A Comparative Effectiveness Study of Newborn Screening Methods for Four Lysosomal Storage Disorders †

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Study Protocol
2.3. Statistical Analysis
3. Results
3.1. Participants
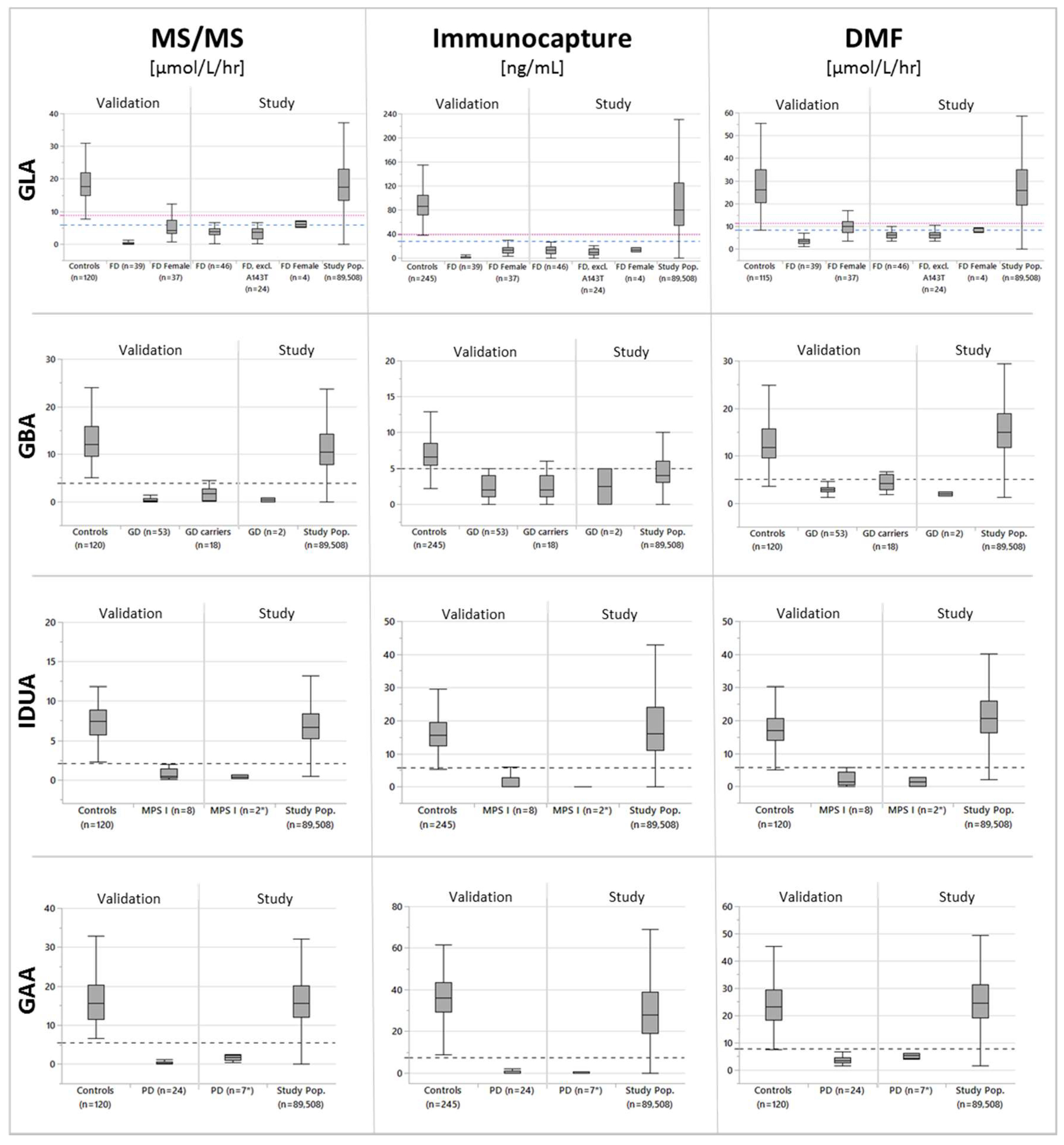
3.2. Cutoff Determination and Assay Performance
3.3. Performance When Using CLIR’s Postanalytical Multivariate Pattern Recognition Tools
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Advisory Committee on Heritable Disorders in Newborns and Children. Available online: http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/index.html (accessed on 4 April 2020).
