
Future Perspectives of Newborn Screening for Inborn Errors of Immunity



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Newborn Screening for (X-Linked) Agammaglobulinemia
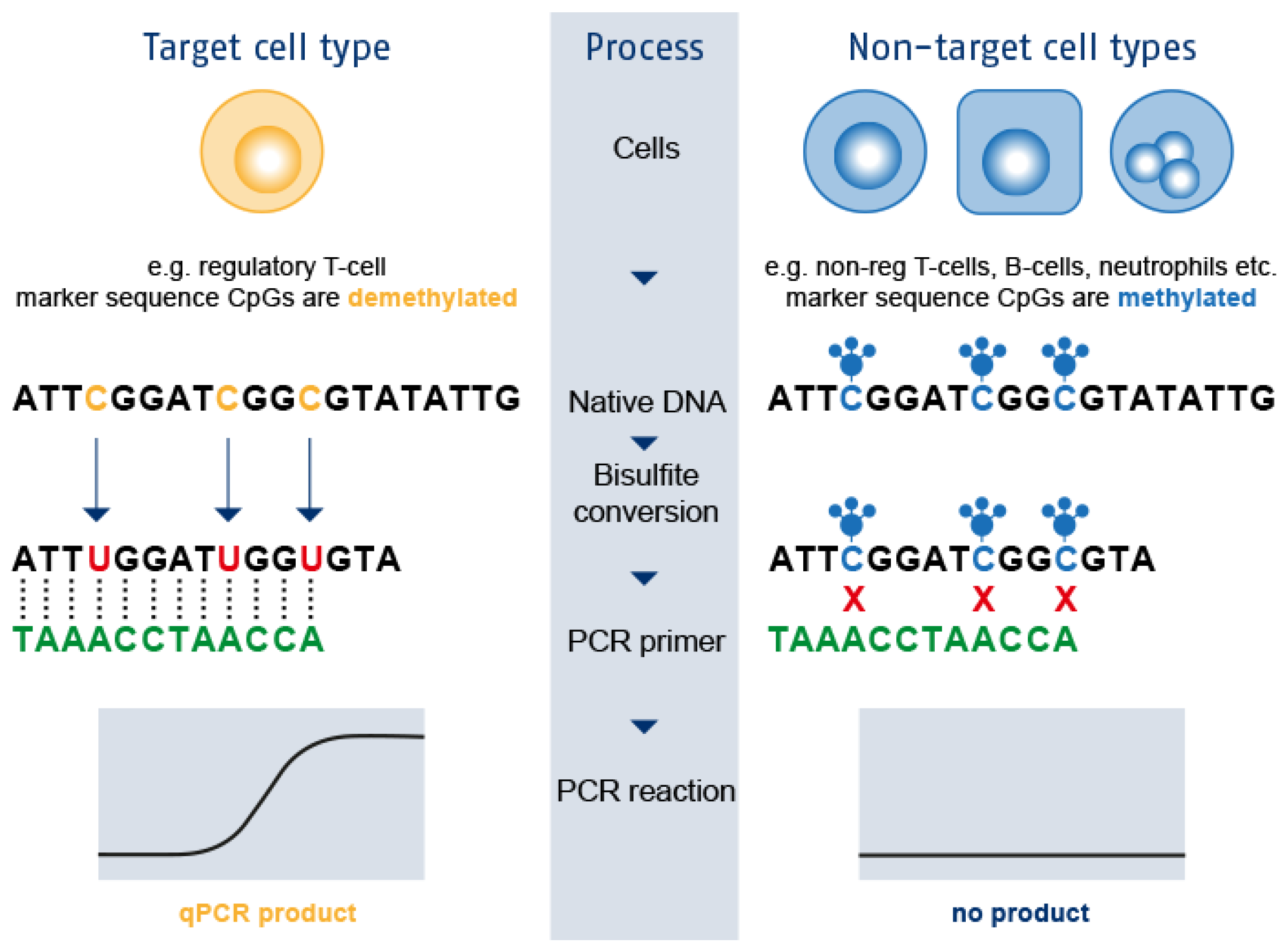
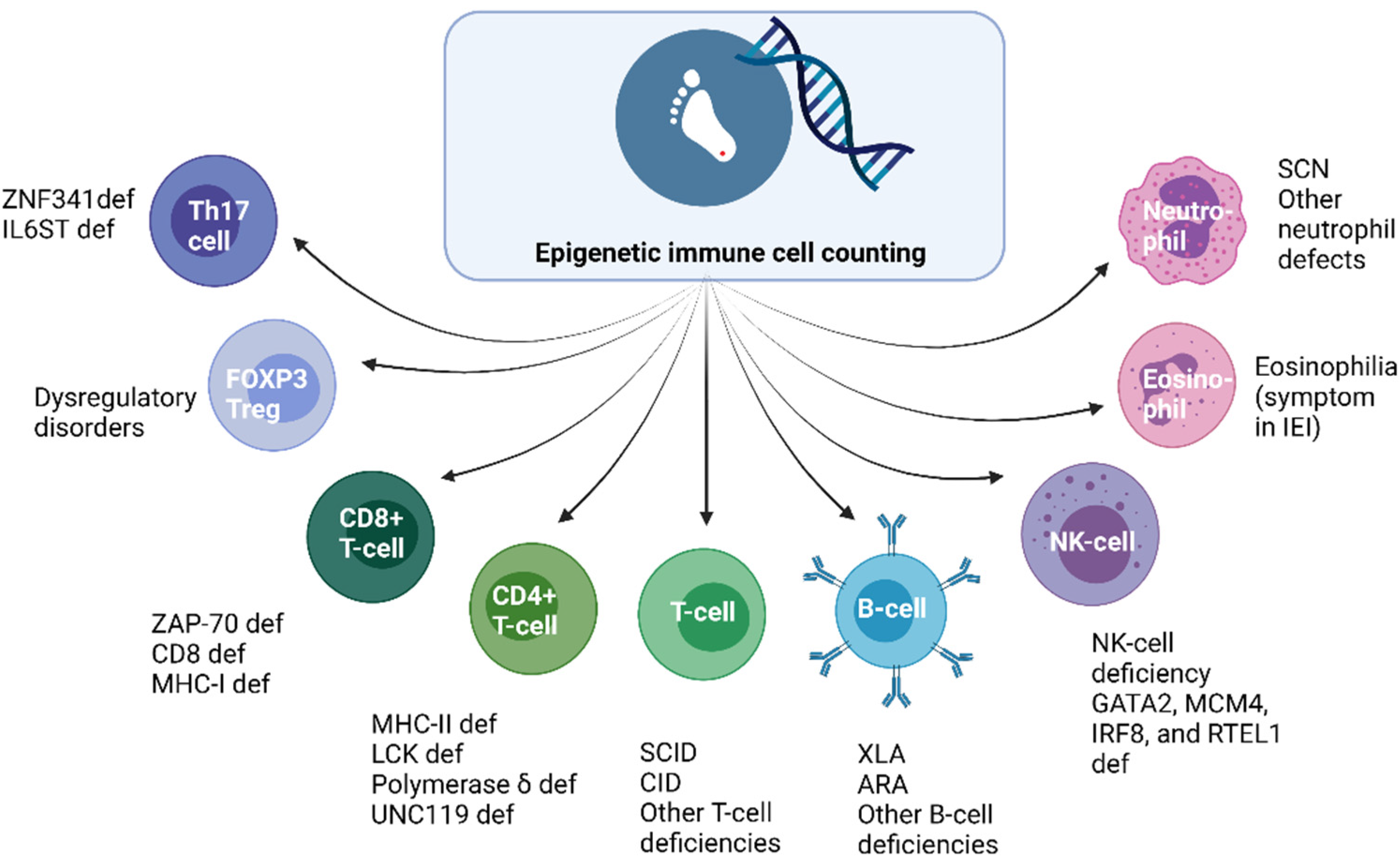
3. Epigenetic Immune Cell Counting: A New Player in the Field
4. Newborn Screening for Interferonopathies
5. Protein-Based Newborn Screening for IEI
6. Genomic-Based Newborn Screening for IEI
7. Future Newborn Screening for SCID
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Fischer, A. Severe combined immunodeficiencies (SCID). Clin. Exp. Immunol. 2000, 122, 143–149. [Google Scholar] [CrossRef]
- Fischer, A.; Notarangelo, L.D.; Neven, B.; Cavazzana, M.; Puck, J.M. Severe combined immunodeficiencies and related disorders. Nat. Rev. Dis. Prim. 2015, 1, 15061. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [Green Version]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Al-Herz, W.; Ailal, F.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J. Clin. Immunol. 2020, 40, 66–81. [Google Scholar] [CrossRef] [Green Version]
- Pai, S.Y.; Logan, B.R.; Griffith, L.M.; Buckley, R.H.; Parrott, R.E.; Dvorak, C.C.; Kapoor, N.; Hanson, I.C.; Filipovich, A.H.; Jyonouchi, S.; et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N. Engl. J. Med. 2014, 371, 434–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimall, J.; Logan, B.R.; Cowan, M.J.; Notarangelo, L.D.; Griffith, L.M.; Puck, J.M.; Kohn, D.B.; Pulsipher, M.A.; Parikh, S.; Martinez, C.; et al. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: A PIDTC natural history study. Blood 2017, 130, 2718–2727. [Google Scholar] [CrossRef] [Green Version]
- Van Zelm, M.C.; van der Burg, M.; Langerak, A.W.; van Dongen, J.J. PID comes full circle: Applications of V(D)J recombination excision circles in research, diagnostics and newborn screening of primary immunodeficiency disorders. Front. Immunol. 2011, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazenberg, M.D.; Verschuren, M.C.; Hamann, D.; Miedema, F.; Dongen, J.J. T cell receptor excision circles as markers for recent thymic emigrants: Basic aspects, technical approach, and guidelines for interpretation. J. Mol. Med. 2001, 79, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Puck, J.M. Development of population-based newborn screening for severe combined immunodeficiency. J. Allergy Clin. Immunol. 2005, 115, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Notarangelo, L.D.; Bacchetta, R.; Casanova, J.L.; Su, H.C. Human inborn errors of immunity: An expanding universe. Sci. Immunol. 2020, 5, eabb1662. [Google Scholar] [CrossRef]
- Fischer, A.; Provot, J.; Jais, J.P.; Alcais, A.; Mahlaoui, N. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J. Allergy Clin. Immunol. 2017, 140, 1388–1393.e1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee. J. Clin. Immunol. 2021, 41, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Jungner, Y.G. Principles and Practice of Screening for Disease. Available online: https://apps.who.int/iris/handle/10665/37650 (accessed on 7 June 2021).
