Performance of Expanded Newborn Screening in Norway Supported by Post-Analytical Bioinformatics Tools and Rapid Second-Tier DNA Analyses

 ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Definitions
2.2. Newborn Screening Methods
2.2.1. First-Tier Methods
2.2.2. Second-Tier Methods
2.2.3. Second-Tier DNA Sequencing
2.3. Diagnostics
3. Results
3.1. Targeted Diagnostic Screening in the Decade before NBS
3.2. Clinical Outcomes
3.2.1. Fatty Acid Oxidation Defects
3.2.2. Aminoacidopathies
3.2.3. Organic Acidurias
3.3. Missed Cases during eNBS 2012–2020
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hall, P.L.; Marquardt, G.; McHugh, D.M.; Currier, R.J.; Tang, H.; Stoway, S.D.; Rinaldo, P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef]
- Marquardt, G.; Currier, R.; McHugh, D.M.; Gavrilov, D.; Magera, M.J.; Matern, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Smith, E.H.; et al. Enhanced interpretation of newborn screening results without analyte cutoff values. Genet. Med. 2012, 14, 648–655. [Google Scholar] [CrossRef]
- Norwegian Institute of Public Health. The Medical Birth Registry of Norway. Available online: https://www.fhi.no/en/hn/health-registries/medical-birth-registry-of-norway/ (accessed on 28 October 2019).
- Wastell, H.; Dale, G.; Bartlett, K. A sensitive fluorimetric rate assay for biotinidase using a new derivative of biotin, biotinyl-6-aminoquinoline. Anal. Biochem. 1984, 140, 69–73. [Google Scholar] [CrossRef]
- Alodaib, A.N.; Carpenter, K.; Wiley, V.; Wotton, T.; Christodoulou, J.; Wilcken, B. Homocysteine measurement in dried blood spot for neonatal detection of homocystinurias. JIMD Rep. 2012, 5, 1–6. [Google Scholar] [CrossRef]
- Fu, X.; Xu, Y.K.; Chan, P.; Pattengale, P.K. Simple, Fast, and Simultaneous Detection of Plasma Total Homocysteine, Methylmalonic Acid, Methionine, and 2-Methylcitric Acid Using Liquid Chromatography and Mass Spectrometry (LC/MS/MS). JIMD Rep. 2013, 10, 69–78. [Google Scholar] [CrossRef]
- Lundman, E.; Gaup, H.J.; Bakkeheim, E.; Olafsdottir, E.J.; Rootwelt, T.; Storrosten, O.T.; Pettersen, R.D. Implementation of newborn screening for cystic fibrosis in Norway. Results from the first three years. J. Cyst. Fibros 2016, 15, 318–324. [Google Scholar] [CrossRef]
- Heath, E.M.; O’Brien, D.P.; Banas, R.; Naylor, E.W.; Dobrowolski, S. Optimization of an Automated DNA Purification Protocol for Neonatal Screening. Arch. Pathol. Lab. Med. 1999, 123, 1154–1160. [Google Scholar]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Wolf, B.; Grier, R.E.; Allen, R.J.; Goodman, S.I.; Kien, C.L. Biotinidase deficiency: The enzymatic defect in late-onset multiple carboxylase deficiency. Clin. Chim. Acta 1983, 131, 273–281. [Google Scholar] [CrossRef]
- Demaugre, F.; Bonnefont, J.P.; Mitchell, G.; Nguyen-Hoang, N.; Pelet, A.; Rimoldi, M.; Di Donato, S.; Saudubray, J.M. Hepatic and muscular presentations of carnitine palmitoyl transferase deficiency: Two distinct entities. Pediatr. Res. 1988, 24, 308–311. [Google Scholar] [CrossRef]
- Wanders, R.J.; Ruiter, J.P.; Ljist, I.J.; Waterham, H.R.; Houten, S.M. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef]
- Alcaide, P.; Krijt, J.; Ruiz-Sala, P.; Jesina, P.; Ugarte, M.; Kozich, V.; Merinero, B. Enzymatic diagnosis of homocystinuria by determination of cystathionine-ss-synthase activity in plasma using LC-MS/MS. Clin. Chim. Acta 2015, 438, 261–265. [Google Scholar] [CrossRef]
- Krijt, J.; Kopecka, J.; Hnizda, A.; Moat, S.; Kluijtmans, L.A.; Mayne, P.; Kozich, V. Determination of cystathionine beta-synthase activity in human plasma by LC-MS/MS: Potential use in diagnosis of CBS deficiency. J. Inherit. Metab. Dis. 2011, 34, 49–55. [Google Scholar] [CrossRef]
- Mosegaard, S.; Bruun, G.H.; Flyvbjerg, K.F.; Bliksrud, Y.T.; Gregersen, N.; Dembic, M.; Annexstad, E.; Tangeraas, T.; Olsen, R.K.J.; Andresen, B.S. An intronic variation in SLC52A1 causes exon skipping and transient riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Mol. Genet. Metab. 2017, 122, 182–188. [Google Scholar] [CrossRef]
