
Validation of a Custom Next-Generation Sequencing Assay for Cystic Fibrosis Newborn Screening

 ,
,
Abstract
1. Introduction
2. Materials and Methods
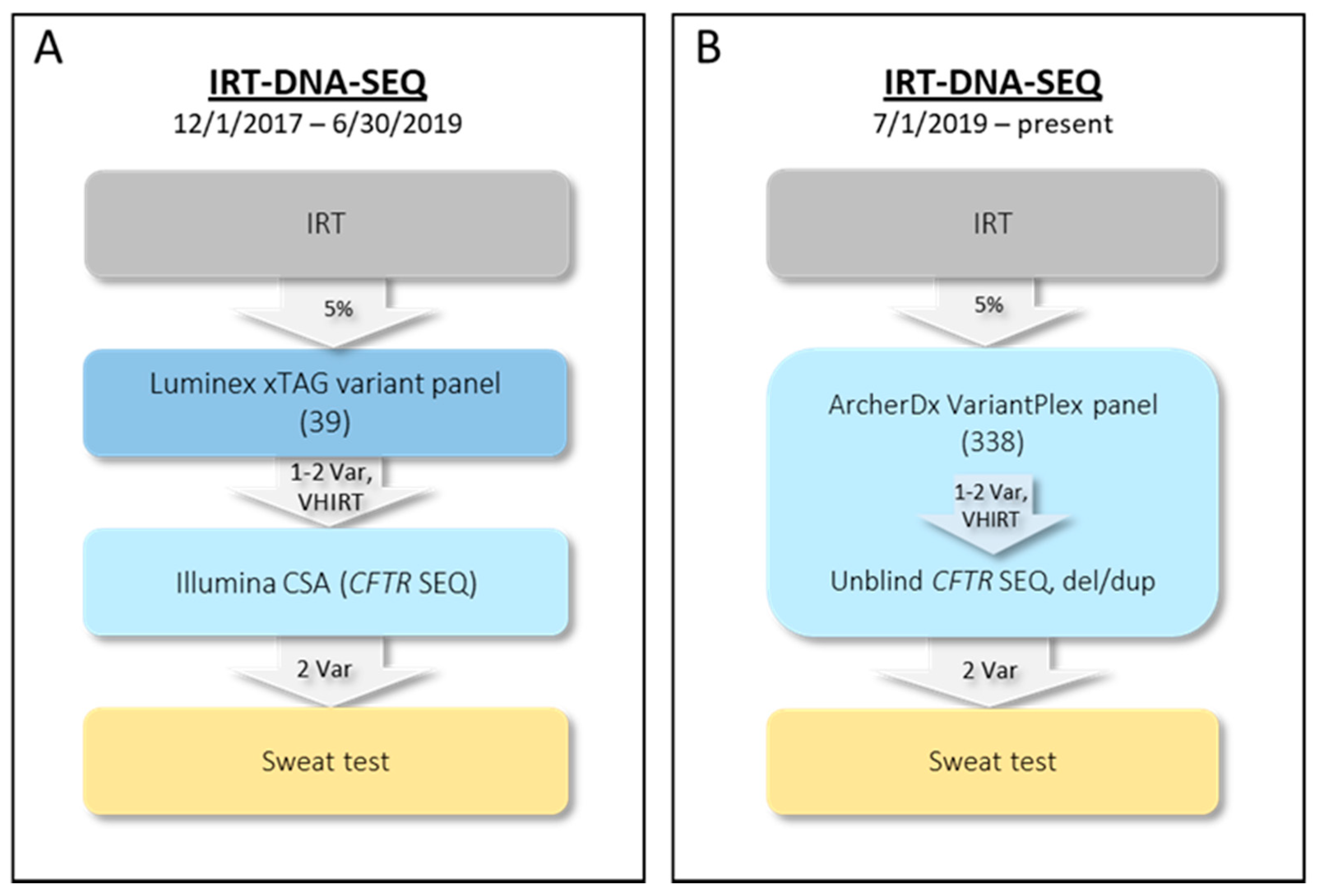
2.1. Initial NYS CF IRT-DNA-SEQ Algorithm (1 December 2017–30 June 2019)
2.2. Current NYS CF IRT-DNA-SEQ Algorithm (1 July 2019–Present)
2.3. Assay Validation—Samples
2.4. Assay Validation—DNA Extraction
2.5. Assay Validation—NGS
2.6. Assay Validation—Bioinformatics Analyses
2.7. Variant Validation
2.8. Variant Interpretation
3. Results
3.1. Archer NGS Assay
3.2. NGS Validation
3.3. Del/dup qPCR Assay Validation
3.4. Prospective Validation Study
3.5. First Year of Clinical Testing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Castellani, C.; Massie, J.; Sontag, M.; Southern, K.W. Newborn screening for cystic fibrosis. Lancet Respir. Med. 2016, 4, 653–661. [Google Scholar] [CrossRef]
- Kay, D.M.; Maloney, B.; Hamel, R.; Pearce, M.; DeMartino, L.; McMahon, R.; McGrath, E.C.; Krein, L.M.; Vogel, B.H.; Saavedra-Matiz, C.A.; et al. Screening for cystic fibrosis in New York State: Considerations for algorithm improvements. Eur. J. Pediatr. 2016, 175, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Kay, D.M.; Langfelder-Schwind, E.; DeCelie-Germana, J.; Sharp, J.K.; Maloney, B.; Tavakoli, N.P.; Saavedra-Matiz, C.A.; Krein, L.M.; Caggana, M.; Kier, C.; et al. Utility of a very high IRT/No mutation referral category in cystic fibrosis newborn screening. Pediatr. Pulmonol. 2015, 50, 771–780. [Google Scholar] [CrossRef]
- Prach, L.; Koepke, R.; Kharrazi, M.; Keiles, S.; Salinas, D.B.; Reyes, M.C.; Pian, M.; Opsimos, H.; Otsuka, K.N.; Hardy, K.A.; et al. Novel CFTR variants identified during the first 3 years of cystic fibrosis newborn screening in California. J. Mol. Diagn. 2013, 15, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Kharrazi, M.; Yang, J.; Bishop, T.; Lessing, S.; Young, S.; Graham, S.; Pearl, M.; Chow, H.; Ho, T.; Currier, R.; et al. Newborn Screening for Cystic Fibrosis in California. Pediatrics 2015, 136, 1062–1072. [Google Scholar] [CrossRef]