- King, J.R.; Notarangelo, L.D.; Hammarström, L. An appraisal of the Wilson & Jungner criteria in the context of genomic-based newborn screening for inborn errors of immunity. J. Allergy Clin. Immunol. 2021, 147, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Lankester, A.C.; Albert, M.H.; Booth, C.; Gennery, A.R.; Güngör, T.; Hönig, M.; Morris, E.C.; Moshous, D.; Neven, B.; Schulz, A.; et al. EBMT/ESID inborn errors working party guidelines for hematopoietic stem cell transplantation for inborn errors of immunity. Bone Marrow Transpl. 2021, 56, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, Z.A.; Abramova, I.; Aldave, J.C.; Al-Herz, W.; Bezrodnik, L.; Boukari, R.; Bousfiha, A.A.; Cancrini, C.; Condino-Neto, A.; Dbaibo, G.; et al. X-linked agammaglobulinemia (XLA):Phenotype, diagnosis, and therapeutic challenges around the world. World Allergy Organ. J. 2019, 12, 100018. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.C.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S.; et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. [Google Scholar] [CrossRef]
- Hagemann, T.L.; Chen, Y.; Rosen, F.S.; Kwan, S.P. Genomic organization of the Btk gene and exon scanning for mutations in patients with X-linked agammaglobulinemia. Hum. Mol. Genet. 1994, 3, 1743–1749. [Google Scholar] [CrossRef]
- Vetrie, D.; Vorechovsky, I.; Sideras, P.; Holland, J.; Davies, A.; Flinter, F.; Hammarstrom, L.; Kinnon, C.; Levinsky, R.; Bobrow, M.; et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. [Google Scholar] [CrossRef]
- Abolhassani, H.; Hirbod-Mobarakeh, A.; Shahinpour, S.; Panahi, M.; Mohammadinejad, P.; Mirminachi, B.; Shakari, M.S.; Samavat, B.; Aghamohammadi, A. Mortality and morbidity in patients with X-linked agammaglobulinaemia. Allergol. Immunopathol. 2015, 43, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F.; Wang, W.F.; Zhang, Y.D.; Zhao, W.; Wu, J.; Chen, T.X. Clinical characteristics and genetic profiles of 174 patients with X-linked agammaglobulinemia: Report from Shanghai, China (2000–2015). Medicine 2016, 95, e4544. [Google Scholar] [CrossRef]
- Bazregari, S.; Azizi, G.; Tavakol, M.; Asgardoon, M.H.; Kiaee, F.; Tavakolinia, N.; Valizadeh, A.; Abolhassani, H.; Aghamohammadi, A. Evaluation of infectious and non-infectious complications in patients with primary immunodeficiency. Cent. Eur. J. Immunol. 2017, 42, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Bearden, D.; Collett, M.; Quan, P.L.; Costa-Carvalho, B.T.; Sullivan, K.E. Enteroviruses in X-Linked Agammaglobulinemia: Update on Epidemiology and Therapy. J. Allergy Clin. Immunol. Pract. 2016, 4, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, J.A.; Marino, M.C.; Lederman, H.M.; Jones, S.M.; Sullivan, K.; Burks, A.W.; Conley, M.E.; Cunningham-Rundles, C.; Ochs, H.D. X-linked agammaglobulinemia: Report on a United States registry of 201 patients. Medicine 2006, 85, 193–202. [Google Scholar] [CrossRef]