- Bremer, S.; Bliksrud, Y.T.; Rootwelt, H.; Woldseth, B.; Tangeraas, T.; Sæves, I.; Watle, S.S.V. Identification of a novel BCKDHA deletion causing maple syrup urine disease. Meta Gene 2016, 10, 86–89. [Google Scholar] [CrossRef]
- Lund, A.M.; Hougaard, D.M.; Simonsen, H.; Andresen, B.S.; Christensen, M.; Duno, M.; Skogstrand, K.; Olsen, R.K.; Jensen, U.G.; Cohen, A.; et al. Biochemical screening of 504,049 newborns in Denmark, the Faroe Islands and Greenland—Experience and development of a routine program for expanded newborn screening. Mol. Genet. Metab. 2012, 107, 281–293. [Google Scholar] [CrossRef]
- Lund, A.; Wibrand, F.; Skogstrand, K.; Cohen, A.; Christensen, M.; Japelt, R.B.; Duno, M.; Skovby, F.; Norgaard-Pedersen, B.; Gregersen, N.; et al. Danish expanded newborn screening is a successful preventive public health programme. Dan. Med. J. 2020, 67, A06190341. [Google Scholar]
- Bollestad, M.; Vik, I.; Grude, N.; Blix, H.S.; Brekke, H.; Lindbaek, M. Bacteriology in uncomplicated urinary tract infections in Norwegian general practice from 2001-2015. BJGP Open 2018, 1, bjgpopen17X101145. [Google Scholar] [CrossRef]
- Gallant, N.M.; Leydiker, K.; Wilnai, Y.; Lee, C.; Lorey, F.; Feuchtbaum, L.; Tang, H.; Carter, J.; Enns, G.M.; Packman, S.; et al. Biochemical characteristics of newborns with carnitine transporter defect identified by newborn screening in California. Mol. Genet. Metab. 2017, 122, 76–84. [Google Scholar] [CrossRef]
- Wilson, C.; Knoll, D.; de Hora, M.; Kyle, C.; Glamuzina, E.; Webster, D. The decision to discontinue screening for carnitine uptake disorder in New Zealand. J. Inherit. Metab. Dis. 2019, 42, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Longo, N. Primary Carnitine Deficiency and Newborn Screening for Disorders of the Carnitine Cycle. Ann. Nutr. Metab. 2016, 68 (Suppl. S3), 5–9. [Google Scholar] [CrossRef]
- Winter, S.C.; Linn, L.S.; Helton, E. Plasma carnitine concentrations in pregnancy, cord blood, and neonates and children. Clin. Chim. Acta 1995, 243, 87–93. [Google Scholar] [CrossRef]
- Lee, N.C.; Tang, N.L.; Chien, Y.H.; Chen, C.A.; Lin, S.J.; Chiu, P.C.; Huang, A.C.; Hwu, W.L. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol. Genet. Metab. 2010, 100, 46–50. [Google Scholar] [CrossRef]
- Frigeni, M.; Balakrishnan, B.; Yin, X.; Calderon, F.R.O.; Mao, R.; Pasquali, M.; Longo, N. Functional and molecular studies in primary carnitine deficiency. Hum. Mutat. 2017, 38, 1684–1699. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Te Brinke, H.; Ruiter, J.P.N.; Haasjes, J.; Oostheim, W.; van Lenthe, H.; Ljist, I.J.; Ebberink, M.S.; Wanders, R.J.A.; Vaz, F.M.; et al. A mutation creating an upstream translation initiation codon in SLC22A5 5′UTR is a frequent cause of primary carnitine deficiency. Hum. Mutat. 2019, 40, 1899–1904. [Google Scholar] [CrossRef]
- Lund, A.M.; Joensen, F.; Hougaard, D.M.; Jensen, L.K.; Christensen, E.; Christensen, M.; Norgaard-Petersen, B.; Schwartz, M.; Skovby, F. Carnitine transporter and holocarboxylase synthetase deficiencies in The Faroe Islands. J. Inherit. Metab. Dis. 2007, 30, 341–349. [Google Scholar] [CrossRef]
- Jorgensen, T.H.; Degn, B.; Wang, A.G.; Vang, M.; Gurling, H.; Kalsi, G.; McQuillin, A.; Kruse, T.A.; Mors, O.; Ewald, H. Linkage disequilibrium and demographic history of the isolated population of the Faroe Islands. Eur. J. Hum. Genet. 2002, 10, 381–387. [Google Scholar] [CrossRef]
- Rinaldo, P.; Stanley, C.A.; Hsu, B.Y.; Sanchez, L.A.; Stern, H.J. Sudden neonatal death in carnitine transporter deficiency. J. Pediatr. 1997, 131, 304–305. [Google Scholar] [CrossRef]
- Turgeon, C.T.; Magera, M.J.; Cuthbert, C.D.; Loken, P.R.; Gavrilov, D.K.; Tortorelli, S.; Raymond, K.M.; Oglesbee, D.; Rinaldo, P.; Matern, D. Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin. Chem. 2010, 56, 1686–1695. [Google Scholar] [CrossRef]
- Landau, Y.E.; Waisbren, S.E.; Chan, L.M.; Levy, H.L. Long-term outcome of expanded newborn screening at Boston children’s hospital: Benefits and challenges in defining true disease. J. Inherit. Metab. Dis. 2017, 40, 209–218. [Google Scholar] [CrossRef]