- Currier, R.J.; Sciortino, S.; Liu, R.; Bishop, T.; Alikhani Koupaei, R.; Feuchtbaum, L. Genomic sequencing in cystic fibrosis newborn screening: What works best, two-tier predefined CFTR mutation panels or second-tier CFTR panel followed by third-tier sequencing? Genet. Med. 2017, 19, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Shen, P.D.; Odegaard, J.I.; Fung, E.L.; Chiang, T.; Peng, G.; Davis, R.W.; Wang, W.; Kharrazi, M.; Schrijver, I.; et al. Next-Generation Molecular Testing of Newborn Dried Blood Spots for Cystic Fibrosis. J. Mol. Diagn. 2016, 18, 267–282. [Google Scholar] [CrossRef]
- Baker, M.W.; Atkins, A.E.; Cordovado, S.K.; Hendrix, M.; Earley, M.C.; Farrell, P.M. Improving newborn screening for cystic fibrosis using next-generation sequencing technology: A technical feasibility study. Genet. Med. 2016, 18, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Furnier, S.M.; Durkin, M.S.; Baker, M.W. Translating Molecular Technologies into Routine Newborn Screening Practice. Int. J. Neonatal Screen. 2020, 6, 80. [Google Scholar] [CrossRef]
- Saavedra-Matiz, C.A.; Isabelle, J.T.; Biski, C.K.; Duva, S.J.; Sweeney, M.L.; Parker, A.L.; Young, A.J.; Diantonio, L.L.; Krein, L.M.; Nichols, M.J.; et al. Cost-effective and scalable DNA extraction method from dried blood spots. Clin. Chem. 2013, 59, 1045–1051. [Google Scholar] [CrossRef]
- Sicko, R.J. NBS_NGS. Available online: https://github.com/rjsicko/NBS_NGS/tree/master/CFTR (accessed on 3 March 2021).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Clinical and Functional Translation of CFTR (CFTR2). Available online: www.cftr2.org/index.php (accessed on 3 March 2021).
- Granell, R.; Solera, J.; Carrasco, S.; Molano, J. Identification of a nonframeshift 84-bp deletion in exon 13 of the cystic fibrosis gene. Am. J. Hum. Genet. 1992, 50, 1022–1026. [Google Scholar]
- Watson, M.S.; Cutting, G.R.; Desnick, R.J.; Driscoll, D.A.; Klinger, K.; Mennuti, M.; Palomaki, G.E.; Popovich, B.W.; Pratt, V.M.; Rohlfs, E.M.; et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet. Med. 2004, 6, 387–391. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. 2012, pp. 1–9. Available online: http://clavius.bc.edu/~erik/haplotype_paper/haplotype_based_variant_detection.pdf (accessed on 3 March 2021).
