A Multicentre Pilot Study of a Two-Tier Newborn Sickle Cell Disease Screening Procedure with a First Tier Based on a Fully Automated MALDI-TOF MS Platform


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Definition of Corrected Phenotypes
2.3. Sample Processing and Analysis
2.4. Sample Preparation for MS Measurements
2.5. Mass Spectrometry Measurements
2.6. Data Processing
2.7. Analytical Data Flow
2.8. Visual Assessment of MS Profiles
2.9. The Data Collector
3. Results
3.1. Optimization of Preanalytical and Analytical Procedures
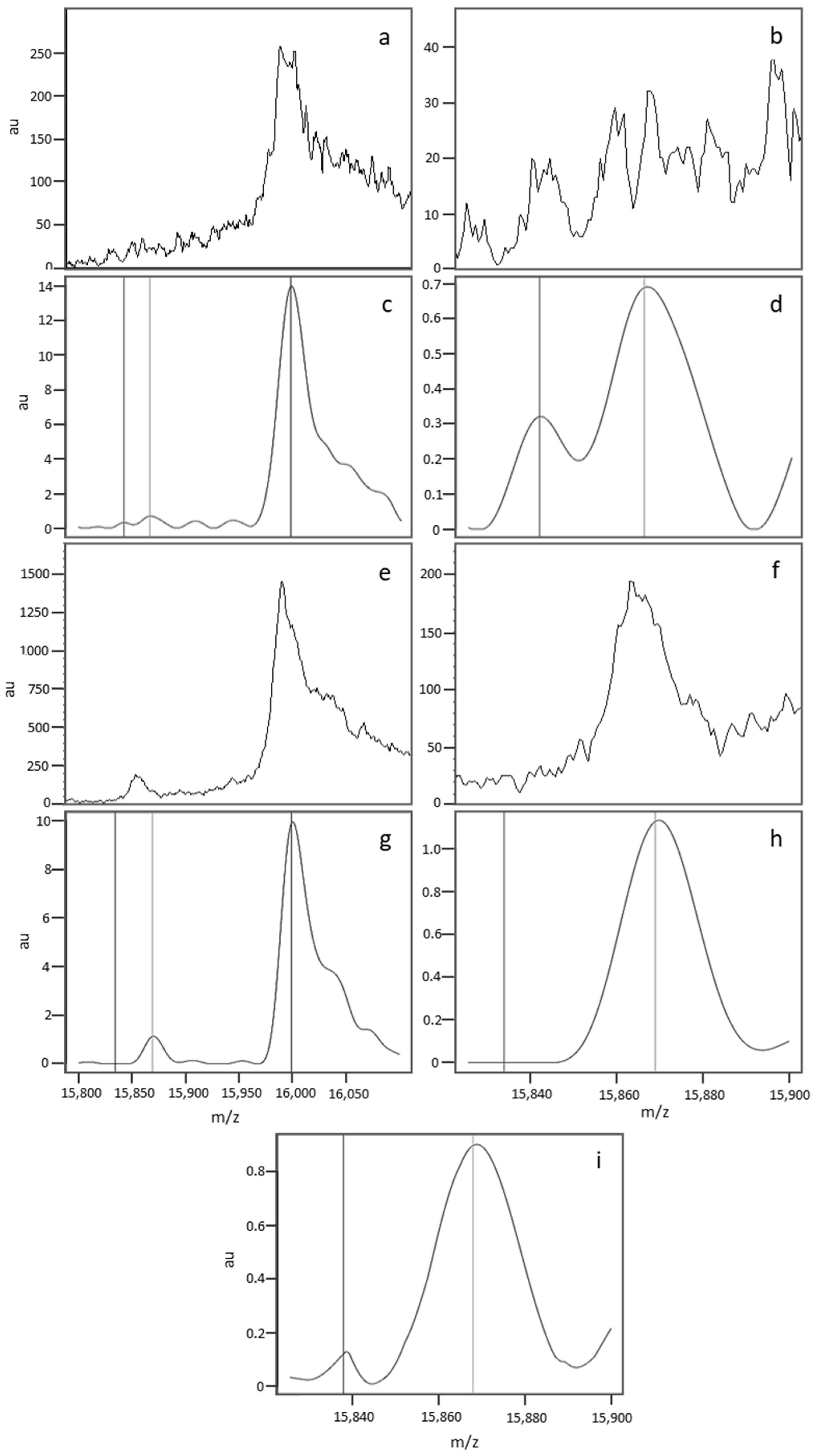
3.2. Spectral Quality of MALDI-TOF Data
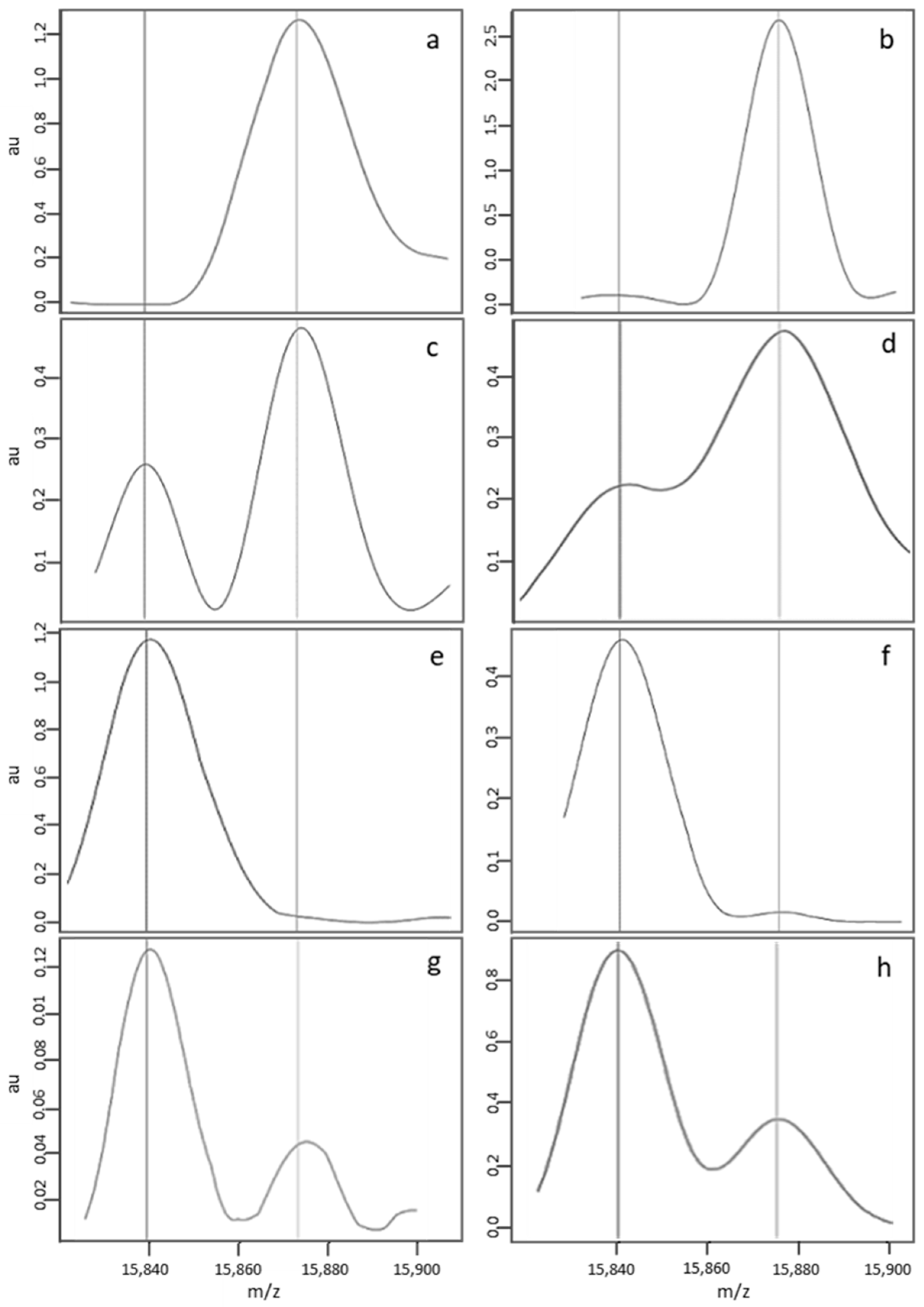
3.2.1. Criteria Used to Define a Standard MALDI-TOF Profile
3.2.2. Classification by the Algorithm of “Standard” Profiles, as a Function of the Newborn’s Corrected Phenotype
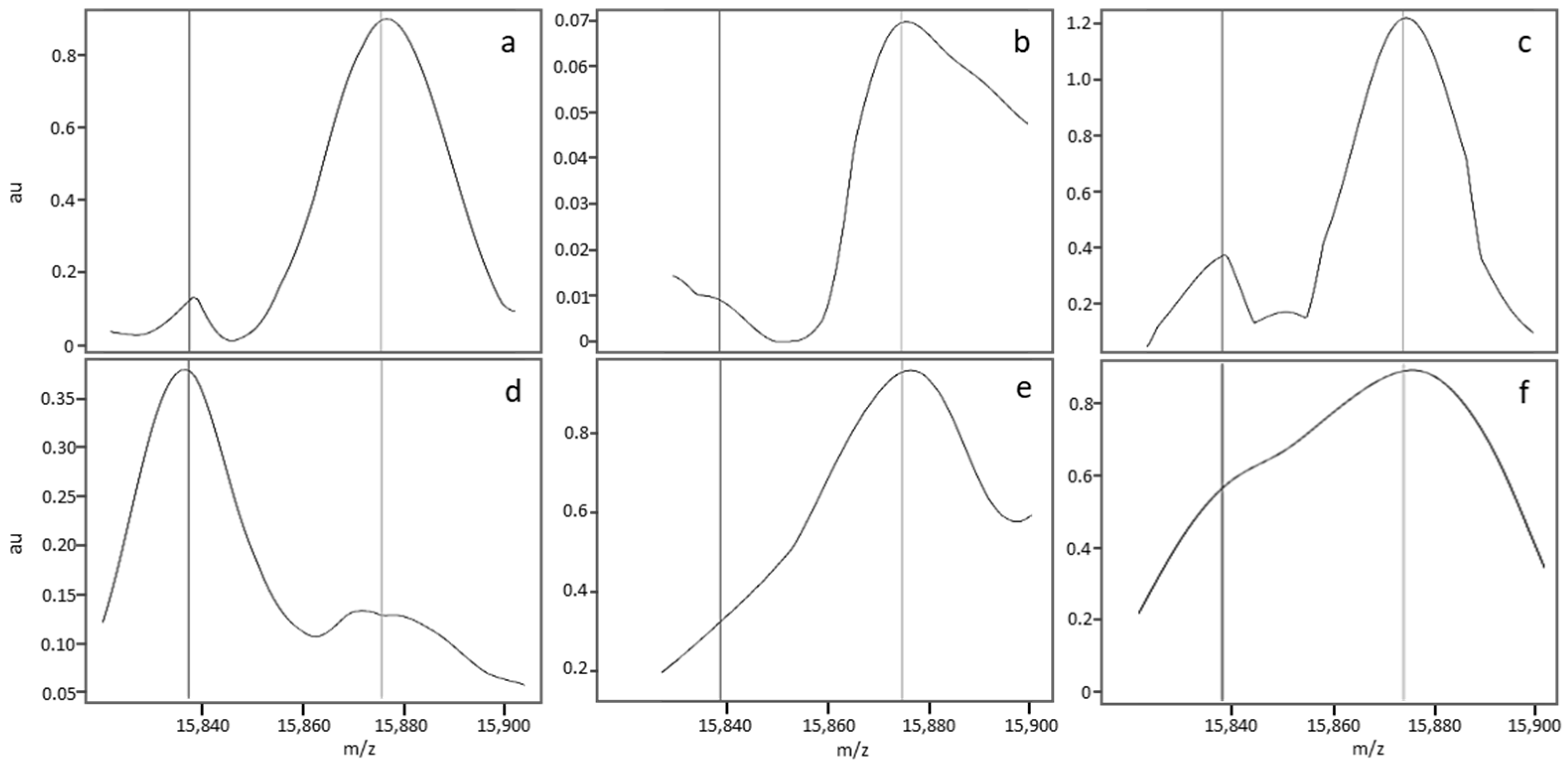
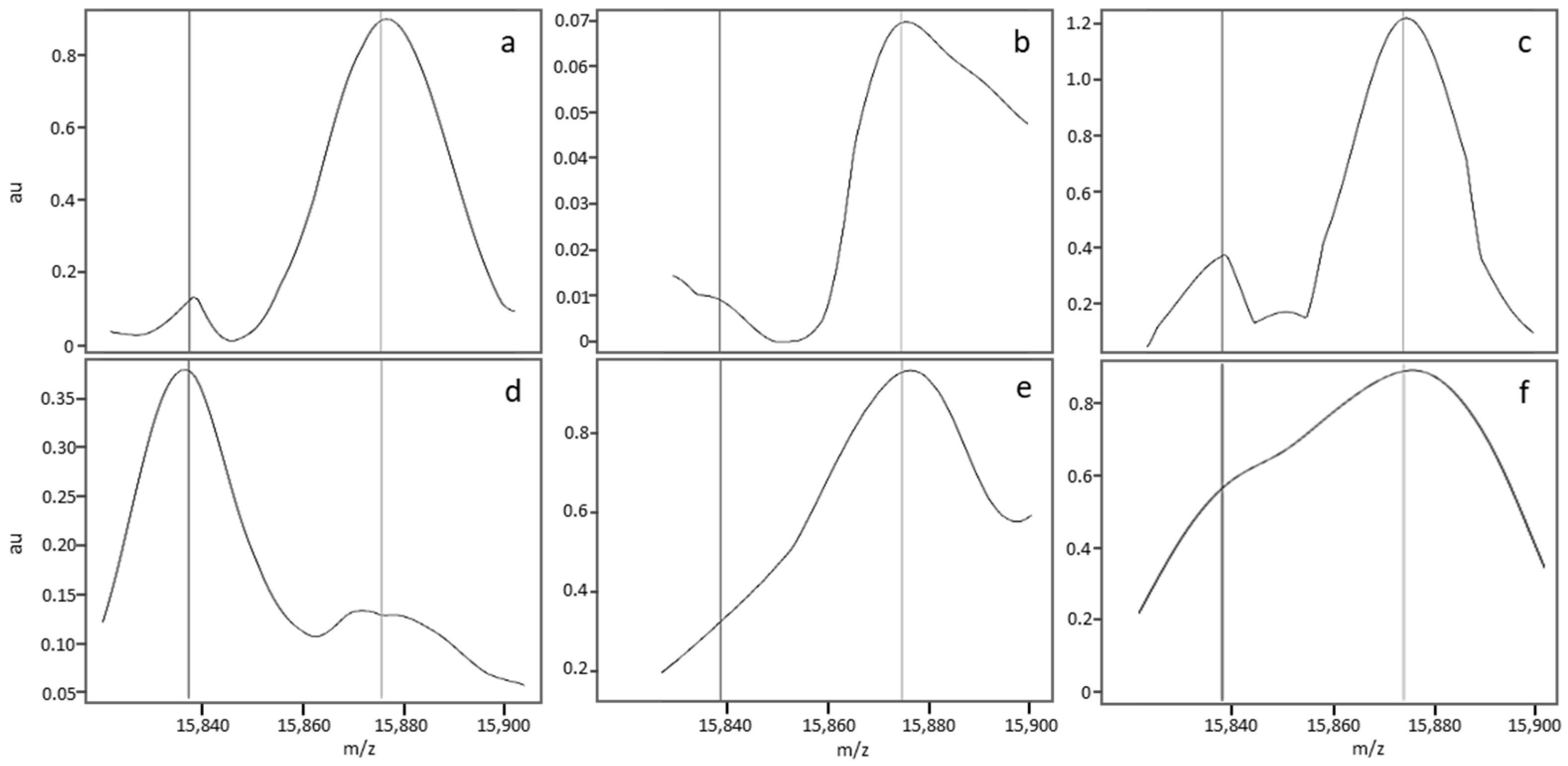
3.2.3. Description of a Nonstandard MALDI-MS Profile
3.2.4. Classification by the Algorithm of Profiles Considered to be Nonstandard in a Visual Assessment, as a Function of the Newborn’s Corrected Phenotype
3.2.5. Causes of Abnormal Spectral Features in Nonstandard MS Profiles
4. Occasional Misclassifications
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Rai, D.K.; Griffiths, W.J.; Landin, B.; Wild, B.J.; Alvelius, G.; Green, B.N. Accurate mass measurement by electrospray ionization quadrupole mass spectrometry: Detection of variants differing by <6 Da from normal in human hemoglobin heterozygotes. Anal. Chem. 2003, 75, 1978–1982. [Google Scholar] [CrossRef] [PubMed]