- Wilcken, B.; Haas, M.; Joy, P.; Wiley, V.; Bowling, F.; Carpenter, K.; Christodoulou, J.; Cowley, D.; Ellaway, C.; Fletcher, J.; et al. Expanded newborn screening: Outcome in screened and unscreened patients at age 6 years. Pediatrics 2009, 124, e241–e248. [Google Scholar] [CrossRef]
- Jager, E.A.; Kuijpers, M.M.; Bosch, A.M.; Mulder, M.F.; Gozalbo, E.R.; Visser, G.; de Vries, M.; Williams, M.; Waterham, H.R.; van Spronsen, F.J.; et al. A nationwide retrospective observational study of population newborn screening for medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in the Netherlands. J. Inherit. Metab. Dis. 2019. [Google Scholar] [CrossRef]
- David, J.; Chrastina, P.; Peskova, K.; Kozich, V.; Friedecky, D.; Adam, T.; Hlidkova, E.; Vinohradska, H.; Novotna, D.; Hedelova, M.; et al. Epidemiology of rare diseases detected by newborn screening in the Czech Republic. Cent. Eur. J. Public Health 2019, 27, 153–159. [Google Scholar] [CrossRef]
- Andresen, B.S.; Lund, A.M.; Hougaard, D.M.; Christensen, E.; Gahrn, B.; Christensen, M.; Bross, P.; Vested, A.; Simonsen, H.; Skogstrand, K.; et al. MCAD deficiency in Denmark. Mol. Genet. Metab. 2012, 106, 175–188. [Google Scholar] [CrossRef]
- De Sain-van der Velden, M.G.M.; Diekman, E.F.; Jans, J.J.; van der Ham, M.; Prinsen, B.H.C.M.T.; Visser, G.; Verhoeven-Duif, N.M. Differences between acylcarnitine profiles in plasma and bloodspots. Mol. Genet. Metab. 2013, 110, 116–121. [Google Scholar] [CrossRef]
- Bleeker, J.C.; Kok, I.L.; Ferdinandusse, S.; van der Pol, W.L.; Cuppen, I.; Bosch, A.M.; Langeveld, M.; Derks, T.G.J.; Williams, M.; de Vries, M.; et al. Impact of newborn screening for very-long-chain acyl-CoA dehydrogenase deficiency on genetic, enzymatic, and clinical outcomes. J. Inherit. Metab. Dis. 2019, 42, 414–423. [Google Scholar] [CrossRef]
- Pena, L.D.; van Calcar, S.C.; Hansen, J.; Edick, M.J.; Walsh Vockley, C.; Leslie, N.; Cameron, C.; Mohsen, A.W.; Berry, S.A.; Arnold, G.L.; et al. Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol. Genet. Metab. 2016, 118, 272–281. [Google Scholar] [CrossRef]
- Merinero, B.; Alcaide, P.; Martin-Hernandez, E.; Morais, A.; Garcia-Silva, M.T.; Quijada-Fraile, P.; Pedron-Giner, C.; Dulin, E.; Yahyaoui, R.; Egea, J.M.; et al. Four Years’ Experience in the Diagnosis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency in Infants Detected in Three Spanish Newborn Screening Centers. JIMD Rep. 2018, 39, 63–74. [Google Scholar] [CrossRef]
- Spiekerkoetter, U. Mitochondrial fatty acid oxidation disorders: Clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J. Inherit. Metab. Dis. 2010, 33, 527–532. [Google Scholar] [CrossRef]
- Hesse, J.; Braun, C.; Behringer, S.; Matysiak, U.; Spiekerkoetter, U.; Tucci, S. The diagnostic challenge in very-long chain acyl-CoA dehydrogenase deficiency (VLCADD). J. Inherit. Metab. Dis. 2018, 41, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, L.; Haussmann, U.; Mueller, M.; Spiekerkoetter, U. VLCAD enzyme activity determinations in newborns identified by screening: A valuable tool for risk assessment. J. Inherit. Metab. Dis. 2012, 35, 269–277. [Google Scholar] [CrossRef]
- Ohlsson, A.; Guthenberg, C.; Holme, E.; von Dobeln, U. Profound biotinidase deficiency: A rare disease among native Swedes. J. Inherit. Metab. Dis. 2010, 33 (Suppl. S3), S175–S180. [Google Scholar] [CrossRef]
- Jay, A.M.; Conway, R.L.; Feldman, G.L.; Nahhas, F.; Spencer, L.; Wolf, B. Outcomes of individuals with profound and partial biotinidase deficiency ascertained by newborn screening in Michigan over 25 years. Genet. Med. 2015, 17, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B. Why screen newborns for profound and partial biotinidase deficiency? Mol. Genet. Metab. 2015, 114, 382–387. [Google Scholar] [CrossRef]