- Conley, M.E.; Howard, V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J. Pediatr. 2002, 141, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Plebani, A.; Soresina, A.; Rondelli, R.; Amato, G.M.; Azzari, C.; Cardinale, F.; Cazzola, G.; Consolini, R.; De Mattia, D.; Dell’Erba, G.; et al. Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: An Italian multicenter study. Clin. Immunol. 2002, 104, 221–230. [Google Scholar] [CrossRef]
- Quinti, I.; Soresina, A.; Guerra, A.; Rondelli, R.; Spadaro, G.; Agostini, C.; Milito, C.; Trombetta, A.C.; Visentini, M.; Martini, H.; et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: Results from a multicenter prospective cohort study. J. Clin. Immunol. 2011, 31, 315–322. [Google Scholar] [CrossRef]
- Chun, J.K.; Lee, T.J.; Song, J.W.; Linton, J.A.; Kim, D.S. Analysis of clinical presentations of Bruton disease: A review of 20 years of accumulated data from pediatric patients at Severance Hospital. Yonsei Med. J. 2008, 49, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Serana, F.; Chiarini, M.; Zanotti, C.; Sottini, A.; Bertoli, D.; Bosio, A.; Caimi, L.; Imberti, L. Use of V(D)J recombination excision circles to identify T- and B-cell defects and to monitor the treatment in primary and acquired immunodeficiencies. J. Transl. Med. 2013, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Borte, S.; von Döbeln, U.; Fasth, A.; Wang, N.; Janzi, M.; Winiarski, J.; Sack, U.; Pan-Hammarström, Q.; Borte, M.; Hammarström, L. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood 2012, 119, 2552–2555. [Google Scholar] [CrossRef]
- Barbaro, M.; Ohlsson, A.; Borte, S.; Jonsson, S.; Zetterström, R.H.; King, J.; Winiarski, J.; von Döbeln, U.; Hammarström, L. Newborn Screening for Severe Primary Immunodeficiency Diseases in Sweden-a 2-Year Pilot TREC and KREC Screening Study. J. Clin. Immunol. 2017, 37, 51–60. [Google Scholar] [CrossRef] [Green Version]
- de Felipe, B.; Olbrich, P.; Lucenas, J.M.; Delgado-Pecellin, C.; Pavon-Delgado, A.; Marquez, J.; Salamanca, C.; Soler-Palacin, P.; Gonzalez-Granado, L.I.; Antolin, L.F.; et al. Prospective neonatal screening for severe T- and B-lymphocyte deficiencies in Seville. Pediatr. Allergy Immunol. 2016, 27, 70–77. [Google Scholar] [CrossRef]
- Kanegae, M.P.P.; Barreiros, L.A.; Sousa, J.L.; Brito, M.A.S.; Oliveira, E.B.J.; Soares, L.P.; Mazzucchelli, J.T.L.; Fernandes, D.Q.; Hadachi, S.M.; Holanda, S.M.; et al. Newborn screening for severe combined immunodeficiencies using trecs and krecs: Second pilot study in brazil. Rev. Paul. Pediatr. 2017, 35, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trück, J.; Prader, S.; Natalucci, G.; Hagmann, C.; Brotschi, B.; Kelly, J.; Bassler, D.; Steindl, K.; Rauch, A.; Baumgartner, M.; et al. Swiss newborn screening for severe T and B cell deficiency with a combined TREC/KREC assay-management recommendations. Swiss. Med. Wkly 2020, 150, w20254. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Imai, K.; Kanegane, H.; Sato, H.; Yamada, M.; Kondoh, K.; Okada, S.; Kobayashi, M.; Agematsu, K.; Takada, H.; et al. Quantification of κ-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J. Allergy Clin. Immunol. 2011, 128, 223–225.e222. [Google Scholar] [CrossRef]