- Wild, B.J.; Green, B.N.; Cooper, E.K.; Lalloz, M.R.; Erten, S.; Stephens, A.D.; Layton, D.M. Rapid identification of hemoglobin variants by electrospray ionization mass spectrometry. Blood Cells Mol. Dis. 2001, 27, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Wild, B.J.; Green, B.N.; Stephens, A.D. The potential of electrospray ionization mass spectrometry for the diagnosis of hemoglobin variants found in newborn screening. Blood Cells Mol. Dis. 2004, 33, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, U.A.; Black, J.A.; Williams, P.; Nelson, R.W. High-throughput analysis of hemoglobin from neonates using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin. Chem. 2002, 48, 947–949. [Google Scholar] [PubMed]
- Hachani, J.; Duban-Deweer, S.; Pottiez, G.; Renom, G.; Flahaut, C.; Périni, J.M. MALDI-TOF MS profiling as the first-tier screen for sickle cell disease in neonates: Matching throughput to objectives. Proteomics Clin. Appl. 2011, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Daniel, Y.A.; Turner, C.; Haynes, R.M.; Hunt, B.J.; Dalton, R.N. Rapid and specific detection of clinically significant haemoglobinopathies using electrospray mass spectrometry-mass spectrometry. Br. J. Haematol. 2005, 130, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Boemer, F.; Ketelslegers, O.; Minon, J.M.; Bours, V.; Schoos, R. Newborn screening for sickle cell disease using tandem mass spectrometry. Clin. Chem. 2008, 54, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Moat, S.J.; Rees, D.; King, L.; Ifederu, A.; Harvey, K.; Hall, K.; Lloyd, G.; Morrell, C.; Hillier, S. Newborn blood spot screening for sickle cell disease by using tandem mass spectrometry: Implementation of a protocol to identify only the disease states of sickle cell disease. Clin. Chem. 2014, 60, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.L.; Griffiths, P.; Bunch, J.; Cooper, H.J. Compound heterozygotes and beta-thalassemia: Top-down mass spectrometry for detection of hemoglobinopathies. Proteomics 2014, 14, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.L.; Griffiths, P.; Bunch, J.; Cooper, H.J. Top-down proteomics and direct surface sampling of neonatal dried blood spots: Diagnosis of unknown hemoglobin variants. J. Am. Soc. Mass Spectrom. 2012, 23, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.L.; Creese, A.J.; Baumert, M.; Griffiths, P.; Bunch, J.; Cooper, H.J. Hemoglobin variant analysis via direct surface sampling of dried blood spots coupled with high-resolution mass spectrometry. Anal. Chem. 2011, 15, 2265–2270. [Google Scholar] [CrossRef] [PubMed]