- Wiltink, R.C.; Kruijshaar, M.E.; van Minkelen, R.; Onkenhout, W.; Verheijen, F.W.; Kemper, E.A.; van Spronsen, F.J.; van der Ploeg, A.T.; Niezen-Koning, K.E.; Saris, J.J.; et al. Neonatal screening for profound biotinidase deficiency in the Netherlands: Consequences and considerations. Eur. J. Hum. Genet. 2016, 24, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Gramer, G.; Haege, G.; Fang-Hoffmann, J.; Schwab, K.O.; Tacke, U.; Trefz, F.K.; Mengel, E.; Wendel, U.; Leichsenring, M.; et al. Efficacy and outcome of expanded newborn screening for metabolic diseases—Report of 10 years from South-West Germany. Orphanet J. Rare Dis. 2011, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Couce, M.L.; Castineiras, D.E.; Boveda, M.D.; Bana, A.; Cocho, J.A.; Iglesias, A.J.; Colon, C.; Alonso-Fernandez, J.R.; Fraga, J.M. Evaluation and long-term follow-up of infants with inborn errors of metabolism identified in an expanded screening programme. Mol. Genet. Metab. 2011, 104, 470–475. [Google Scholar] [CrossRef]
- Tal, G.; Pitt, J.; Morrisy, S.; Tzanakos, N.; Boneh, A. An audit of newborn screening procedure: Impact on infants presenting clinically before results are available. Mol. Genet. Metab. 2015, 114, 403–408. [Google Scholar] [CrossRef]
- Refsum, H.; Fredriksen, A.; Meyer, K.; Ueland, P.M.; Kase, B.F. Birth prevalence of homocystinuria. J. Pediatr. 2004, 144, 830–832. [Google Scholar] [CrossRef]
- Estrella, J.; Wilcken, B.; Carpenter, K.; Bhattacharya, K.; Tchan, M.; Wiley, V. Expanded newborn screening in New South Wales: Missed cases. J. Inherit. Metab. Dis. 2014, 37, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Skovby, F.; Gaustadnes, M.; Mudd, S.H. A revisit to the natural history of homocystinuria due to cystathionine beta-synthase deficiency. Mol. Genet. Metab. 2010, 99, 1–3. [Google Scholar] [CrossRef]
- Edmondson, A.C.; Salant, J.; Ierardi-Curto, L.A.; Ficicioglu, C. Missed Newborn Screening Case of Carnitine Palmitoyltransferase-II Deficiency. JIMD Rep. 2017, 33, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Tajima, G.; Hara, K.; Yuasa, M. Carnitine palmitoyltransferase II deficiency with a focus on newborn screening. J. Hum. Genet. 2019, 64, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Puckett, R.L.; Lorey, F.; Rinaldo, P.; Lipson, M.H.; Matern, D.; Sowa, M.E.; Levine, S.; Chang, R.; Wang, R.Y.; Abdenur, J.E. Maple syrup urine disease: Further evidence that newborn screening may fail to identify variant forms. Mol. Genet. Metab. 2010, 100, 136–142. [Google Scholar] [CrossRef]
- Brodtkorb, E.; Strand, J.; Backe, P.H.; Lund, A.M.; Bjoras, M.; Rootwelt, T.; Rootwelt, H.; Woldseth, B.; Eide, L. Four novel mutations identified in Norwegian patients result in intermittent maple syrup urine disease when combined with the R301C mutation. Mol. Genet. Metab. 2010, 100, 324–332. [Google Scholar] [CrossRef]
- Sperk, A.; Mueller, M.; Spiekerkoetter, U. Outcome in six patients with mitochondrial trifunctional protein disorders identified by newborn screening. Mol. Genet. Metab. 2010, 101, 205–207. [Google Scholar] [CrossRef]
- Heringer, J.; Valayannopoulos, V.; Lund, A.M.; Wijburg, F.A.; Freisinger, P.; Baric, I.; Baumgartner, M.R.; Burgard, P.; Burlina, A.B.; Chapman, K.A.; et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J. Inherit. Metab. Dis. 2016, 39, 341–353. [Google Scholar] [CrossRef]


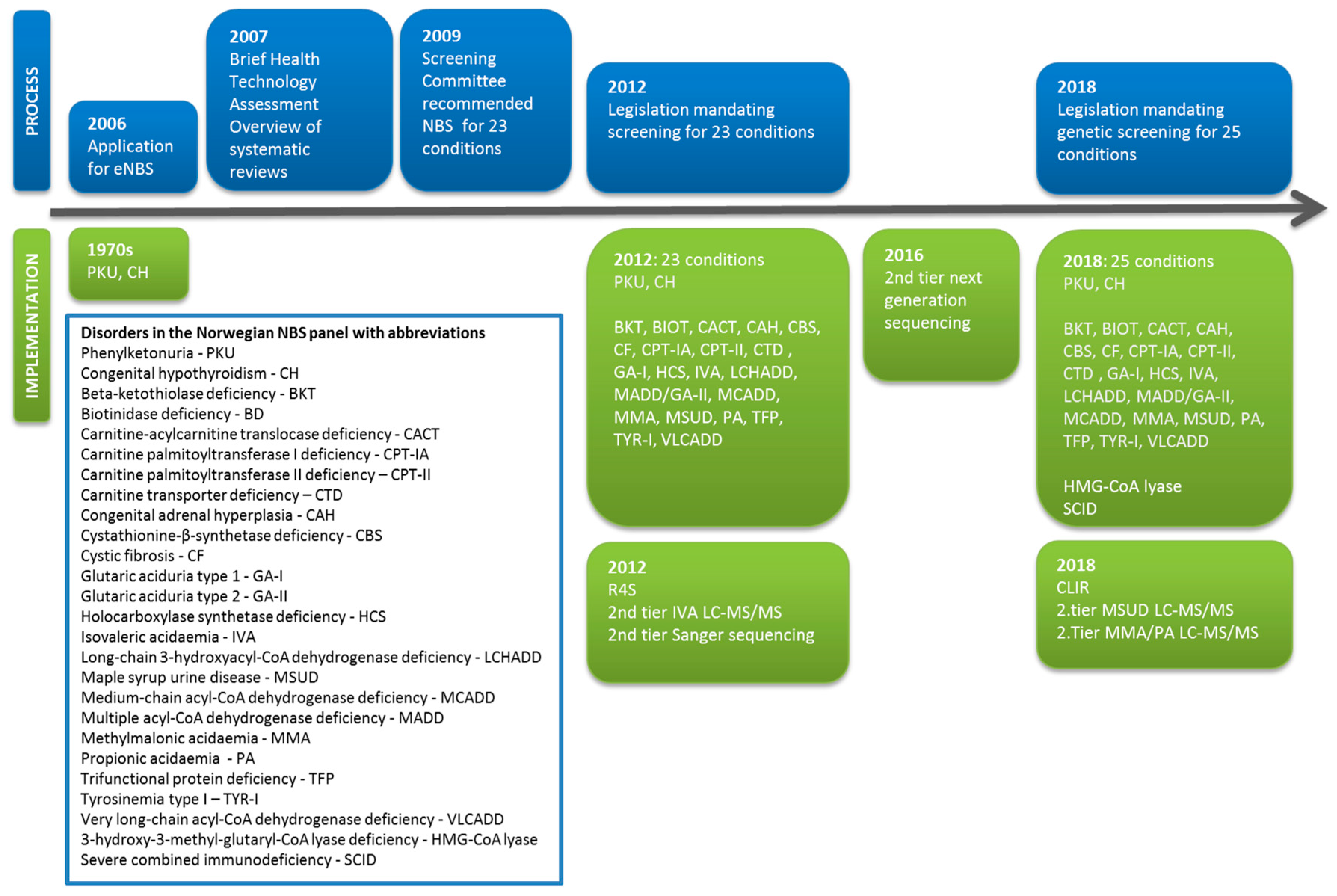{kind=link}
| Condition | Cut-Off Values | Screening Results Values (µmol/L or Ratio) | Molecular Analyses (Start Year) | |
|---|---|---|---|---|
| µmol/L or ratio | True-positive cases | False-positive cases | ||
| Median (range) | Median (range) | |||
| MMA/PA | C3 > 4.75 | 14.1 (10.1–27) | 8.9 (3.3–18.5) | 2016 (NGS) |
| MMA/PA | C3/C2 > 0.25 | 0.74 (0.45–1.16) | 0.27 (0.11–0.40) | |
| PA | C4/C3 < 0.05 | 0.02 (0.01–0.02) | 0.05 (0.02–0.2) | |
| IVA | C5 > 1 | 8.2 (3.4–12.9) | - | 2012 |
| C5/C0 > 0.04 | 0.9 (0.2–1.6) | - | ||
| GA-I | C5DC > 0.4 | 4.25 (2.2–5.4) | - | 2012 |
| C5DC/C16 > 0.1 | 1.12 (0.34–1.22) | - | ||
| MSUD | LEU\ILE\PRO-OH > 250 | 1540 (1300–1790) | 290 (284–296) | 2016 (NGS) |
| LEU\ILE\PRO-OH/ALA > 1.3 | 4.2 (0.84–7.6) | 1.65 (1.55–1.75) | ||
| CBS | MET > 40 | 93.8 (69.1–119.0) | 78.3 (56.7–99.9) | 2014 |