- Kemper, A.R.; Hwu, W.-L.; Lloyd-Puryear, M.; Kishnani, P.S. Newborn Screening for Pompe Disease: Synthesis of the Evidence and Development of Screening Recommendations. Pediatrics 2007, 120, e1327–e1334. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.-L.; Chien, Y.-H.; Lee, N.-C.; Chiang, S.-C.; Dobrovolny, R.; Huang, A.-C.; Yeh, H.-Y.; Chao, M.-C.; Lin, S.-J.; Kitagawa, T.; et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Gelb, M.H.; Turecek, F.; Scott, C.R.; Chamoles, N.A. Direct multiplex assay of enzymes in dried blood spots by tandem mass spectrometry for the newborn screening of lysosomal storage disorders. J. Inherit. Metab. Dis. 2006, 29, 397–404. [Google Scholar] [CrossRef]
- Orsini, J.J.; Martin, M.M.; Showers, A.L.; Bodamer, O.A.; Zhang, X.K.; Gelb, M.H.; Caggana, M. Lysosomal storage disorder 4 + 1 multiplex assay for newborn screening using tandem mass spectrometry: Application to a small-scale population study for five lysosomal storage disorders. Clin. Chim. Acta 2012, 413, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- Tortorelli, S.; Turgeon, C.T.; Gavrilov, D.K.; Oglesbee, D.; Raymond, K.M.; Rinaldo, P.; Matern, D. Simultaneous Testing for 6 Lysosomal Storage Disorders and X-Adrenoleukodystrophy in Dried Blood Spots by Tandem Mass Spectrometry. Clin. Chem. 2016, 62, 1248–1254. [Google Scholar] [CrossRef]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, R.K.; Nash, C.; Shryock, H.; et al. Newborn Screening for Lysosomal Storage Disorders in Illinois: The Initial 15-Month Experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal Storage Disorder Screening Implementation: Findings from the First Six Months of Full Population Pilot Testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef]
- Parkinson-Lawrence, E.; Fuller, M.; Hopwood, J.J.; Meikle, P.J.; Brooks, D.A. Immunochemistry of Lysosomal Storage Disorders. Clin. Chem. 2006, 52, 1660–1668. [Google Scholar] [CrossRef]
- Meikle, P.J.; Grasby, D.J.; Dean, C.J.; Lang, D.L.; Bockmann, M.; Whittle, A.M.; Fietz, M.J.; Simonsen, H.; Fuller, M.; Brooks, D.A.; et al. Newborn screening for lysosomal storage disorders. Mol. Genet. Metab. 2006, 88, 307–314. [Google Scholar] [CrossRef]
- Stapleton, M.; Kubaski, F.; Mason, R.W.; Shintaku, H.; Kobayashi, H.; Yamaguchi, S.; Taketani, T.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Newborn screening for mucopolysaccharidoses: Measurement of glycosaminoglycans by LC-MS/MS. Mol. Genet. Metab. Rep. 2020, 22, 100563. [Google Scholar] [CrossRef]
- Fuller, M.; Tucker, J.; Lang, D.L.; Dean, C.J.; Fietz, M.J.; Meikle, P.J.; Hopwood, J.J. Screening patients referred to a metabolic clinic for lysosomal storage disorders. J. Med Genet. 2011, 48, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Matern, D.; Oglesbee, D.; Tortorelli, S. Newborn screening for lysosomal storage disorders and other neuronopathic conditions. Dev. Disabil. Res. Rev. 2013, 17, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Sista, R.S.; Wang, T.; Wu, N.; Graham, C.; Eckhardt, A.; Winger, T.; Srinivasan, V.; Bali, D.; Millington, D.S.; Pamula, V.K. Multiplex newborn screening for Pompe, Fabry, Hunter, Gaucher, and Hurler diseases using a digital microfluidic platform. Clin. Chim. Acta 2013, 424, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta 2001, 308, 195–196. [Google Scholar] [CrossRef]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Diagnosis of α-l-Iduronidase Deficiency in Dried Blood Spots on Filter Paper: The Possibility of Newborn Diagnosis. Clin. Chem. 2001, 47, 780–781. [Google Scholar] [CrossRef] [PubMed]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D.; Casentini, C. Gaucher and Niemann–Pick diseases—enzymatic diagnosis in dried blood spots on filter paper: Retrospective diagnoses in newborn-screening cards. Clin. Chim. Acta 2002, 317, 191–197. [Google Scholar] [CrossRef]