- Keenan, K.; Dupuis, A.; Griffin, K.; Castellani, C.; Tullis, E.D.; Gonska, T. Phenotypic Spectrum of Patients with Cystic Fibrosis and Cystic Fibrosis-Related Disease Carrying p.Arg117His. J. Cyst. Fibros. 2019, 18, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Salinas, D.B.; Azen, C.; Young, S.; Keens, T.G.; Kharrazi, M.; Parad, R.B. Phenotypes of California CF Newborn Screen-Positive Children with CFTR 5T Allele by TG Repeat Length. Genet. Test. Mol. Biomark. 2016, 20, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Groman, J.D.; Hefferon, T.W.; Casals, T.; Bassas, L.; Estivill, X.; Des Georges, M.; Guittard, C.; Koudova, M.; Fallin, M.D.; Nemeth, K.; et al. Variation in a Repeat Sequence Determines Whether a Common Variant of the Cystic Fibrosis Transmembrane Conductance Regulator Gene Is Pathogenic or Benign. Am. J. Hum. Genet. 2004, 74, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Pagin, A.; Devos, A.; Figeac, M.; Truant, M.; Willoquaux, C.; Broly, F.; Lalau, G. Applicability and Efficiency of NGS in Routine Diagnosis: In-Depth Performance Analysis of a Complete Workflow for CFTR Mutation Analysis. PLoS ONE 2016, 11, e0149426. [Google Scholar] [CrossRef]
- Cleary, J.G.; Braithwaite, R.; Gaastra, K.; Hilbush, B.S.; Inglis, S.; Irvine, S.A.; Jackson, A.; Littin, R.; Rathod, M.; Ware, D.; et al. Comparing Variant Call Files for Performance Benchmarking of Next-Generation Sequencing Variant Calling Pipelines. bioRxiv 2015. [Google Scholar] [CrossRef]
- Hughes, E.E.; Stevens, C.F.; Saavedra-Matiz, C.A.; Tavakoli, N.P.; Krein, L.M.; Parker, A.; Zhang, Z.; Maloney, B.; Vogel, B.; DeCelie-Germana, J.; et al. Clinical sensitivity of cystic fibrosis mutation panels in a diverse population. Hum. Mutat. 2016, 37, 201–208. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Tluczek, A.; Orland, K.M.; Cavanagh, L. Psychosocial consequences of false-positive newborn screens for cystic fibrosis. Qual. Health Res. 2011, 21, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Beucher, J.; Leray, E.; Deneuville, E.; Roblin, M.; Pin, I.; Bremont, F.; Turck, D.; Giniès, J.-L.; Foucaud, P.; Rault, G.; et al. Psychological Effects of False-Positive Results in Cystic Fibrosis Newborn Screening: A Two-Year Follow-Up. J. Pediatr. 2010, 156, 771–776.e1. [Google Scholar] [CrossRef]
- Grosse, S.D.; Boyle, C.A.; Botkin, J.R.; Comeau, A.M.; Kharrazi, M.; Rosenfeld, M.; Wilfond, B.S. CDC Newborn screening for cystic fibrosis: Evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm. Rep. 2004, 53, 1–36. [Google Scholar]
- Dorfman, R. Cystic Fibrosis Mutation Database (SickKids). Available online: http://www.genet.sickkids.on.ca/cftr/Home.html (accessed on 3 March 2021).
