1. Introduction
Gaucher disease (GD) is the most prevalent lysosomal storage disorder, with a notably higher incidence among individuals of Ashkenazi Jewish descent. It is an autosomal recessive metabolic condition caused by mutations in the GBA1 gene, which result in the deficient activity of the enzyme glucocerebrosidase. This enzymatic deficiency leads to the accumulation of glucocerebroside within lysosomes, causing widespread multisystem involvement. Clinical manifestations include hepatosplenomegaly, pancytopenia, osteoporosis, and avascular necrosis, with severity varying based on the disease subtype.
This review focuses on the diagnosis and management of GD, emphasizing the critical role of a multidisciplinary approach in optimizing patient care and outcomes. It also underscores the value of early prenatal diagnosis through advanced techniques such as prenatal WES.
GD presents with a broad clinical spectrum, ranging from mild to severe symptoms, and can affect individuals across all age groups and backgrounds. As an inborn error of metabolism, it is particularly relevant in pediatrics, often manifesting during the neonatal period but not exclusively so. The disease is classified into five subtypes: type 1 (non-neuropathic), type 2 (acute neuropathic), type 3 (chronic neuropathic), perinatal lethal, and cardiovascular type. Among these, the perinatal lethal form is the most severe, with complications arising as early as fetal development or during early infancy [
1,
2].
Recognizing the major manifestations of inborn errors of metabolism (IEMs) is crucial for accurate diagnosis. These disorders typically stem from deficiencies or the insufficient activity of specific enzymes that play key roles in metabolic processes. The primary enzymatic defects in IEMs can be categorized into two main types: enzymes required for converting fats or carbohydrates into energy and enzymes necessary for breaking down amino acids or other metabolites.
GD falls into the category of “toxic accumulation” IEMs caused by the buildup of glucocerebroside lipids. Toxic accumulation disorders can be further classified into three subtypes: localized toxicity, circulating toxicity, and a combination of both.
The underlying cause of all forms of Gaucher disease is mutations in the GBA1 gene. These mutations result in a lysosomal deficiency of glucocerebrosidase activity, leading to the toxic accumulation of glucocerebroside lipids. Glucocerebroside consists of a glucose molecule linked to the oxygen atom on carbon 1 of the sphingosine moiety of ceramide. This complex lipid structure accumulates in lysosomes due to enzymatic deficiency, causing cellular dysfunction and tissue damage. The accumulation of glucocerebroside primarily affects the liver, spleen, and bone marrow.
This localized toxicity leads to the characteristic clinical manifestations of Gaucher disease, including hepatosplenomegaly, pancytopenia, and bone abnormalities. The severity and progression of symptoms can vary widely among individuals, depending on the specific GBA1 mutations and other genetic and environmental factors.
Understanding the biochemical and genetic basis of GD is essential for developing targeted therapies and improving patient outcomes. Current treatment approaches, such as enzyme replacement therapy and substrate reduction therapy, aim to address underlying enzymatic deficiency or reduce the accumulation of glucocerebroside. GD belongs to the group of lysosomal storage disorders (LSDs), which includes more than 40 conditions, such as mucopolysaccharidoses, Tay–Sachs disease, and Fabry disease. All LSDs are progressive because lysosomes—intracellular organelles responsible for degrading macromolecules—fail to clear accumulated substrates. In macrophages, where lysosomes are abundant, undigested glucocerebroside causes the progressive enlargement of these organelles. Electron microscopy reveals lipid-filled lysosomes with a characteristic “crumpled tissue paper” appearance. Without treatment, hepatosplenomegaly can become severe, with the liver expanding up to 2–3 times and the spleen up to 15 times its normal size.
Despite the well-characterized genetic basis of Gaucher disease, its clinical presentation remains highly variable. Patients with the same mutation can exhibit different symptoms and disease severity, while those with similar phenotypes may carry distinct genetic mutations. This variability suggests that environmental factors and individual genetic backgrounds play a crucial role in disease expression.