- Zhang, H.; Kallwass, H.; Young, S.P.; Carr, C.; Dai, J.; Kishnani, P.S.; Millington, D.; Keutzer, J.; Chen, Y.-T.; Bali, D. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid α-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet. Med. 2006, 8, 302–306. [Google Scholar] [CrossRef]
- De Ruijter, J.; De Ru, M.H.; Wagemans, T.; Ijlst, L.; Lund, A.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; Van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef]
- Rolfs, A.; Giese, A.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Böttcher, T.; Lukas, J.; Hübner, R.; Gölnitz, U.; et al. Glucosylsphingosine Is a Highly Sensitive and Specific Biomarker for Primary Diagnostic and Follow-Up Monitoring in Gaucher Disease in a Non-Jewish, Caucasian Cohort of Gaucher Disease Patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef]
- Tortorelli, S.; Eckerman, J.S.; Orsini, J.J.; Stevens, C.; Hart, J.; Hall, P.L.; Alexander, J.J.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; et al. Moonlighting newborn screening markers: The incidental discovery of a second-tier test for Pompe disease. Genet. Med. 2017, 20, 840–846. [Google Scholar] [CrossRef]
- Johnson, B.; Mascher, H.; Mascher, D.; Legnini, E.; Hung, C.Y.; Dajnoki, A.; Chien, Y.-H.; Maródi, L.; Hwu, W.-L.; Bodamer, O.A. Analysis of Lyso-Globotriaosylsphingosine in Dried Blood Spots. Ann. Lab. Med. 2013, 33, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, G.; Currier, R.; McHugh, D.M.; Gavrilov, D.; Magera, M.J.; Matern, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Smith, E.H.; et al. Enhanced interpretation of newborn screening results without analyte cutoff values. Genet. Med. 2012, 14, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.L.; Marquardt, G.; McHugh, D.M.; Currier, R.; Tang, H.; Stoway, S.D.; Rinaldo, P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef]
- Mørkrid, L.; Rowe, A.D.; Elgstoen, K.B.P.; Olesen, J.H.; Ruijter, G.; Hall, P.L.; Tortorelli, S.; Schulze, A.; Kyriakopoulou, L.; Wamelink, M.M.; et al. Continuous Age- and Sex-Adjusted Reference Intervals of Urinary Markers for Cerebral Creatine Deficiency Syndromes: A Novel Approach to the Definition of Reference Intervals. Clin. Chem. 2015, 61, 760–768. [Google Scholar] [CrossRef]
- McHugh, D.M.S.; Cameron, C.A.; Abdenur, J.E.; Abdulrahman, M.; Adair, O.; Al Nuaimi, S.A.; Åhlman, H.; Allen, J.J.; Antonozzi, I.; Archer, S.; et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: A worldwide collaborative project. Genet. Med. 2011, 13, 230–254. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Albers, S.; Amato, S.; Ampola, M.; Brewster, T.G.; Demmer, L.; Eaton, R.B.; Greenstein, R.; Korson, M.; Larson, C.; et al. Effect of Expanded Newborn Screening for Biochemical Genetic Disorders on Child Outcomes and Parental Stress. JAMA 2003, 290, 2564–2572. [Google Scholar] [CrossRef]
- Tarini, B.A.; Christakis, D.A.; Welch, H.G. State Newborn Screening in the Tandem Mass Spectrometry Era: More Tests, More False-Positive Results. Pediatrics 2006, 118, 448–456. [Google Scholar] [CrossRef]
- California Dept. of Public Health. Births, by Mother’s Race/Ethnicity. Available online: https://www.kidsdata.org/topic/31/births-race/table#fmt=146&loc=2,127,347,1763,331,348,336,171,321,345,357,332,324,369,358,362,360,337,327,364,356,217,353,328,354,323,352,320,339,334,365,343,330,367,344,355,366,368,265,349,361,4,273,59,370,326,333,322,341,338,350,342,329,325,359,351,363,340,335&tf=84&ch=7,11,8,507,9,73,74 (accessed on 4 April 2020).
- Rinaldo, P.; Zafari, S.; Tortorelli, S.; Matern, D. Making the case for objective performance metrics in newborn screening by tandem mass spectrometry. Ment. Retard. Dev. Disabil. Res. Rev. 2006, 12, 255–261. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000042454.12 (accessed on 28 April 2020).