- Health Council of the Netherlands. Neonatal Screening: New Recommendations; Publication no. 2015/08; Health Council of the Netherlands: Hague, The Netherlands, 2015. [Google Scholar]
- Baron, U.; Werner, J.; Schildknecht, K.; Schulze, J.J.; Mulu, A.; Liebert, U.G.; Sack, U.; Speckmann, C.; Gossen, M.; Wong, R.J.; et al. Epigenetic immune cell counting in human blood samples for immunodiagnostics. Sci. Transl. Med. 2018, 10, eaan3508. [Google Scholar] [CrossRef] [Green Version]
- Kalina, T.; Bakardjieva, M.; Blom, M.; Perez-Andres, M.; Barendregt, B.; Kanderová, V.; Bonroy, C.; Philippé, J.; Blanco, E.; Pico-Knijnenburg, I.; et al. EuroFlow Standardized Approach to Diagnostic Immunopheneotyping of Severe PID in Newborns and Young Children. Front. Immunol. 2020, 11, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, S.; Etzioni, A. MHC class I and II deficiencies. J. Allergy Clin. Immunol. 2014, 134, 269–275. [Google Scholar] [CrossRef]
- Lum, S.H.; Neven, B.; Slatter, M.A.; Gennery, A.R. Hematopoietic Cell Transplantation for MHC Class II Deficiency. Front. Pediatrics 2019, 7, 516. [Google Scholar] [CrossRef] [Green Version]
- Cepika, A.-M.; Sato, Y.; Liu, J.M.-H.; Uyeda, M.J.; Bacchetta, R.; Roncarolo, M.G. Tregopathies: Monogenic diseases resulting in regulatory T-cell deficiency. J. Allergy Clin. Immunol. 2018, 142, 1679–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzaghi, F.; Passerini, L. IPEX Syndrome: Improved Knowledge of Immune Pathogenesis Empowers Diagnosis. Front. Pediatrics 2021, 9, 612760. [Google Scholar] [CrossRef]
- d’Hennezel, E.; Ben-Shoshan, M.; Ochs, H.D.; Torgerson, T.R.; Russell, L.J.; Lejtenyi, C.; Noya, F.J.; Jabado, N.; Mazer, B.; Piccirillo, C.A. FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. N. Engl. J. Med. 2009, 361, 1710–1713. [Google Scholar] [CrossRef]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Prim. 2017, 3, 17032. [Google Scholar] [CrossRef] [PubMed]
- Fioredda, F.; Iacobelli, S.; van Biezen, A.; Gaspar, B.; Ancliff, P.; Donadieu, J.; Aljurf, M.; Peters, C.; Calvillo, M.; Matthes-Martin, S.; et al. Stem cell transplantation in severe congenital neutropenia: An analysis from the European Society for Blood and Marrow Transplantation. Blood 2015, 126, 1885–1892. [Google Scholar] [CrossRef] [Green Version]
- Spoor, J.; Farajifard, H.; Rezaei, N. Congenital neutropenia and primary immunodeficiency diseases. Crit. Rev. Oncol. /Hematol. 2019, 133, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, A. Neutropenia in the newborn. Curr. Opin. Hematol. 2014, 21, 43–49. [Google Scholar] [CrossRef] [Green Version]