Types of Gaucher Disease and Their Clinical Characteristics (Table 1)
With appropriate treatment, individuals with Type 1 Gaucher disease can achieve a near-normal life expectancy. The most common manifestations include hepatosplenomegaly, anemia, and thrombocytopenia, which typically improve with enzyme replacement therapy (ERT). Bone disease may progress more slowly or stabilize with treatment, though some skeletal abnormalities may persist.
Type 2 GD (the acute neuropathic form) is a severe, rapidly progressive form that primarily affects infants. The prognosis is poor, with significantly reduced life expectancy. Affected children often experience neurological decline and severe complications in infancy or early childhood.
Milder than Type 2, Type 3 GD (the chronic neuropathic form) has a later onset of symptoms, typically emerging in childhood or adolescence. Prognosis varies; some individuals maintain a relatively stable course while others develop significant neurological involvement. Life expectancy is reduced compared to Type 1 but is less severe than Type 2.
Perinatal lethal GD is the rarest and most severe form of GD, with symptoms appearing before birth or in the neonatal period. The prognosis is extremely poor, with most cases being fatal shortly after birth. Due to the severity of the disease, ERT and substrate reduction therapy (SRT) are ineffective. Palliative care is often the primary approach, focusing on comfort and support.
Cardiovascular-type Gaucher disease is a rare, atypical variant that primarily affects the cardiovascular system. It is characterized by glucocerebroside deposition in the heart valves, arteries, and vascular structures. The severity varies widely, with some individuals experiencing mild symptoms, while others develop significant cardiac complications. Cardiac involvement may not respond as effectively to ERT or SRT compared to other forms of Gaucher disease.
Prognosis depends on early diagnosis, the extent of cardiac involvement, and access to specialized cardiac care [
1,
2,
3].
All types of GD have been summarized in
Table 1 as follows:
Table 1.
Types of Gaucher disease and their clinical characteristics.
Table 1.
Types of Gaucher disease and their clinical characteristics.
| Type | Main Characteristics | Clinical Features | Prognosis |
|---|
| Type 1 (Non-Neuropathic) | Most common form; no central nervous system involvement | - -
Hepatosplenomegaly - -
Anemia - -
Thrombocytopenia - -
Bone abnormalities - -
Joint conditions
| Near-normal life expectancy with appropriate treatment |
| Type 2 (Acute Neuropathic) | Severe form affecting infants; rapid progression | - -
Neurological decline - -
Seizures - -
Abnormal eye movements - -
Brain damage - -
Hepatosplenomegaly
| Poor prognosis; significantly reduced life expectancy |
| Type 3 (Chronic Neuropathic) | Milder than Type 2; later onset | - -
Progressive encephalopathy - -
Systemic manifestations similar to Type 1 - -
Ophthalmoplegia - -
Myoclonic epilepsy - -
Cerebellar ataxia
| Reduced life expectancy compared to Type 1 but less severe than Type 2 |
| Perinatal Lethal | Rarest and most severe form | - -
Hydrops fetalis - -
Ichthyosis - -
Hepatosplenomegaly - -
Distinctive facial features - -
Severe neurological problems
| Extremely poor; fatal shortly after birth |
| Cardiovascular Type | Primarily affects the heart | - -
Heart valve calcification - -
Eye abnormalities - -
Bone disease - -
Mild splenomegaly
| Varies widely, from mild symptoms to significant cardiac complications |
2. Materials and Methods
In this study, we present the case of a 35-year-old Romanian Caucasian patient who underwent non-invasive prenatal testing (NIPT) using the Veragene platform.
NIPT represents a groundbreaking advancement in prenatal screening, leveraging next-generation sequencing (NGS) or other high-throughput technologies to analyze cell-free placental DNA circulating in maternal blood. This approach offers a safer alternative to traditional invasive procedures, such as chorionic villus sampling (CVS) or amniocentesis, as it poses no direct risk to the fetus.
Despite its widespread promotion and commercialization, it is crucial to critically evaluate the benefits, limitations, and risks of NIPT compared to both invasive and other non-invasive testing methods. While NIPT is highly effective for screening common aneuploidies, such as trisomy 21 (Down syndrome), trisomy 18 (Edwards syndrome), and trisomy 13 (Patau syndrome), its clinical utility is limited by its inability to provide a definitive diagnosis. Additionally, false-positive and false-negative results may occur due to factors such as placental mosaicism, maternal health conditions, or fetal-specific variables.