- Macklin, S.; Laney, D.; Lisi, E.; Atherton, A.; Smith, E. The Psychosocial Impact of Carrying a Debated Variant in the GLA Gene. J. Genet. Couns. 2017, 27, 217–224. [Google Scholar] [CrossRef]
- Liao, H.-C.; Chiang, C.-C.; Niu, D.-M.; Wang, C.-H.; Kao, S.-M.; Tsai, F.-J.; Huang, Y.-H.; Liu, H.-C.; Huang, C.-K.; Gao, H.-J.; et al. Detecting multiple lysosomal storage diseases by tandem mass spectrometry—A national newborn screening program in Taiwan. Clin. Chim. Acta 2014, 431, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Orsini, J.J.; the New York State Krabbe Disease Consortium; Kay, D.M.; Saavedra-Matiz, C.A.; Wenger, D.A.; Duffner, P.K.; Erbe, R.W.; Biski, C.; Martin, M.; Krein, L.M.; et al. Newborn screening for Krabbe disease in New York State: The first eight years’ experience. Genet. Med. 2016, 18, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Matern, D.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Tortorelli, S. Newborn screening for lysosomal storage disorders. Semin. Perinatol. 2015, 39, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-H.; Chiang, S.-C.; Zhang, X.K.; Keutzer, J.; Lee, N.-C.; Huang, A.-C.; Chen, C.-A.; Wu, M.-H.; Huang, P.-H.; Tsai, F.-J.; et al. Early Detection of Pompe Disease by Newborn Screening Is Feasible: Results from the Taiwan Screening Program. Pediatrics 2008, 122, e39–e45. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Chong, K.-W.; Hsu, J.-H.; Yu, H.-C.; Shih, C.-C.; Huang, C.-H.; Lin, S.-J.; Chen, C.-H.; Chiang, C.-C.; Ho, H.-J.; et al. High Incidence of the Cardiac Variant of Fabry Disease Revealed by Newborn Screening in the Taiwan Chinese Population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef]
- Lin, S.-P.; Lin, H.-Y.; Wang, T.-J.; Chang, C.-Y.; Lin, K.-J.; Huang, S.-F.; Tsai, C.-C.; Liu, H.-L.; Keutzer, J.; Chuang, C.-K. A pilot newborn screening program for Mucopolysaccharidosis type I in Taiwan. Orphanet J. Rare Dis. 2013, 8, 147. [Google Scholar] [CrossRef]
- Hopkins, P.V.; Klug, T.; Vermette, L.; Raburn-Miller, J.; Kiesling, J.; Rogers, S. Incidence of 4 Lysosomal Storage Disorders from 4 Years of Newborn Screening. JAMA Pediatr. 2018, 172, 696. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet. Med. 2018, 21, 631–640. [Google Scholar] [CrossRef]
- Chan, M.-J.; Liao, H.-C.; Gelb, M.H.; Chuang, C.-K.; Liu, M.-Y.; Chen, H.-J.; Kao, S.-M.; Lin, H.-Y.; Huang, Y.-H.; Kumar, A.B.; et al. Taiwan National Newborn Screening Program by Tandem Mass Spectrometry for Mucopolysaccharidoses Types I, II, and VI. J. Pediatr. 2019, 205, 176–182. [Google Scholar] [CrossRef]
- Tang, H.; Feuchtbaum, L.; Sciortino, S.; Matteson, J.; Mathur, D.; Bishop, T.; Olney, R.S. The First Year Experience of Newborn Screening for Pompe Disease in California. Int. J. Neonatal Screen. 2020, 6, 9. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Andriola, M.; Arnold, G.; Aron, A.; Duffner, P.; Erbe, R.W.; Escolar, M.L.; Estrella, L.; Galvin-Parton, P.; Iglesias, A.; et al. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet. Med. 2016, 18, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision newborn screening for lysosomal disorders. Genet. Med. 2017, 20, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Peck, D.S.; Lacey, J.M.; White, A.L.; Pino, G.; Studinski, A.L.; Fisher, R.; Ahmad, A.; Spencer, L.; Viall, S.; Shallow, N.; et al. Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I. Int. J. Neonatal Screen. 2020, 6, 10. [Google Scholar] [CrossRef]
- Freedenberg, D.; Berry, S.A.; Dimmock, D.; Gibson, J.; Greene, C.; Kronn, D.; Tanksley, S. SIMD Position Statement: Identifying Abnormal Newborn Screens Requiring Immediate Notification of the Health Care Provider. Available online: https://www.simd.org/Issues/SIMD%20NBS%20Critical%20Conditions%20policy%20statement.pdf (accessed on 26 May 2020).