| MET/PHE > 0.7 | 1.74 (1.18–2.3) | 1.23 (0.71–1.80) | ||
| TYR-I | SUAC > 2 | 9.1 (7.5–16.2) | 5.2 (3.1–5.4) | 2013 |
| HCS | C3 > 1.57 | 3.1 (3.1–3.1) | 1.7(1.3–2.2) | 2012 |
| C5OH > 0.85 | 2.38 (2.38–2.38) | 5.49 (1.3–9.7) | ||
| HMG-CoA lyase | C5OH > 0.85 | - | - | 2018 |
| BKT | C5:1 > 0.1 | - | - | 2013 |
| C3DC\C4OH > 0.5 | - | - | ||
| BD | <60 U/dL | 32 (7–58) | 43 (31–57) | 2013 |
| CTD | C0 < 6 | 3.9 (1.7–6.2) | 4.7 (2.5–22) | 2013 |
| C3 + C16 > 2 | 1.48 (1.27–1.61) | 1.94 (0.37–11.6) | ||
| CPT-IA | C0/C16 + C18 > 40 | 277 (277–277) | 41.8 (41.8–41.8) | 2012 |
| C16 + C18:1/C2 < 0.15 | 0.14 (0.014–0.014) | 0.098 (0.098–0.098) | ||
| MCADD | C8 > 0.4 | 10.71 (2.4–28.0) | 0.65 (0.41–0.69) | 2012 |
| CPT-II /CACT | C16 + C18:1/C2 > 0.52 | 1.42 (1.26–10.8) | 0.44 (0.31–0.57) | 2012 |
| C16 > 5.5 | 5.1 (4.64–13.1) | 8.8 (7.8–9.8) | ||
| VLCADD | C14:1 > 0.5 | 1.9 (1.0–2.0) | 1.4 (1.0–2.0) | 2012 |
| C14:1/C2 > 0.02 | 0.11 (0.03–0.18) | 0.06 (0.04–0.07) | ||
| LCHADD | C16OH > 0.1 | 1.6 (1.6–1.6) | 0.14 (0.14–0.14) | 2012 |
| C18OH > 0.1 | 0.92 (0.92–0.92) | 0.35 (0.35–0.35) | ||
| TFP | C16OH > 0.1 | 1.1 (.57–1.6) | - | |
| C18OH > 0.1 | 0.81 (0.62–1.0) | - | ||
| MADD/GA-II | C14:1/C2 > 0.02 | 0.07 (0.05–0.09) | 0.1 (0.06–0.14) | 2016 (NGS) |
| C12 > 0.4 | 1.9 (1.9–1.9) | 2.0 (2.0–2.1) | ||
| 2002–2012 | eNBS 2012–2020 | ||||
|---|---|---|---|---|---|
| Clinically Presenting Cases | True-Positive Cases | False-Positive Cases | |||
| n | Incidence | n | Incidence | n | |
| MMA | 5 | 1:119,318 | 4 * | 1:115,342 | 56 |
| PA | - | - | 3 | 1:153,789 | |
| IVA | 4 | 1:149,147 | 2 | 1:230,684 | - |
| GA-I | 6 | 1:99,431 | 4 | 1:115,342 | - |
| MSUD | 5 | 1:119,318 | 2 | 1:230,684 | 2 |
| CBS | 1 | 1:596,591 | 2 | 1:230,684 | 2 |
| TYR-I | 8 | 1:74,573 | 5 | 1:92,273 | 3 |
| HCS | 2 | 1:298,295 | 1 | 1:461,369 | 2 |
| HMG-CoA lyase | - | - | -a | - | - |
| BKT | 1 | 1:596,591 | - | - | - |
| BD | - | - | 13 | 1:35,489 | 5 |
| CTD | - | - | 3 | 1:153,789 | 22 |
| CPT-IA | - | - | 1 | 1:461,369 | 1 |
| MCADD | 8 | 1:74,573 | 17 | 1:27,139 | 4 |
| CPT-II | - | - | 4 | 1:115,342 | 2 |
| CACT | 2 | 1:298,295 | 1 | 1:461,369 | - |
| VLCADD | 1 | 1:596,591 | 6 | 1:76,894 | 5 |
| LCHADD | 4 | 1:149,147 | 1 | 1:461,369 | 1 |
| TFP | 3 | 1:198,863 | 4 | 1:115,342 | - |
| MADD/GA-II | 2 | 1:298,295 | 2 | 1:230,684 | 2 |
| Total number | 52 | 1:11 472 | 75 | 1:6151 | 107 |
| Condition | Age at Presentation (Days) | Age at Final Diagnosis (Days) | Clinical and Biochemical Findings | Mode of First Detection |
|---|---|---|---|---|
| MMA | 2 | 2 | Lethargy, metabolic acidosis, hyperammonemia (260 µmol/L) | TD |
| MMA | 2 | 6 | Encephalopathy, hypoglycemia, metabolic acidosis, bulging fontanel hyperammonemia (1400 µmol/L) | NBS |
| PA | 3 | 4 | Encephalopathy, vomiting, metabolic acidosis, hyperammonemia (372 µmol/L) | TD |
| PA | 3 | 4 | Encephalopathy, vomiting, metabolic acidosis, hyperammonemia (740 µmol/L) | TD |
| PA | 3 | 4 | Encephalopathy, metabolic acidosis, seizures, hyperammonemia (1400 µmol/L) | NBS |
| IVA | 3 | 4 | Encephalopathy, metabolic acidosis, seizures, hyperammonemia (769 µmol/L) | TD |
| MSUD | 3 | 5 | Encephalopathy, abnormal movements, seizures (leucine 2560 µmol/L) | NBS |
| MSUD | 5 | 5 | Subtle encephalopathy, abnormal movements (leucine 2200 µmol/L) | NBS |
| CPT-IA | 1 | 74 * | Hypoglycemia, lactic acidosis | NBS |
| MCADD | 2 | 6 | Severe hypoglycemia (p-glucose 0.1 mmol/l) with MRI correlate | NBS |
| CPT-II | 0 | 7 | Multi-organ failure from birth (microgyria, renal failure, cardiomyopathy) | NBS |
| VLCADD | 1 | 6 | Hypoglycemia, lactic acidosis, CK 10 000 U/L at 24 h of age. | NBS |
| TFP | 1 | 9 | Heart failure (dilated cardiomyopathy), respiratory distress | NBS |
| TFP | 0 | 5 | Heart failure (dilated cardiomyopathy), respiratory distress | TD |
| MADD/GA-II | 4 | 704 | Encephalopathy, metabolic acidosis, respiratory distress, hyperammonemia (740 µmol/L) | NBS |
| MADD/GA-II | 1 | 4 | Lethargy, hypoglycemia, lactic acidosis, hypoglycemia, arrhythmia | NBS |