- Lobitz, S.; Telfer, P.; Cela, E.; Allaf, B.; Angastiniotis, M.; Backman Johansson, C.; Badens, C.; Bento, C.; Bouva, M.J.; Canatan, D.; et al. Newborn screening for sickle cell disease in Europe: Recommendations from a Pan-European consensus conference. Br. J. Haematol. 2018. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics of the Neonates and Samples | ||||||||
|---|---|---|---|---|---|---|---|---|
| Phenotype | Number | Term | Sample Collection Date | |||||
| 23–32 weeks | 33–36 weeks | >37 weeks | 3–5 day | 6–10 day | 11–30 day | >30 day | ||
| FS | 71 | 0 | 5 | 66 | 67 | 3 | 1 | 0 |
| F/A | 2834 | 203 | 809 | 1822 | 2651 | 90 | 77 | 16 |
| F/AC | 576 | 13 | 50 | 513 | 547 | 20 | 2 | 7 |
| F/AE | 141 | 2 | 9 | 130 | 138 | 2 | 0 | 1 |
| F/AO-Arab | 21 | 0 | 2 | 19 | 19 | 2 | 0 | 0 |
| F/AD | 17 | 0 | 0 | 17 | 17 | 0 | 0 | 0 |
| F/AKorle-Bu | 10 | 0 | 0 | 10 | 10 | 0 | 0 | 0 |
| F/AX | 86 | 4 | 3 | 79 | 81 | 3 | 0 | 2 |
| F/C | 7 | 0 | 1 | 6 | 6 | 0 | 0 | 1 |
| F/E | 3 | 0 | 0 | 3 | 2 | 1 | 0 | 0 |
| F/O-Arab | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
| Corrected FA | 3696 | 222 | 874 | 2600 | 3472 | 118 | 79 | |
| F/AS | 2894 | 57 | 247 | 2590 | 2763 | 89 | 23 | 19 |
| F/SC | 23 | 0 | 1 | 22 | 22 | 1 | 0 | 0 |
| F/SE | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
| F/SO-Arab | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| Corrected FAS | 2919 | 57 | 249 | 2613 | 2787 | 90 | 23 | 19 |
| S-β+-thalassaemia | 15 | 0 | 1 | 14 | 15 | 0 | 0 | 0 |
| Lille MS Facility | Dijon MS Facility | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Standard Profiles | 6181 | 5698 | ||||||||||
