Merocyanine and Oxonole Dyes. Patent WO/2009/080689, 2 July 2009. [Google Scholar]
- Yesudas, K.; Jemmis, E.D.; Bhanuprakash, K. Ketocyanine dyes: Impact of conjugation length on optical absorption and third-order polarizabilities. Phys. Chem. Chem. Phys. 2015, 17, 12988–12999. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Akiba, M.; Tani, T. Two-Photon Absorbing Polymerizable Composition and Polymerization Process Thereof. Patent US/2004/0204513/A1, 14 October 2004. [Google Scholar]
- Kim, J.H.; Kim, S.H.; Moon, G.S.; Shin, M.Y.; Won, D.H.; Jeon, H.S. Photosensitive Resin Compositions with Good Contrast and Brightness for Color Filters. Patent KR 10-2014-0075414, 19 June 2014. [Google Scholar]
- Abu-Shanab, F.A.; Sherif, S.M.; Mousa, S.A.S. Dimethylformamide dimethyl acetal as a building block in heterocyclic synthesis. J. Heterocycl. Chem. 2009, 46, 801–827. [Google Scholar] [CrossRef]
- Zhang, W.; Henry, Y. A new synthetic route to 3,4-bridged 1,6,6a lambda (4)-trithiapentalenes. Synlett 2001, 7, 1129–1130. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
S. Martins, I.; A. S. Coelho, J. Solvent-Free Synthesis of 2,5-Bis((dimethylamino)methylene)cyclopentanone. Methods Protoc. 2019, 2, 69. https://doi.org/10.3390/mps2030069
S. Martins I, A. S. Coelho J. Solvent-Free Synthesis of 2,5-Bis((dimethylamino)methylene)cyclopentanone. Methods and Protocols. 2019; 2(3):69. https://doi.org/10.3390/mps2030069
Chicago/Turabian StyleS. Martins, Inês, and Jaime A. S. Coelho. 2019. "Solvent-Free Synthesis of 2,5-Bis((dimethylamino)methylene)cyclopentanone" Methods and Protocols 2, no. 3: 69. https://doi.org/10.3390/mps2030069
APA StyleS. Martins, I., & A. S. Coelho, J. (2019). Solvent-Free Synthesis of 2,5-Bis((dimethylamino)methylene)cyclopentanone. Methods and Protocols, 2(3), 69. https://doi.org/10.3390/mps2030069