- McNulty, S.N.; Evenson, M.J.; Riley, M.; Yoest, J.M.; Corliss, M.M.; Heusel, J.W.; Duncavage, E.J.; Pfeifer, J.D. A Next-Generation Sequencing Test for Severe Congenital Neutropenia: Utility in a Broader Clinicopathologic Spectrum of Disease. J. Mol. Diagn. 2021, 23, 200–211. [Google Scholar] [CrossRef]
- Livingston, J.H.; Crow, Y.J. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutières Syndrome and Beyond. Neuropediatrics 2016, 47, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Volpi, S.; Picco, P.; Caorsi, R.; Candotti, F.; Gattorno, M. Type I interferonopathies in pediatric rheumatology. Pediatr. Rheumatol. Online J. 2016, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; Melki, I.; Frémond, M.L.; Briggs, T.A.; Rodero, M.P.; Kitabayashi, N.; Oojageer, A.; Bader-Meunier, B.; Belot, A.; Bodemer, C.; et al. Assessment of Type I Interferon Signaling in Pediatric Inflammatory Disease. J. Clin. Immunol. 2017, 37, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Armangue, T.; Orsini, J.J.; Takanohashi, A.; Gavazzi, F.; Conant, A.; Ulrick, N.; Morrissey, M.A.; Nahhas, N.; Helman, G.; Gordish-Dressman, H.; et al. Neonatal detection of Aicardi Goutières Syndrome by increased C26:0 lysophosphatidylcholine and interferon signature on newborn screening blood spots. Mol. Genet. Metab. 2017, 122, 134–139. [Google Scholar] [CrossRef]
- Crow, Y.J.; Shetty, J.; Livingston, J.H. Treatments in Aicardi-Goutières syndrome. Dev. Med. Child Neurol. 2020, 62, 42–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janzi, M.; Sjöberg, R.; Wan, J.; Fischler, B.; von Döbeln, U.; Isaac, L.; Nilsson, P.; Hammarström, L. Screening for C3 deficiency in newborns using microarrays. PLoS ONE 2009, 4, e5321. [Google Scholar] [CrossRef] [Green Version]
- Hamsten, C.; Skattum, L.; Truedsson, L.; von Döbeln, U.; Uhlén, M.; Schwenk, J.M.; Hammarström, L.; Nilsson, P.; Neiman, M. Heat differentiated complement factor profiling. J. Proteom. 2015, 126, 155–162. [Google Scholar] [CrossRef]
- Dezfouli, M.; Bergström, S.; Skattum, L.; Abolhassani, H.; Neiman, M.; Torabi-Rahvar, M.; Franco Jarava, C.; Martin-Nalda, A.; Ferrer Balaguer, J.M.; Slade, C.A.; et al. Newborn Screening for Presymptomatic Diagnosis of Complement and Phagocyte Deficiencies. Front. Immunol. 2020, 11, 455. [Google Scholar] [CrossRef] [Green Version]
- Kerfoot, S.A.; Jung, S.; Golob, K.; Torgerson, T.R.; Hahn, S.H. Tryptic peptide screening for primary immunodeficiency disease by LC/MS-MS. Proteom. Clin. Appl. 2012, 6, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Massaad, M.J.; Ramesh, N.; Geha, R.S. Wiskott-Aldrich syndrome: A comprehensive review. Ann. N. Y. Acad. Sci. 2013, 1285, 26–43. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.J.; Chang, I.J.; Jung, S.; Dayuha, R.; Whiteaker, J.R.; Segundo, G.R.S.; Torgerson, T.R.; Ochs, H.D.; Paulovich, A.G.; Hahn, S.H. Rapid Multiplexed Proteomic Screening for Primary Immunodeficiency Disorders From Dried Blood Spots. Front. Immunol. 2018, 9, 2756. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.J.; Yi, F.; Dayuha, R.; Whiteaker, J.R.; Ochs, H.D.; Freeman, A.; Su, H.C.; Paulovich, A.G.; Segundo, G.R.S.; Torgerson, T.; et al. Multiplexed Proteomic Analysis for Diagnosis and Screening of Five Primary Immunodeficiency Disorders From Dried Blood Spots. Front. Immunol. 2020, 11, 464. [Google Scholar] [CrossRef] [Green Version]