| Corrected Phenotype | FA | FAS | FS | FA | FAS | FS | ||||||
| Number of samples | 3386 | 2753 | 37 | 3161 | 2507 | 35 | ||||||
| classification by the algorithm | + | − | + | − | + | − | + | − | + | − | + | − |
| 3386 | 0 | 2751 | 2 | 37 | 0 | 3161 | 0 | 2490 | 17 | 35 | 0 | |
| Corrected Phenotypes | |||||
|---|---|---|---|---|---|
| FA | FAS | FS | |||
| MALDI-TOF MS classification of non-standard profiles | Lille | FA | 282 (54.7%) | 30 (5.9%) | 0 (0%) |
| FAS | 7 (1.4%) | 118 (23.2%) | 0 (0%) | ||
| FS | 0 (0%) | 1 (0.2%) | 34 (6.7%) | ||
| IN | 21 (4.5%) | 17 (3.4%) | 0 (0%) | ||
| Dijon | FA | 446 (45%) | 22 (2.2%) | 0 (0%) | |
| FAS | 21 (2.2%) | 323 (32.7%) | 1 (0.2%) | ||
| FS | 0 (0%) | 1 (0.2%) | 36 (3.7%) | ||
| IN | 68 (7.2%) | 67 (6.8%) | 0 (0%) | ||
| A | Corrected Phenoztype | B | Correctly Classified | Misclassified | ||||
|---|---|---|---|---|---|---|---|---|
| FA | FAS | FS | ||||||
| Lille | Cause 1 | 179 (37%) | 90 (19%) | 3 (0%) | Lille | Cause 1 | 252 (94%) | 17 (6%) |
| Cause 2 | 98 (21%) | 28 (5%) | 0 (0%) | Cause 2 | 105 (84%) | 21 (16%) | ||
| Cause 3 | 12 (3%) | 31 (7%) | 31 (7%) | Cause 3 | 77 (100%) | 0 (0%) | ||
| Dijon | Cause 1 | 193 (23%) | 69 (8%) | 0 (0%) | Dijon | Cause 1 | 248 (95%) | 14 (5%) |
| Cause 2 | 103 (12%) | 24 (3%) | 0 (0%) | Cause 2 | 107 (84%) | 20 (16%) | ||
| Cause 3 | 171 (20%) | 253 (30%) | 36 (4%) | Cause 3 | 449 (98%) | 11 (2%) | ||