- Yang, C.-F.; Yang, C.C.; Liao, H.-C.; Huang, L.-Y.; Chiang, C.-C.; Ho, H.-C.; Lai, C.-J.; Chu, T.-H.; Yang, T.-F.; Hsu, T.-R.; et al. Very Early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes. J. Pediatr. 2016, 169, 174–180.e1. [Google Scholar] [CrossRef]
- Carducci, C.; Santagata, S.; Leuzzi, V.; Carducci, C.; Artiola, C.; Giovanniello, T.; Battini, R.; Antonozzi, I. Quantitative determination of guanidinoacetate and creatine in dried blood spot by flow injection analysis-electrospray tandem mass spectrometry. Clin. Chim. Acta 2006, 364, 180–187. [Google Scholar] [CrossRef]
- Hall, P.L.; Sanchez, R.; Hagar, A.; Jerris, S.C.; Wittenauer, A.; Wilcox, W.R. Two-Tiered Newborn Screening with Post-Analytical Tools for Pompe Disease and Mucopolysaccharidosis Type I Results in Performance Improvement and Future Direction. Int. J. Neonatal Screen. 2020, 6, 2. [Google Scholar] [CrossRef]
- Liao, H.-C.; Chan, M.-J.; Yang, C.-F.; Chiang, C.-C.; Niu, D.-M.; Huang, C.-K.; Gelb, M.H. Mass Spectrometry but Not Fluorimetry Distinguishes Affected and Pseudodeficiency Patients in Newborn Screening for Pompe Disease. Clin. Chem. 2017, 63, 1271–1277. [Google Scholar] [CrossRef]
- Millington, D.S.; Bali, D.M. Misinformation regarding tandem mass spectrometric vs fluorometric assays to screen newborns for LSDs. Mol. Genet. Metab. Rep. 2017, 11, 72–73. [Google Scholar] [CrossRef]
- Gelb, M.H.; Scott, C.R.; Turecek, F.; Liao, H.-C. Comparison of tandem mass spectrometry to fluorimetry for newborn screening of LSDs. Mol. Genet. Metab. Rep. 2017, 12, 80–81. [Google Scholar] [CrossRef]
- Millington, D.S. Response to Gelb et al.: “Comparison of tandem mass spectrometry to fluorimetry for newborn screening of LSDs”. Mol. Genet. Metab. Rep. 2017, 12, 98. [Google Scholar] [CrossRef]
- Gelb, M.H.; Lukacs, Z.; Ranieri, E.; Schielen, P. Newborn Screening for Lysosomal Storage Disorders: Methodologies for Measurement of Enzymatic Activities in Dried Blood Spots. Int. J. Neonatal Screen. 2018, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Wadman, M. Minnesota starts to destroy stored blood spots. Nature 2012. [Google Scholar] [CrossRef]
- Gelb, M.H. A universal newborn and diagnostic screening platform for lysosomal diseases and beyond. Mol. Genet. Metab. 2020, 129, S61. [Google Scholar] [CrossRef]


{kind=link}
| Race/Ethnicity | Study Population | California Birth Data * |
|---|---|---|
| White | 37.8% | 27.6% |
| Hispanic/Latina | 41.6% | 48.8% |
| African American/ Black | 5.4% | 5.3% |
| Asian/Pacific Islander | 11.0% | 13.6% |
| American Indian/ Alaska Native | 0.14% | 0.4% |
| Multiracial | - | 2.2% |
| Other/Unknown | 4.2% | 2.2% |
| Enzyme | DMF [μmol/L/h] | Immunocapture [ng/mL] | MS/MS [μmol/L/h] |
|---|---|---|---|
| GAA | 7.71 (n = 412, 0.412%) | 7.36 (n = 4,478; 5.003%) | 5.55 (n = 900, 1.005%) |
| GBA | 5.02 1 (n = 376, 0.420%) | 5.00 1 (n = 45,105; 50.392%) | 3.95 (n = 1521; 1.699%) |