The NIPT used was Veragene testing, a validated Laboratory-Developed Test (LDT) from Medicover Genetics Ltd. for prenatal screening. It can test for common chromosomal aneuploidies such as trisomies 13, 21, 18, X, and selected microdeletions through the multiplexed parallel analysis of specific regions of interest. It can also detect 100 monogenic disorders, such as GD. Veragene is created based on the Target Capture Sequences (TACSs) approach, which consists of long synthetic DNA probes that bind to specific DNA regions of the genome and then utilize NGS technology. This particular methodology is designed to reduce the current described limitations of whole-genome-based NIPT [
4,
5,
6,
7,
8,
9,
10,
11,
12]. TACS enrichment has been successfully used to screen for fetal risk for monogenic disorders based on parental carrier screening results.
Prenatal WES was performed through NGS technology on Illumina’s NovaSeq platform. The bioinformatic pipeline used was Illumina Dragen Bio-IT, and an interpretation of the result was made with the help of VarSeq software version 2.3.0 from GoldenHelix. The data were aligned to Human Reference Genome hg38 (NCBI GRCh38). The classification and interpretation of variants were performed following the ACMG (American College of Medical Genetics) and Association for Clinical Genomic Science (ACGS) standards [
13,
14].
This case underscores the necessity of understanding both the advantages and limitations of non-invasive prenatal testing (NIPT) in prenatal care, particularly the importance of confirmatory diagnostic testing when abnormalities are identified. Given these constraints, the medical indications for NIPT remain surprisingly narrow, raising critical questions about what individual pregnant women can genuinely learn from the test and how the results should be interpreted in clinical practice.
3. Case Presentation
We present the case of a 35-year-old pregnant woman who underwent Veragene NIPT in our clinic, with the indication for testing given by her gynecologist. The couple is non-consanguineous, has no family history, and they have a 5-year-old daughter, who is affirmatively healthy. Our patient took the test at the gestational age of 14 weeks, and the results were as follows: a very high-risk result for GD and very low risk for all other conditions tested, such as chromosomal aneuploidies and microdeletions. The variants that the NIPT test was able to discover and report were the GBA1 gene, NM_00157.4:c.1448T>C p.(Leu483Pro) (in the mother/fetus) and NM_00157.4:c.1504C>T p.(Arg502Cys) (in the father’s buccal swab). These two variants are reportedly both pathogenic (class 5), and if transmitted to the fetus, they would create the phenotype of GD. The first one, c.1448T>C, found in maternal blood, is a missense variant that results in a non-conservative amino acid change in the encoded protein. In silico tools predict the damaging effect of the variant on protein function. The second variant is also a missense variant, which results in a non-conservative amino acid substitution that causes damage to the protein. All in silico tools predict a deleterious effect on the protein function.
This very high risk of GD reported was explained to the patients, including the fact that NIPT technology could not report whether the variants would be transmitted from the parents, which is a limitation of NIPT. It was then decided to continue with an invasive prenatal diagnosis, looking closer at the two known variants. The NIPT test did not detect the presence of the Y chromosome.