- Grody, W.W.; Cutting, G.R.; Klinger, K.W.; Richards, C.S.; Watson, M.S.; Desnick, R.J. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet. Med. 2001, 3, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Férec, C.; Casals, T.; Chuzhanova, N.; Macek, M.; Bienvenu, T.; Holubová, A.; King, C.; McDevitt, T.; Castellani, C.; Farrell, P.M.; et al. Gross genomic rearrangements involving deletions in the CFTR gene: Characterization of six new events from a large cohort of hitherto unidentified cystic fibrosis chromosomes and meta-analysis of the underlying mechanisms. Eur. J. Hum. Genet. 2006, 14, 567–576. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hantash, F.M.; Redman, J.B.; Starn, K.; Anderson, B.; Buller, A.; McGinniss, M.J.; Quan, F.; Peng, M.; Sun, W.; Strom, C.M. Novel and recurrent rearrangements in the CFTR gene: Clinical and laboratory implications for cystic fibrosis screening. Hum. Genet. 2006, 119, 126–136. [Google Scholar] [CrossRef]
- Tang, S.; Moonnumakal, S.P.; Stevens, B.; Douglas, G.; Mason, S.; Schmitt, E.S.; Eng, C.M.; Katz, M.; Fang, P. Characterization of a recurrent 3.8kb deletion involving exons 17a and 17b within the CFTR gene. J. Cyst. Fibros. 2013, 12, 290–294. [Google Scholar] [CrossRef][Green Version]
- Nectoux, J.; Audrézet, M.-P.; Viel, M.; Leroy, C.; Raguenes, O.; Férec, C.; Lesure, J.F.; Davy, N.; Renouil, M.; Cartault, F.; et al. A frequent large rearrangement in the CFTR gene in cystic fibrosis patients from Reunion Island. Genet. Test. 2006, 10, 208. [Google Scholar] [CrossRef]
- Dörk, T.; Macek, M.; Mekus, F.; Tümmler, B.; Tzountzouris, J.; Casals, T.; Krebsová, A.; Koudová, M.; Sakmaryová, I.; Vávrová, V.; et al. Characterization of a novel 21-kb deletion, CFTRdele2,3(21 kb), in the CFTR gene: A cystic fibrosis mutation of Slavic origin common in Central and East Europe. Hum. Genet. 2000, 106, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Hantash, F.M.; Redman, J.B.; Goos, D.; Kammesheidt, A.; McGinniss, M.J.; Sun, W.; Strom, C.M. Characterization of a recurrent novel large duplication in the cystic fibrosis transmembrane conductance regulator gene. J. Mol. Diagn. 2007, 9, 556–560. [Google Scholar] [CrossRef][Green Version]
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 2013, 45, 1160–1167. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Run | DBS 1 | Samples 2 | Library Prep | MiSeq Reagent Kit | Sensitivity 3 | Adjusted Sensitivity 4 | Specificity |
|---|---|---|---|---|---|---|---|
| A | 1 | 39 | M | S | 100% | 100% | 100% |
| B | 1 | 76 | M and A | S | 100% | 100% | 100% |
| C | 2 | 78 | A | S | 100% | 100% | 100% |
| D | 2 | 78 | A | S | 98.9% | 100% | 100% |
| E | 2 | 38 | M | S | 100% | 100% | 100% |
| F | 2 | 19 | M | Mi | 100% | 100% | 100% |
| Algorithm | TP | FP | TN | FN | Sensitivity | Specificity |
|---|---|---|---|---|---|---|
| Combined SV and CNV | 22 | 8 | 152 | 0 | 100% | 95.0% |
| Structural variant (SV) | 18 | 1 | 158 | 4 | 81.8% | 99.4% |
| Copy number variant (CNV) | 11 | 7 | 153 | 11 | 50.0% | 95.6% |
| Level of Algorithm Reached | Description | Action Required | Number |
|---|---|---|---|
| IRT | Total infants screened | NA | 220,878 |
| Second-tier variant panel (TMF) | Top 5% IRT | NA | 9557 |
| Third-tier comprehensive CFTR analysis | VHIRT with no variants | Screen negative | 165 |
| One variant | Carrier letter | 439 | |
| Two or more variants | Screen positive referral | 113 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sicko, R.J.; Stevens, C.F.; Hughes, E.E.; Leisner, M.; Ling, H.; Saavedra-Matiz, C.A.; Caggana, M.; Kay, D.M. Validation of a Custom Next-Generation Sequencing Assay for Cystic Fibrosis Newborn Screening. Int. J. Neonatal Screen. 2021, 7, 73. https://doi.org/10.3390/ijns7040073
Sicko RJ, Stevens CF, Hughes EE, Leisner M, Ling H, Saavedra-Matiz CA, Caggana M, Kay DM. Validation of a Custom Next-Generation Sequencing Assay for Cystic Fibrosis Newborn Screening. International Journal of Neonatal Screening. 2021; 7(4):73. https://doi.org/10.3390/ijns7040073
Chicago/Turabian StyleSicko, Robert J., Colleen F. Stevens, Erin E. Hughes, Melissa Leisner, Helen Ling, Carlos A. Saavedra-Matiz, Michele Caggana, and Denise M. Kay. 2021. "Validation of a Custom Next-Generation Sequencing Assay for Cystic Fibrosis Newborn Screening" International Journal of Neonatal Screening 7, no. 4: 73. https://doi.org/10.3390/ijns7040073
APA StyleSicko, R. J., Stevens, C. F., Hughes, E. E., Leisner, M., Ling, H., Saavedra-Matiz, C. A., Caggana, M., & Kay, D. M. (2021). Validation of a Custom Next-Generation Sequencing Assay for Cystic Fibrosis Newborn Screening. International Journal of Neonatal Screening, 7(4), 73. https://doi.org/10.3390/ijns7040073