| IDUA | 5.72 1 (n = 268, 0.299%) | 5.79 1 (n = 4372; 4.884%) | 2.1 (n = 312, 0.349%) |
| GLA 2 | 11.4 3 (n = 2357, 2.633%) | 39.36 3 (n = 9764; 10.909%) | 8.90 3 (n = 3637; 4.063%) |
| GLA (males only) 4 | 8.36 (n = 473, 0.528%) | 27.41 (n = 2931; 3.275%) | 5.95 (n = 354; 0.395%) |
| Case | Enzyme/ Gene | DMF [µmol/L/h] | Immunocapture [ng/mL] | MS/MS [µmol/L/h] | 2TT 1 [µmol/L/h] | Genotype Interpretation 2 | Variant 1 | Variant 2 |
|---|---|---|---|---|---|---|---|---|
| FD #1 | GLA | 9.1 * | 21.0 | 6.3 | 2.5 | VUS | c.352C>T (p.R118C) | NA |
| FD #2 | GLA | 4.4 | 12.0 | 3.3 | 1.1 | VUS | c.352C>T (p.R118C) | NA |
| FD #3 | GLA | 7.5 | 15.0 | 3.6 | 1.8 | VUS | c.352C>T (p.R118C) | NA |
| FD #4 | GLA | 5.8 | 0.0 | 0.2 | 0.0 | P | c.1023A>C (p.E341D) | NA |
| FD #5 | GLA | 7.0 | 19.0 * | 4.7 | 1.7 | VUS | c.419A>C (p.K140T) | NA |
| FD #6 | GLA | 10.6 * | 10.0 * | 5.9 * | 2.0 | VUS | c.352C>T (p.R118C) | NA |
| FD #7 | GLA | 5.5 | 3.0 | 0.7 | 0.0 | P | c.644A>G (p.N215S) | NA |
| FD #8 | GLA | 5.6 | 21.0 | 4.8 | 2.5 | VUS | c.352C>T (p.R118C) | NA |
| FD #9 | GLA | 7.1 | 9.0 | 2.8 | 0.6 | LP | c.197A>G (p.E66G) | NA |
| FD #10 | GLA | 3.9 | 7.0 | 1.5 | 0.0 | LP | c.1088G>A (p.R363H) | NA |
| FD #11 | GLA | 6.1 | 6.0 | 2.3 | 0.7 | LP | c.197A>G (p.E66G) | NA |
| FD #12 | GLA | 4.3 | 8.0 | 0.5 | 0.0 | LP | c.1088G>A (p.R363H) | NA |
| FD #13 | GLA | 5.5 | 5.0 | 2.4 * | 0.6 | VUS | c.70T>A (p.W24R), c.1255A>G (p.N419D) | NA |
| FD #14 | GLA | 5.1 | 15.0 | 6.7 * | 2.7 | VUS | c.352C>T (p.R118C) | NA |
| FD #15 | GLA | 3.8 | 1.0 | 0.8 | 0.0 | LP | c.593T>C (p.I198T) | NA |
| FD #16 | GLA | 5.5 | 0.0 | 1.0 | 0.0 | LP | c.593T>C (p.I198T) | NA |
| FD #17 | GLA | 7.7 | 15.0 | 4.8 | 1.4 | VUS | c.352C>T (p.R118C) | NA |
| FD #18 | GLA | 3.6 | 9.0 | 3.7 | 1.0 | VUS | c.473C>A (p.T158N) | NA |
| FD #19 | GLA | 9.8 | 3.0 | 3.0 | 0.5 | P | c.124A>C (p.M42L) | NA |
| FD #20 | GLA | 6.7 | 7.0 | 3.9 | 0.9 | VUS | c.313A>G (p.R105G) | NA |
| FD #21 | GLA | 6.7 | 9.0 | 4.5 | 1.2 | VUS | c.352C>T (p.R118C) | NA |
| FD #22 | GLA | 6.6 | 16.0 | 6.3 * | 1.0 | VUS | c.419A>C (p.K140T) | NA |
| FD #23 | GLA | 7.0 | 6.0 | 4.6 | 1.1 | P | c.639+919G>A | NA |
| FD #24 | GLA | 10.0 * | 17.0 * | 4.6 * | 1.1 | VUS | c.122C>G (p.T41S) | NA |
| FD #25 | GLA | 6.8 * | 22.0 * | 5.9 * | 1.3 | PD | c.427G>A (p.A143T) | NA |
| FD #26 | GLA | 11.2 * | 22.0 * | 5.7 * | 1.3 | PD | c.427G>A (p.A143T) | NA |
| FD #27 | GLA | 9.8 * | 22.0 * | 4.8 * | 1.6 | PD | c.427G>A (p.A143T) | NA |
| FD #28 | GLA | 6.2 * | 27.0 * | 3.4 * | 0.7 | PD | c.427G>A (p.A143T) | NA |
| FD #29 | GLA | 7.2 * | 19.0 * | 3.1 * | 0.7 | PD | c.427G>A (p.A143T) | NA |
| FD #30 | GLA | 11.4 * | 15.0 * | 3.9 * | 1.1 | PD | c.427G>A (p.A143T) | NA |
| FD #31 | GLA | 6.6 * | 19.0 * | 4.7 * | 1.4 | PD | c.427G>A (p.A143T) | NA |
| FD #32 | GLA | 5.4 * | 20.0 * | 4.6 * | 1.2 | PD | c.427G>A (p.A143T) | NA |