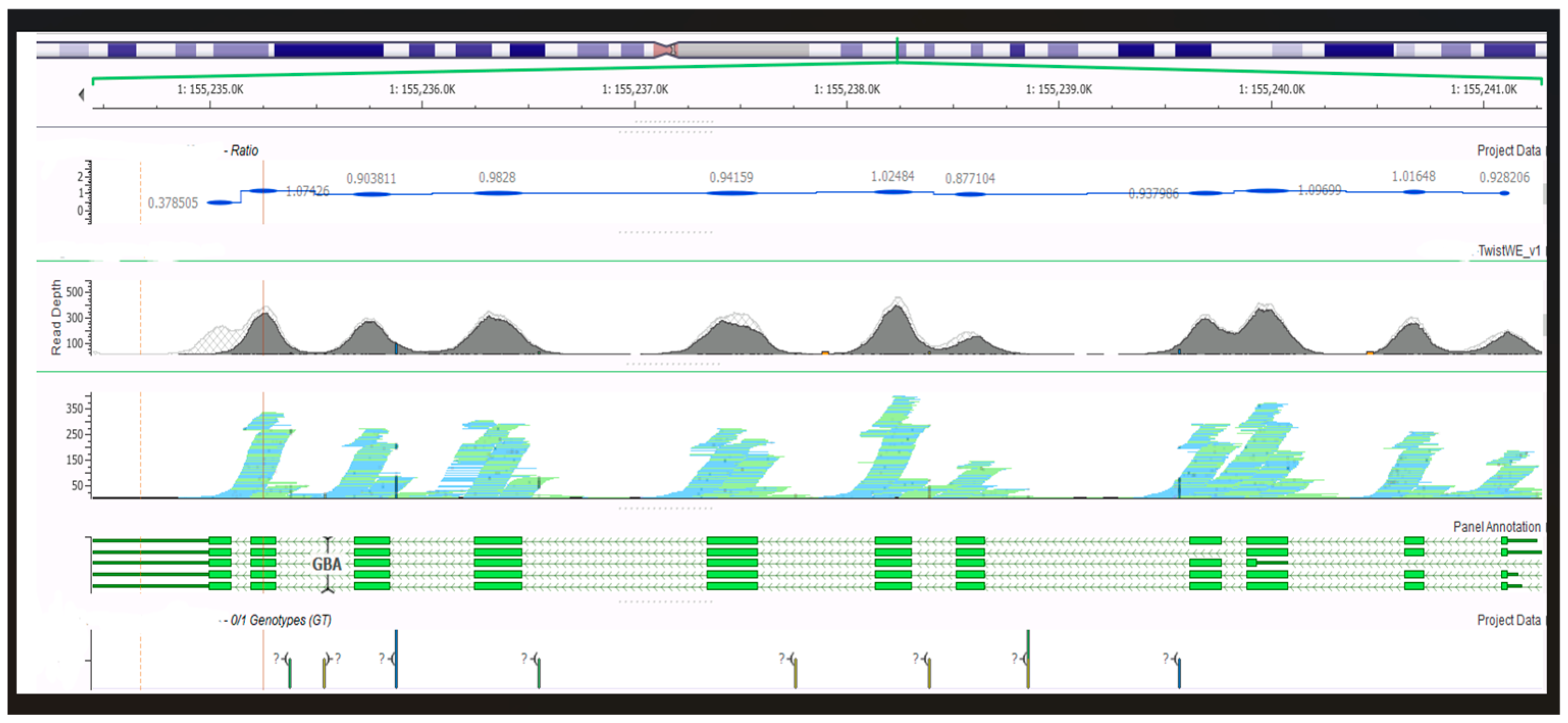
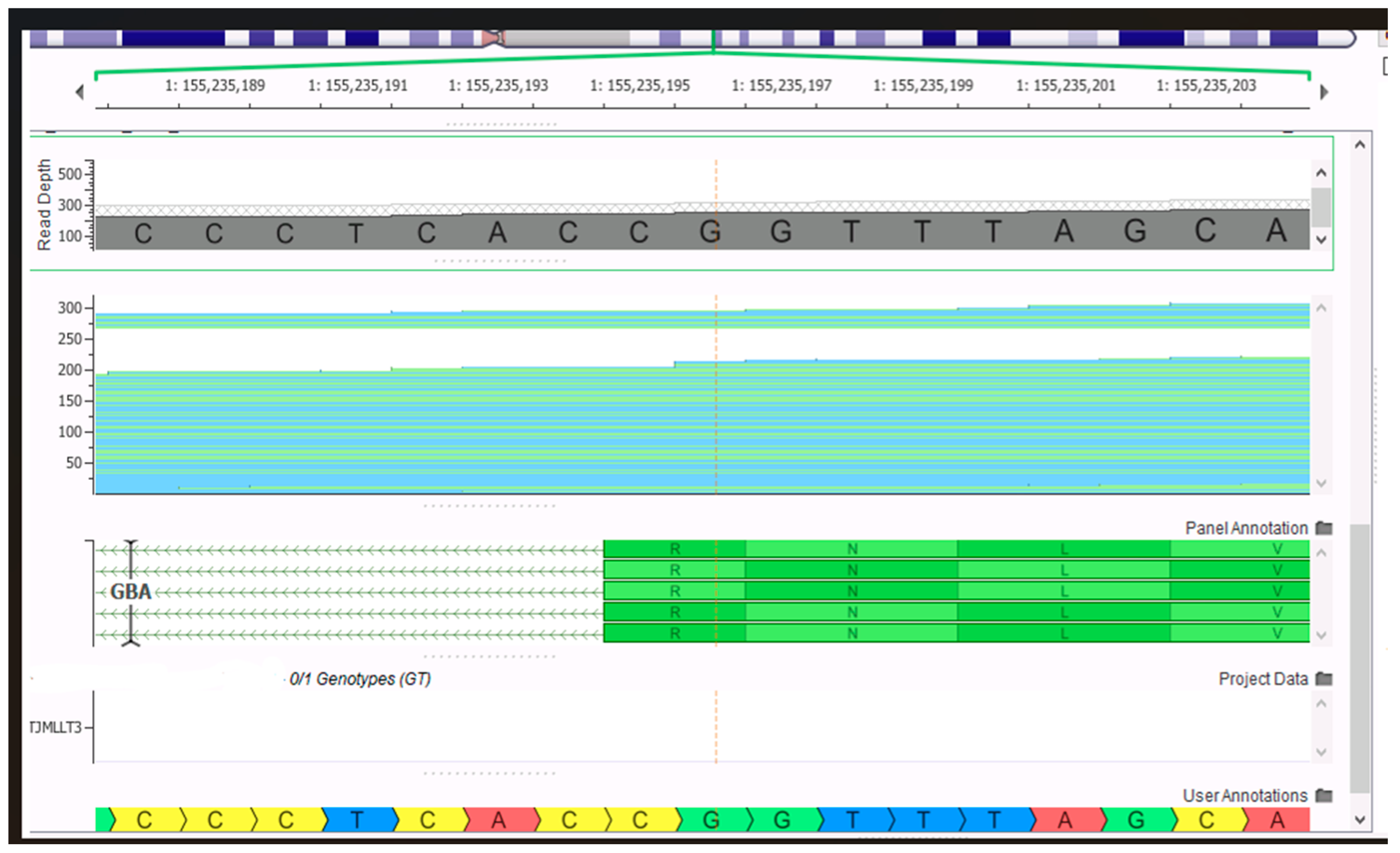
The couple was advised to receive immediate genetic counseling and thus receive interpretation information, and the couple was informed again that NIPT is a screening test and not a diagnostic test, requiring confirmation by invasive prenatal testing. The couple accepted the prenatal diagnosis; the patient underwent amniocentesis, and we performed prenatal WES with a special focus on the known familial variants in the GBA1 gene and classical chromosomal analysis since the woman was over 35 years of age. Prenatal WES yielded no pathogenic or likely pathogenic variants, so there was a normal female WES report for this fetus, with none of the parental variants inherited; therefore, GD was ruled out as observed in the bioinformatic data shown in
Figure 1 and
Figure 2.