- Barben, J.; Castellani, C.; Dankert-Roelse, J.; Gartner, S.; Kashirskaya, N.; Linnane, B.; Mayell, S.; Munck, A.; Sands, D.; Sommerburg, O.; et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J. Cyst. Fibros. 2017, 16, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Bergougnoux, A.; Lopez, M.; Girodon, E. The Role of Extended CFTR Gene Sequencing in Newborn Screening for Cystic Fibrosis. Int. J. Neonatal. Screen 2020, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Borte, S.; Meeths, M.; Liebscher, I.; Krist, K.; Nordenskjöld, M.; Hammarström, L.; von Döbeln, U.; Henter, J.I.; Bryceson, Y.T. Combined newborn screening for familial hemophagocytic lymphohistiocytosis and severe T- and B-cell immunodeficiencies. J. Allergy Clin. Immunol. 2014, 134, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Filipovich, A.H. The expanding spectrum of hemophagocytic lymphohistiocytosis. Curr. Opin. Allergy Clin. Immunol. 2011, 11, 512–516. [Google Scholar] [CrossRef]
- Henter, J.I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef]
- Stray-Pedersen, A.; Sorte, H.S.; Samarakoon, P.; Gambin, T.; Chinn, I.K.; Coban Akdemir, Z.H.; Erichsen, H.C.; Forbes, L.R.; Gu, S.; Yuan, B.; et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J. Allergy Clin. Immunol. 2017, 139, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Pavey, A.R.; Bodian, D.L.; Vilboux, T.; Khromykh, A.; Hauser, N.S.; Huddleston, K.; Klein, E.; Black, A.; Kane, M.S.; Iyer, R.K.; et al. Utilization of genomic sequencing for population screening of immunodeficiencies in the newborn. Genet. Med. 2017, 19, 1367–1375. [Google Scholar] [CrossRef]
- King, J.; Ludvigsson, J.F.; Hammarström, L. Newborn Screening for Primary Immunodeficiency Diseases: The Past, the Present and the Future. Int. J. Neonatal Screen. 2017, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- King, J.R.; Hammarström, L. Newborn Screening for Primary Immunodeficiency Diseases: History, Current and Future Practice. J. Clin. Immunol. 2018, 38, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Strand, J.; Gul, K.A.; Erichsen, H.C.; Lundman, E.; Berge, M.C.; Trømborg, A.K.; Sørgjerd, L.K.; Ytre-Arne, M.; Hogner, S.; Halsne, R.; et al. Second-Tier Next Generation Sequencing Integrated in Nationwide Newborn Screening Provides Rapid Molecular Diagnostics of Severe Combined Immunodeficiency. Front. Immunol. 2020, 11, 1417. [Google Scholar] [CrossRef] [PubMed]