| FD #33 | GLA | 4.2 * | 13.0 * | 2.8 * | 1.4 | PD | c.427G>A (p.A143T) | NA |
| FD #34 | GLA | 6.5 * | 15.0 * | 4.1 * | 1.4 | PD | c.427G>A (p.A143T) | NA |
| FD #35 | GLA | 4.9 * | 10.0 * | 2.9 * | 0.7 | PD | c.427G>A (p.A143T) | NA |
| FD #36 | GLA | 6.4 * | 10.0 * | 4.0 * | 1.8 | PD | c.427G>A (p.A143T) | NA |
| FD #37 | GLA | 4.6 * | 13.0 * | 4.0 * | 0.5 | PD | c.427G>A (p.A143T) | NA |
| FD #38 | GLA | 3.7 * | 16.0 * | 3.2 * | 0.9 | PD | c.427G>A (p.A143T) | NA |
| FD #39 | GLA | 8.8 * | 16.0 * | 5.2 * | 0.9 | PD | c.427G>A (p.A143T) | NA |
| FD #40 | GLA | 5.6 * | 7.0 * | 3.1 * | 0.6 | PD | c.427G>A (p.A143T) | NA |
| FD #41 | GLA | 6.0 * | 13.0 * | 3.4 * | 0.6 | PD | c.427G>A (p.A143T) | NA |
| FD #42 | GLA | 5.5 * | 11.0 * | 3.8 * | 0.6 | PD | c.427G>A (p.A143T) | NA |
| FD #43 | GLA | 6.5 * | 14.0 * | 6.3 * | 1.3 | PD | c.427G>A (p.A143T) | NA |
| FD #44 | GLA | 6.4 * | 19.0 * | 4.4 * | 1.1 | PD | c.427G>A (p.A143T) | NA |
| FD #45 | GLA | 4.1 * | 18.0 * | 2.1 * | 0.3 | PD | c.427G>A (p.A143T) | NA |
| FD #46 | GLA | 7.6 * | 14.0 * | 4.8 * | 0.9 | PD | c.427G>A (p.A143T) | NA |
| FD het #1 | GLA | 7.4 * | 18.0 | 7.2 * | 2.2 | P/nd | c.870G>GC,p.M290MI | - |
| FD het #2 | GLA | 9.5* | 11.0 | 4.2 | 0.7 | P/nd | c.870G>A,p.M290I | - |
| FD het #3 | GLA | 7.5 | 9.0 | 5.5 | 1.6 | P/B | c.937G>T,p.D313Y | c.1000-22C>T |
| FD het #4 | GLA | 8.0 * | 16.0 * | 6.5 * | 2.0 | PD/nd | c.427G>A (p.A143T) | - |
| GD #1 | GBA | 2.4 | 5.0* | 0.8 | 0 | P/P | c.1226A>G (p.N409S) | c.1226A>G (p.N409S) |
| GD #2 | GBA | 1.5 | 0 | 0 | 0 | P/P | c.680A>G (p.N227S) | c.680A>G (p.N227S) |
| MPS I | IDUA | 2.9 | 0 | 0.2 | 0 | P/P | c.1205G>A (p.W402X) | c.46_57del (p.(Ser16_Ala19del)) |
| PD #1 | GAA | 4.3 | 4.0* | 1.6 | 0.8 | P/VUS | c.-32-13T>G | c.1909C>A (p.L637M) |
| PD #2 | GAA | 5.3 | 0 | 1.7 | 1.1 | LP/VUS | c.1292_1295dup p.(Gln433Alafs *74) | c.1019A>G (p.Y340C) |
| PD #3 | GAA | 3.8 * | 0 | 0.5 | 0 | P/P | c.-32-13T>G | [c.752C>T (p.S251L); c.761C>T (p.S254L)] |
| PD #4 | GAA | 4.4 | 0 | 2.5 | 2.6 | VUS/VUS | c.257C>G (p.P86R) | c.1306C>T (p.R436W) |
| PD #5 | GAA | 5.4 | 0 | 2.4 | 0 | P/P | c.752C>T (p.S251L) | c.761C>T (p.S254L) |
| PD #6 | GAA | 8.8 | 0 | 2.2 | 1.5 | P/P | c.752C>T (p.S251L) | c.761C>T (p.S254L) |
| Test Platform | Immunocapture + 2TT 5 | Immunocapture + CLIR Tools | DMF + 2TT 5 | DMF + CLIR Tools | MS/MS + 2TT 5 | MS/MS + CLIR Tools |
|---|---|---|---|---|---|---|
| TP | 27 | 24 | 22 | 22 | 23 | 21 |
| Sensitivity 1 | 100% | 92% | 82% | 82% | 85% | 78% |
| FP 2 | 74 | 2 | 74 | 39 | 74 | 31 |
| Reduction in need for 2TT | - | 97% | - | 47% | - | 58% |
| FPR 3 | 0.085% | 0.002% | 0.085% | 0.045% | 0.085% | 0.036% |
| PPV 4 | 27% | 92% | 23% | 36% | 30% | 40% |
| FN | 0 | 3 | 5 | 5 | 4 | 6 |
| Test Platform | Immunocapture + 2TT | Immunocapture + CLIR Tools | DMF + 2TT | DMF + CLIR Tools | MS/MS + 2TT | MS/MS + CLIR Tools |
|---|---|---|---|---|---|---|