| Profile Abnormalities at 15,850 ± 10 m/z | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Neonates with a Corrected FA Phenotype | |||||||||||
| Automatic Classification of Matched Profiles | Irregular Base Line | Low, Broad Peak | Low Resolution | Regular Base Line | Uninterpretable | ||||||
| Lille | Dijon | Lille | Dijon | Lille | Dijon | Lille | Dijon | Lille | Dijon | Lille | Dijon |
| correctly classified | uninterpretable | 1 | 0 | 0 | 0 | 0 | 0 | 67 | 0 | 0 | 68 |
| correctly classified | Misclassified | 4 | 10 | 7 | 0 | 1 | 5 | 17 | 0 | 0 | 0 |
| uninterpretable | correctly classified | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 21 | 0 | 0 |
| Misclassified | correctly classified | 4 | 0 | 3 | 0 | 0 | 0 | 0 | 7 | 0 | 0 |
| Neonates with a Corrected FAS Phenotype | |||||||||||
| Automatic Classification of Matched Profiles | Irregular Base Line | Low, Broad Peak | Low Resolution | Regular Base Line | Uninterpretable | ||||||
| Lille | Dijon | Lille | Dijon | Lille | Dijon | Lille | Dijon | Lille | Dijon | Lille | Dijon |
| correctly classified | uninterpretable | 0 | 0 | 0 | 0 | 0 | 0 | 68 | 0 | 0 | 68 |
| correctly classified | Misclassified | 0 | 4 | 0 | 14 | 1 | 5 | 23 | 0 | 0 | 0 |
| uninterpretable | correctly classified | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 16 | 17 | 0 |