| ID | Enzyme | Condition | Reference Sequence | Allele 1 | ACMG | Allele 2 | ACMG |
|---|---|---|---|---|---|---|---|
| Organic acidurias | |||||||
| 1 | F Cbl a | MMA | NM_052845.3(MMAB) | c.291–1G > A (splice defect) | 5 | c.571C > T (p.Arg191Trp) | 5 |
| 2 | F MUT b | MMA | NM_000255.4(MMUT) | c.675_677delTAT (p.Phe225_Met226delinsLeu) | 5 | c.1106G > A (p.Arg369His) | 5 |
| 3 | NP | MMA | NM_000255.4(MMUT) | c.1655C > T (p.Ala552Val) | 5 | c.1677-1G > A (splice defect) | 5 |
| 4 | NP | Cbl C | NM_015506.2(MMACHC) | c.271dupA (p.Arg91Lysfs*14) | 5 | c.271dupA (p.Arg91Lysfs*14) | 5 |
| 5 | F c | PA | NM_000532.4(PCCB) | c.319G > A (p.Val107Met) | 4 | c.1281_1282delCA (p.Thr428Serfs*12) | 5 |
| 6 | NP | PA | NM_000532.4(PCCB) | c.331C > T (p.Arg111 *) | 5 | c.838dup (p.Leu280Profs*11) | 5 |
| 7 | NP | PA | NM_000532.4(PCCB) | c.1498 + 2T>C (splice defect) | 5 | c.1498 + 2T>C (splice defect) | 5 |
| 8 | L d | IVA | NM_002225.3(IVD) | c.208G>T (p.Glu70 *) | 5 | c.941C>T (p.Ala314Val) | 5 |
| 9 | NP | IVA | NM_002225.3(IVD) | c.296-2A>G (splice defect) | 4 | c.296-2A > G (splice defect) | 4 |
| 10 | NP | GA-I | NM_000159.2(GCDH) | c.572T>C (p.Met191Thr) | 5 | c.1045G>A (p.Ala349Thr) | 5 |
| 11 | NP | GA-I | NM_000159.3(GCDH) | c.1045G>A (p.Ala349Thr) | 5 | c.1204C>T (p.Arg402Trp) | 5 |
| 12 | NP | GA-I | NM_000159.3(GCDH) | c.1240G>A (p.Glu414Lys) | 5 | c.1240G>A (p.Glu414Lys) | 5 |
| 13 | NP | GA-I | NM_000159.3(GCDH) | c.1240G>A (p.Glu414Lys) | 5 | c.1240G>A (p.Glu414Lys) | 5 |
| 14 | NP | HCS | NM_000411.6(HLCS) | c.1519 + 5G > A(splice defect) | 5 | c.1993C > T (p.Arg665 *) | 5 |
| Aminoacidopathies | |||||||
| 15 | 4.5% e | MSUD | ND | ND | |||
| 16 | NP | MSUD | NM_000709.3(BCKDHA) | c.375 + 648_484 + 520del p.Gly126Valfs*3 (ref below) | 5 | c.375 + 648_484 + 520del p.Gly126Valfs*3 (ref below) | 5 |
| 17 | NP | MSUD Ɨ | NM_001918.3(DBT) | c.901C>T (p.Arg301Cys) | 5 | c.1291C>T (p.Arg 431 *) | 5 |
| 18 | NP | MSUD Ɨ | NM_001918.3(DBT) | c.901C>T (p.Arg301Cys) | 5 | c.1291C>T (p.Arg 431 *) | 5 |
| 19 | P f | CBS | NM_000071.2(CBS) | c.451 + 2T>G (splice defect) | 4 | c.833T>C (p.Ile278Thr) | 5 |
| 20 | NP | CBS | NM_000071.2(CBS) | c.728A>G (p.Gln243Arg) | 4 | c.728A>G (p.Gln243Arg) | 4 |
| 21 | NP | TYR-I | NM_000137.2(FAH) | c.554-1G>T (splice defect) | 5 | c.1062 + 5G>A (splice defect) | 5 |
| 22 | NP | TYR-I | NM_000137.2(FAH) | c.742delG (p.Pro249Hisfs*55) | 5 | c.1062 + 5G>A (splice defect) | 5 |
| 23 | NP | TYR-I | NM_000137.2(FAH) | c.742delG (p.Pro249Hisfs*55) | 5 | c.1062 + 5G>A (splice defect) | 5 |
| 24 | NP | TYR-I | NM_000137.2(FAH) | c.1008C>G (p.Asn336Lys) | 4 | c.1062 + 5G>A (splice defect) | 5 |
| 25 | NP | TYR-I | NM_000137.2(FAH) | c.1062 + 5G>A (splice defect) | 5 | c.1062 + 5G>A (splice defect) | 5 |
| 26 | S16% g | BD | NM_000060.2(BTD) | c.235C>T (p.Arg79Cys) | 5 | c.1330G>C (p.Asp444His) | 5 |
| 27 | S22% g | BD | NM_000060.2(BTD) | c.278A>G (p.Tyr93Cys) | 5 | c.1330G>C (p.Asp444His) | 5 |
| 8 | S2% g | BD | NM_000060.2(BTD) | c.424C>A (p.Pro142Thr) | 4 | c.424C>A (p.Pro142Thr) | 4 |
| 29 | S11% g | BD | NM_000060.3(BTD) | c.470G>A (p.Arg157His) | 5 | c.470G>A (p.Arg157His) | 5 |
| 30 | S18% g | BD | NM_000060.3(BTD) | c.470G>A (p.Arg157His) | 5 | c.1330G>C (p.Asp444His) | 5 |
| 31 | S6% g | BD | NM_000060.2(BTD) | c.470G>A (p.Arg157His) | 5 | c.1333G>A (p.Gly445Arg) | 5 |
| 32 | S23% g | BD | NM_000060.3(BTD) | c.511G>A (p.Ala171Thr) | 5 | c.1330G>C (p.Asp444His) | 5 |
| 33 | S15% g | BD | NM_000060.3(BTD) | c.511G>A (p.Ala171Thr) | 5 | c.1330G>C (p.Asp444His) | 5 |
| 34 | S11% g | BD | NM_000060.3(BTD) | c.605A>T (p.Asn202Ile) | 5 | c.605A>T (p.Asn202Ile) | 5 |
| 35 | S7% g | BD | NM_000060.2(BTD) | c.1006C > T (p.Gln336 *) | 5 | c.1006C > T (p.Gln336 *) | 5 |
| 36 | S < 1% g | BD | NM_000060.2(BTD) | c.1006C > T (p.Gln336 *) | 5 | c.1006C > T (p.Gln336 *) | 5 |
| 37 | S27% g | BD | NM_000060.3(BTD) | c.1330G>C (p.Asp444His) | 5 | c.1368A>C (p.Gln456His) | 5 |
| 38 | S8% g | BD | NM_000060.2(BTD) | c.626G>A (p.Arg209His) | 5 | c.1595C>T (p.Thr532Met) | 5 |