| TP | 1 | 1 | 2 | 2 | 2 | 2 |
| Sensitivity 1 | 50% | 50% | 100% | 100% | 100% | 100% |
| FP 2 | 107 | 1 | 107 | 13 | 107 | 47 |
| Reduction in need for 2TT | - | 99% | - | 88% | - | 56% |
| FPR 3 | 0.123% | 0.001% | 0.123% | 0.015% | 0.123% | 0.056% |
| PPV 4 | 0.9% | 4.1% | 1.8% | 13.3% | 1.8% | 50.0% |
| FN | 1 | 1 | 0 | 0 | 0 | 0 |
| Test Platform | Immunocapture + 2TT | Immunocapture + CLIR Tools | DMF + 2TT | DMF + CLIR Tools | MS/MS + 2TT | MS/MS + CLIR Tools |
|---|---|---|---|---|---|---|
| TP | 1 | 1 | 1 | 1 | 1 | 1 |
| Sensitivity 1 | 100% | 100% | 100% | 100% | 100% | 100% |
| FP 2 | 159 | 15 | 159 | 17 | 159 | 5 |
| Reduction in need for 2TT | - | 91% | - | 89% | - | 97% |
| FPR 3 | 0.183% | 0.017% | 0.183% | 0.020% | 0.183% | 0.006% |
| PPV 4 | 0.6% | 6.3% | 0.6% | 5.6% | 0.6% | 16.7% |
| FN | 0 | 0 | 0 | 0 | 0 | 0 |
| Test Platform | Immunocapture + 2TT | Immunocapture + CLIR Tools | DMF + 2TT | DMF + CLIR Tools | MS/MS + 2TT | MS/MS + CLIR Tools |
|---|---|---|---|---|---|---|
| TP | 6 | 5 | 5 | 5 | 6 | 6 |
| Sensitivity 1 | 100% | 83% | 83% | 83% | 100% | 100% |
| FP 2 | 99 | 7 | 99 | 65 | 99 | 5 |
| Reduction in need for 2TT | - | 93% | - | 32% | - | 95% |
| FPR 3 | 0.109% | 0.008% | 0.109% | 0.075% | 0.109% | 0.006% |
| PPV 4 | 5.9% | 41.7% | 4.8% | 7.1% | 5.9% | 45.5% |
| FN | 0 | 1 | 1 | 1 | 0 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanders, K.A.; Gavrilov, D.K.; Oglesbee, D.; Raymond, K.M.; Tortorelli, S.; Hopwood, J.J.; Lorey, F.; Majumdar, R.; Kroll, C.A.; McDonald, A.M.; et al. A Comparative Effectiveness Study of Newborn Screening Methods for Four Lysosomal Storage Disorders. Int. J. Neonatal Screen. 2020, 6, 44. https://doi.org/10.3390/ijns6020044
Sanders KA, Gavrilov DK, Oglesbee D, Raymond KM, Tortorelli S, Hopwood JJ, Lorey F, Majumdar R, Kroll CA, McDonald AM, et al. A Comparative Effectiveness Study of Newborn Screening Methods for Four Lysosomal Storage Disorders. International Journal of Neonatal Screening. 2020; 6(2):44. https://doi.org/10.3390/ijns6020044
Chicago/Turabian StyleSanders, Karen A., Dimitar K. Gavrilov, Devin Oglesbee, Kimiyo M. Raymond, Silvia Tortorelli, John J. Hopwood, Fred Lorey, Ramanath Majumdar, Charles A. Kroll, Amber M. McDonald, and et al. 2020. "A Comparative Effectiveness Study of Newborn Screening Methods for Four Lysosomal Storage Disorders" International Journal of Neonatal Screening 6, no. 2: 44. https://doi.org/10.3390/ijns6020044
APA StyleSanders, K. A., Gavrilov, D. K., Oglesbee, D., Raymond, K. M., Tortorelli, S., Hopwood, J. J., Lorey, F., Majumdar, R., Kroll, C. A., McDonald, A. M., Lacey, J. M., Turgeon, C. T., Tucker, J. N., Tang, H., Currier, R., Isaya, G., Rinaldo, P., & Matern, D. (2020). A Comparative Effectiveness Study of Newborn Screening Methods for Four Lysosomal Storage Disorders. International Journal of Neonatal Screening, 6(2), 44. https://doi.org/10.3390/ijns6020044