| Misclassified | correctly classified | 12 | 0 | 12 | 0 | 0 | 4 | 6 | 26 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naubourg, P.; El Osta, M.; Rageot, D.; Grunewald, O.; Renom, G.; Ducoroy, P.; Périni, J.-M. A Multicentre Pilot Study of a Two-Tier Newborn Sickle Cell Disease Screening Procedure with a First Tier Based on a Fully Automated MALDI-TOF MS Platform. Int. J. Neonatal Screen. 2019, 5, 10. https://doi.org/10.3390/ijns5010010
Naubourg P, El Osta M, Rageot D, Grunewald O, Renom G, Ducoroy P, Périni J-M. A Multicentre Pilot Study of a Two-Tier Newborn Sickle Cell Disease Screening Procedure with a First Tier Based on a Fully Automated MALDI-TOF MS Platform. International Journal of Neonatal Screening. 2019; 5(1):10. https://doi.org/10.3390/ijns5010010
Chicago/Turabian StyleNaubourg, Pierre, Marven El Osta, David Rageot, Olivier Grunewald, Gilles Renom, Patrick Ducoroy, and Jean-Marc Périni. 2019. "A Multicentre Pilot Study of a Two-Tier Newborn Sickle Cell Disease Screening Procedure with a First Tier Based on a Fully Automated MALDI-TOF MS Platform" International Journal of Neonatal Screening 5, no. 1: 10. https://doi.org/10.3390/ijns5010010
APA StyleNaubourg, P., El Osta, M., Rageot, D., Grunewald, O., Renom, G., Ducoroy, P., & Périni, J.-M. (2019). A Multicentre Pilot Study of a Two-Tier Newborn Sickle Cell Disease Screening Procedure with a First Tier Based on a Fully Automated MALDI-TOF MS Platform. International Journal of Neonatal Screening, 5(1), 10. https://doi.org/10.3390/ijns5010010