| Fatty acid oxidation defects | |||||||
| 39 | NP | CTD | NM_003060.3(SLC22A5) | c.51C>G (p.Phe17Leu) | 5 | c.136C>T (P.Pro46Ser) | 5 |
| 40 | F12% h | CTD Ɨ | NM_003060.3(SLC22A5) | c.136C>T (p.Pro46Ser) | 5 | c.136C>T (p.Pro46Ser) | 5 |
| 41 | NP | CTD Ɨ | NM_003060.3(SLC22A5) | c.136C>T (p.Pro46Ser) | 5 | c.136C>T (p.Pro46Ser) | 5 |
| 42 | NP | CTD | NM_003060.3(SLC22A5) | c.136C>T (p.Pro46Ser) | 5 | c.844C>T (p.Arg282 *) | 5 |
| 43 | NP | CTD | NM_003060.3(SLC22A5) | c.847T>A (p.Trp283Arg) | 4 | c.847T>A (p.Trp283Arg) | 4 |
| 44 | F i | CPT-IA | NM_001031847.2(CPT1A) | c.619C > T (p.Gln207 *) | 5 | c.2215A > G (p.Lys739Glu) | 4 |
| 45 | 14% j | MCADD | NM_000016.4(ACADM) | c.250C>T (p.Leu84Phe) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 46 | 11% j | MCADD | NM_000016.4(ACADM) | c.250C>T (p.Leu84Phe) | 5 | c.985A >G (p.Lys329Glu) | 5 |
| 47 | <1% j | MCADD | NM_000016.4(ACADM) | c.362C>T (p.Thr121Ile) | 5 | c.362C>T (p.Thr121Ile) | 5 |
| 48 | 6% j | MCADD | NM_000016.4(ACADM) | c.388-19T > A (intronic) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 49 | NP | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 50 | NP | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 51 | NP | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 52 | NP | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 53 | <1% j | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 54 | <1% j | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 55 | 3% j | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 56 | <1% j | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 57 | <1% j | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 58 | NP | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.985A>G (p.Lys329Glu) | 5 |
| 59 | <1% j | MCADD | NM_000016.4(ACADM) | c.985A>G (p.Lys329Glu) | 5 | c.1171A>G (p.Met391Val) | 3+ |
| 60 | NP | MCADD | NM_000016.4(ACADM) | c.244dup (p.Trp82Leufs*23) | 5 | c.244dup (p.Trp82Leufs*23) | 5 |
| 61 | NP | MCADD | NM_000016.4(ACADM) | c.244dup (p.Trp82Leufs*23) | 5 | c.244dup (p.Trp82Leufs*23) | 5 |
| 62 | L k | CPT-II | NM_000098.2(CPT2) | c.149C>A (p.Pro50His) | 5 | c.149C>A (p.Pro50His) | 5 |
| 63 | L k | CPT-II | NM_000098.2(CPT2) | c.149C>A (p.Pro50His) | 5 | c.1369A>T (p.Lys457 *) | 5 |
| 64 | NP | CPT-II Ɨ | NM_000098.2(CPT2) | c.338C>T (p.Ser113Leu) | 5 | c.481C>T (p.Arg161Trp) | 4 |
| 65 | L k | CPT-II | NM_000098.2(CPT2) | c.338C>T (p.Ser113Leu) | 5 | c.1444_1447del (p.Thr482Trpfs*49) | 5 |
| 66 | NP | CPT-II | NM_000098.2(CPT2) | c.1798G>A (p.Gly600Arg) | 4 | c.1798G>A (p.Gly600Arg) | 4 |
| 67 | F l | CACT | NM_000387.5(SLC25A20) | c.82G>T (p.Gly28Cys) | 5 | c.82G>T (p.Gly28Cys) | 5 |
| 68 | 15% m | VLCADD | NM_000018.3(ACADVL) | c.533T>C (p.Leu178Pro) | 5 | c.1066A>G (p.Ile356Val) | 3 |
| 69 | 17% m | VLCADD | NM_000018.3(ACADVL) | c.848T>C (p.Val283Ala) | 5 | c.848T>C (p.Val283Ala) | 5 |
| 70 | 12% m | VLCADD | NM_000018.3(ACADVL) | c.848T>C (p.Val283Ala) | 5 | c.848T>C (p.Val283Ala) | 5 |
| 71 | 7% m | VLCADD | NM_000018.3(ACADVL) | c.848T>C (p.Val283Ala) | 5 | c.865G>A (p.Gly289Arg) | 5 |
| 72 | 9% m | VLCADD | NM_000018.3(ACADVL) | c.848T>C (p.Val283Ala) | 5 | c.1177A>G (p.Thr393Ala) | 3+ |
| 73 | <1% m | VLCADD | NM_000018.3(ACADVL) | c.1837C>T (p.Arg613Trp) | 5 | c.1837C>T (p.Arg613Trp) | 5 |
| 74 | NP | LCHADD | NM_000182.4(HADHA) | c.1528G>C (p.Glu510Gln) | 5 | c.1528G>C (p.Glu510Gln) | 5 |
| 75 | L n | TFP | NM_000182.4(HADHA) | c.1678C>T (p.Arg560 *) | 5 | c.1678C>T (p.Arg560 *) | 5 |
| 76 | NP | TFP | NM_000182.4(HADHA) | c.1678C>T (p.Arg560 *) | 5 | c.1678C>T (p.Arg560 *) | 5 |
| 77 | NP | TFP | NM_000183.2(HADHB) | c.209 + 1G>C (splice defect) | 5 | c.255-1G>A (splice defect) | 5 |
| 78 | L o | TFP | NM_000182.4(HADHA) | c.180 + 3A>G (splice defect) | 5 | c.180 + 3A>G (splice defect) | 5 |
| 79 | F p | MADD/GA-II | NM_017986.3(SLC52A1) | c.1134 + 11G>A (intronic) | 1 | wild type | |
| 80 | F22% q | MADD/GA-II | NM_000126.3(ETFA) | c.348A>T (splice defect) | 3+ | c.348A>T (splice defect) | 3+ |