- Al-Mousa, H.; Al-Dakheel, G.; Jabr, A.; Elbadaoui, F.; Abouelhoda, M.; Baig, M.; Monies, D.; Meyer, B.; Hawwari, A.; Dasouki, M. High Incidence of Severe Combined Immunodeficiency Disease in Saudi Arabia Detected Through Combined T Cell Receptor Excision Circle and Next Generation Sequencing of Newborn Dried Blood Spots. Front. Immunol. 2018, 9, 782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, J.S.; Agrawal, P.B.; Bailey, D.B., Jr.; Beggs, A.H.; Brenner, S.E.; Brower, A.M.; Cakici, J.A.; Ceyhan-Birsoy, O.; Chan, K.; Chen, F.; et al. Newborn Sequencing in Genomic Medicine and Public Health. Pediatrics 2017, 139, e20162252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.M.; Cornel, M.C.; Goldenberg, A.J.; Lister, K.J.; Sénécal, K.; Vears, D.F. Genomic newborn screening: Public health policy considerations and recommendations. BMC Med. Genom. 2017, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Joseph, G.; Chen, F.; Harris-Wai, J.; Puck, J.M.; Young, C.; Koenig, B.A. Parental Views on Expanded Newborn Screening Using Whole-Genome Sequencing. Pediatrics 2016, 137 (Suppl. S1), 36–46. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, L.E.; Fergus, K.A.; Ojeda, N.; Au, S.M. Parental attitudes toward ethical and social issues surrounding the expansion of newborn screening using new technologies. Public Health Genom. 2011, 14, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Etchegary, H.; Dicks, E.; Hodgkinson, K.; Pullman, D.; Green, J.; Parfey, P. Public attitudes about genetic testing in the newborn period. J. Obstet. Gynecol. Neonatal Nurs. 2012, 41, 191–200. [Google Scholar] [CrossRef]
- Almond, B. Genetic profiling of newborns: Ethical and social issues. Nat. Rev. Genet. 2006, 7, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Al-Mousa, H.; Al-Saud, B. Primary Immunodeficiency Diseases in Highly Consanguineous Populations from Middle East and North Africa: Epidemiology, Diagnosis, and Care. Front. Immunol. 2017, 8, 678. [Google Scholar] [CrossRef] [Green Version]
- Padilla, C.D.; Krotoski, D.; Therrell, B.L. Newborn Screening Progress in Developing Countries—Overcoming Internal Barriers. Semin. Perinatol. 2010, 34, 145–155. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, Z.A.; Radwan, N. Newborn Screening for Primary Immunodeficiencies: The Gaps, Challenges, and Outlook for Developing Countries. Front. Immunol. 2020, 10, 2987. [Google Scholar] [CrossRef] [Green Version]
- Blom, M.; Knijnenburg, I.-P.; Imholz, S.; Vissers, L.; Schulze, J.; Werner, J.; Bredius, R.; Burg, M.v.d. Second tier testing to reduce the number of non-actionable secondary findings and false positive referrals in newborn screening for severe combined immunodeficiency. J. Clin. Immunol. 2021. [Google Scholar] [CrossRef]
- Dorsey, M.J.; Wright, N.A.M.; Chaimowitz, N.S.; Dávila Saldaña, B.J.; Miller, H.; Keller, M.D.; Thakar, M.S.; Shah, A.J.; Abu-Arja, R.; Andolina, J.; et al. Infections in Infants with SCID: Isolation, Infection Screening, and Prophylaxis in PIDTC Centers. J. Clin. Immunol. 2021, 41, 38–50. [Google Scholar] [CrossRef]
- EURORDIS. Key Principles for Newborn Screening. Available online: https://www.eurordis.org/newbornscreening (accessed on 7 June 2021).