| Condition | False Negatives | False Positives | True Positives | |||
|---|---|---|---|---|---|---|
| BIOT | 5 samples | percentiles: 22 (15–25) | 13 samples | percentiles: 27 (16–86) | ||
| scores: 128 (47–170) | scores: 188 (54–472) | |||||
| CPT-IA | 1 sample | percentiles: 0 (0–0) | 1 sample | percentiles: 57 (57–57) | ||
| scores: 0 (0–0) | scores: 553 (553–553) | |||||
| CPT-II/CACT | 1 sample | percentiles: 0 (0–0) | 2 samples | percentiles: 0 (0–0) | 5 samples | percentiles: 9 (0–82) |
| scores: 1 (1–1) | scores: 11.5 (0–23) | scores: 166 (1–754) | ||||
| CUD | 2 samples | percentiles: 27 (19–35) | 23 samples | percentiles: 39 (0–96) | 3 samples | percentiles: 53 (38–88) |
| scores: 70.5 (54–87) | scores: 97 (0–259) | scores: 128 (95–213) | ||||
| GA-I | 4 samples | percentiles: 61 (27–67) | ||||
| scores: 461 (175–504) | ||||||
| GA-II | 2 samples | percentiles: 37.5 (36–39) | 2 samples | percentiles: 40 (38–42) | ||
| scores: 404 (370–439) | scores: 452 (415–490) | |||||
| CBS | 2 samples | percentiles: 13.5 (0–27) | 2 samples | percentiles: 18 (4–32) | ||
| scores: 71.5 (18–125) | scores: 108 (32–184) | |||||
| IVA | 2 samples | percentiles: 61 (46–76) | ||||
| scores: 416 (286–546) | ||||||
| LCHADD/TFP | 1 sample | percentiles: 0 (0–0) | 5 samples | percentiles: 34 (2–87) | ||
| scores: 39 (39–39) | scores: 459 (132–760) | |||||
| MCADD | 4 samples | percentiles: 1.5 (0–2) | 16 samples | percentiles: 81.5 (25–99) | ||
| scores: 14.5 (3–24) | scores: 888 (265–1010) | |||||
| HCS | 2 samples | percentiles: 50 (0–100) | 1 sample | percentiles: 27 (27–27) | ||
| scores: 331 (83–579) | scores: 180 (180–180) | |||||
| MMA/PA/CblC | 56 samples | percentiles: 13 (0–29) | 7 samples | percentiles: 31 (24–91) | ||
| scores: 91.5 (0–252) | scores: 281 (194–683) | |||||
| MSUD | 2 samples | percentiles: 0 (0–0) | 2 samples | percentiles: 10 (6–14) | 2 samples | percentiles: 57 (37–77) |
| scores: 0 (0–0) | scores: 60.5 (36–85) | scores: 393 (255–531) | ||||
| TYR-I | 3 samples | percentiles: 20 (4–20) | 5 samples | percentiles: 23 (20–65) | ||
| scores: 83 (14–83) | scores: 102 (79–195) | |||||
| VLCADD | 5 samples | percentiles: 20 (5–34) | 6 samples | percentiles: 52 (11–66) | ||
| scores: 129 (63–214) | scores: 312 (94–418) | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tangeraas, T.; Sæves, I.; Klingenberg, C.; Jørgensen, J.; Kristensen, E.; Gunnarsdottir, G.; Hansen, E.V.; Strand, J.; Lundman, E.; Ferdinandusse, S.; et al. Performance of Expanded Newborn Screening in Norway Supported by Post-Analytical Bioinformatics Tools and Rapid Second-Tier DNA Analyses. Int. J. Neonatal Screen. 2020, 6, 51. https://doi.org/10.3390/ijns6030051
Tangeraas T, Sæves I, Klingenberg C, Jørgensen J, Kristensen E, Gunnarsdottir G, Hansen EV, Strand J, Lundman E, Ferdinandusse S, et al. Performance of Expanded Newborn Screening in Norway Supported by Post-Analytical Bioinformatics Tools and Rapid Second-Tier DNA Analyses. International Journal of Neonatal Screening. 2020; 6(3):51. https://doi.org/10.3390/ijns6030051
Chicago/Turabian StyleTangeraas, Trine, Ingjerd Sæves, Claus Klingenberg, Jens Jørgensen, Erle Kristensen, Gunnþórunn Gunnarsdottir, Eirik Vangsøy Hansen, Janne Strand, Emma Lundman, Sacha Ferdinandusse, and et al. 2020. "Performance of Expanded Newborn Screening in Norway Supported by Post-Analytical Bioinformatics Tools and Rapid Second-Tier DNA Analyses" International Journal of Neonatal Screening 6, no. 3: 51. https://doi.org/10.3390/ijns6030051
APA StyleTangeraas, T., Sæves, I., Klingenberg, C., Jørgensen, J., Kristensen, E., Gunnarsdottir, G., Hansen, E. V., Strand, J., Lundman, E., Ferdinandusse, S., Salvador, C. L., Woldseth, B., Bliksrud, Y. T., Sagredo, C., Olsen, Ø. E., Berge, M. C., Trømborg, A. K., Ziegler, A., Zhang, J. H., ... Pettersen, R. D. (2020). Performance of Expanded Newborn Screening in Norway Supported by Post-Analytical Bioinformatics Tools and Rapid Second-Tier DNA Analyses. International Journal of Neonatal Screening, 6(3), 51. https://doi.org/10.3390/ijns6030051