- Puck, J.M. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia. Immunol. Rev. 2019, 287, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, M.J.; Dvorak, C.C.; Cowan, M.J.; Puck, J.M. Treatment of infants identified as having severe combined immunodeficiency by means of newborn screening. J. Allergy Clin. Immunol. 2017, 139, 733–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Mateo, C.; Timonen, A.; Vaahtera, K.; Jaakkola, M.; Hougaard, D.M.; Bybjerg-Grauholm, J.; Baekvad-Hansen, M.; Adamsen, D.; Filippov, G.; Dallaire, S.; et al. Development of a Multiplex Real-Time PCR Assay for the Newborn Screening of SCID, SMA, and XLA. Int. J. Neonatal Screen 2019, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Speckmann, C.; Neumann, C.; Borte, S.; la Marca, G.; Sass, J.O.; Wiech, E.; Fisch, P.; Schwarz, K.; Buchholz, B.; Schlesier, M.; et al. Delayed-onset adenosine deaminase deficiency: Strategies for an early diagnosis. J. Allergy Clin. Immunol. 2012, 130, 991–994. [Google Scholar] [CrossRef] [PubMed]
- La Marca, G.; Canessa, C.; Giocaliere, E.; Romano, F.; Duse, M.; Malvagia, S.; Lippi, F.; Funghini, S.; Bianchi, L.; Della Bona, M.L.; et al. Tandem mass spectrometry, but not T-cell receptor excision circle analysis, identifies newborns with late-onset adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2013, 131, 1604–1610. [Google Scholar] [CrossRef] [Green Version]
- La Marca, G.; Canessa, C.; Giocaliere, E.; Romano, F.; Malvagia, S.; Funghini, S.; Moriondo, M.; Valleriani, C.; Lippi, F.; Ombrone, D.; et al. Diagnosis of immunodeficiency caused by a purine nucleoside phosphorylase defect by using tandem mass spectrometry on dried blood spots. J. Allergy Clin. Immunol. 2014, 134, 155–159. [Google Scholar] [CrossRef] [PubMed]
- La Marca, G.; Giocaliere, E.; Malvagia, S.; Funghini, S.; Ombrone, D.; Della Bona, M.L.; Canessa, C.; Lippi, F.; Romano, F.; Guerrini, R.; et al. The inclusion of ADA-SCID in expanded newborn screening by tandem mass spectrometry. J. Pharm. Biomed. Anal. 2014, 88, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Malvagia, S.; Funghini, S.; Della Bona, M.; Ombrone, D.; Mura, M.; Damiano, R.; Ricci, S.; Cortimiglia, M.; Azzari, C.; la Marca, G. The successful inclusion of ADA SCID in Tuscany expanded newborn screening program. Clin. Chem. Lab. Med. 2021, 59, e401–e404. [Google Scholar] [CrossRef]
- Thaventhiran, J.E.D.; Lango Allen, H.; Burren, O.S.; Rae, W.; Greene, D.; Staples, E.; Zhang, Z.; Farmery, J.H.R.; Simeoni, I.; Rivers, E.; et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature 2020, 583, 90–95. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blom, M.; Bredius, R.G.M.; van der Burg, M. Future Perspectives of Newborn Screening for Inborn Errors of Immunity. Int. J. Neonatal Screen. 2021, 7, 74. https://doi.org/10.3390/ijns7040074
Blom M, Bredius RGM, van der Burg M. Future Perspectives of Newborn Screening for Inborn Errors of Immunity. International Journal of Neonatal Screening. 2021; 7(4):74. https://doi.org/10.3390/ijns7040074
Chicago/Turabian StyleBlom, Maartje, Robbert G. M. Bredius, and Mirjam van der Burg. 2021. "Future Perspectives of Newborn Screening for Inborn Errors of Immunity" International Journal of Neonatal Screening 7, no. 4: 74. https://doi.org/10.3390/ijns7040074
APA StyleBlom, M., Bredius, R. G. M., & van der Burg, M. (2021). Future Perspectives of Newborn Screening for Inborn Errors of Immunity. International Journal of Neonatal Screening, 7(4), 74. https://doi.org/10.3390/ijns7040074