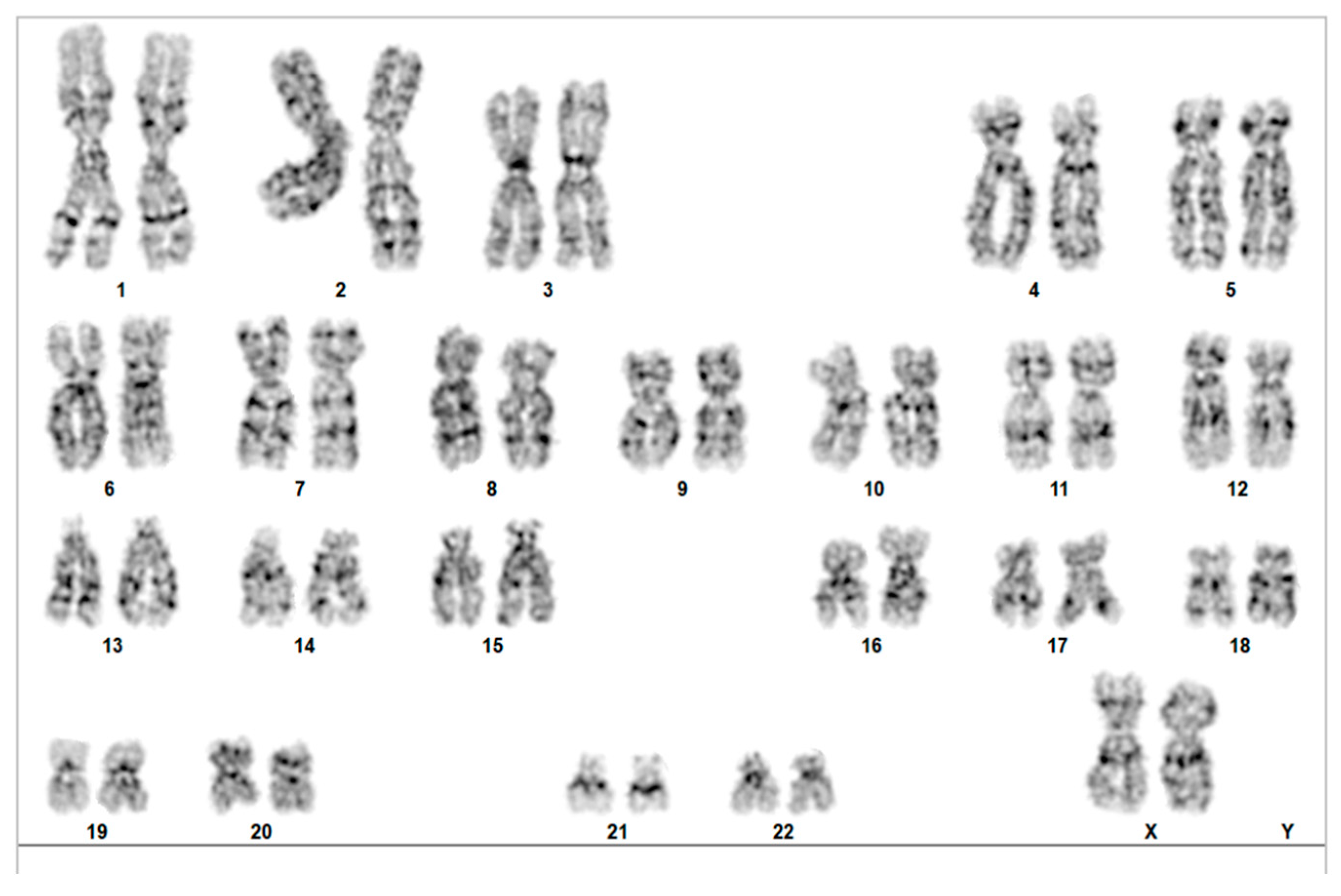
Due to the patient’s advanced maternal age, we also recommended that a classical karyotype analysis be performed from the amniotic fluid, which yielded a normal feminine fetus result, 46, XX,
Figure 3.
The team also recommended genetic counseling post-testing to interpret the results for the expecting family. We also decided to test the 5-year-old child of the couple with the known familial variants only to rule out a possible GD with later onset. The results were negative for both variants, so the child was not carrying either of the known familial variants.
4. Discussion
The global adoption of NIPT has been propelled by collaborative research studies and consortiums, with both governmental and private organizations making substantial efforts to incorporate this testing into national healthcare frameworks. Recognizing the significant potential of NIPT to enhance prenatal care, these entities have invested considerable resources to promote their integration into healthcare systems. By forming partnerships and supporting research initiatives, they aim to facilitate the adoption of NIPT as a standard screening tool, ensuring that it is accessible and effective for expectant parents worldwide.
This collective effort highlights a commitment to leveraging advanced genetic testing technologies to improve prenatal care and achieve better maternal and fetal outcomes on a global scale. In many countries, NIPT is viewed as a promising screening tool and is actively being integrated into routine practice. In this article, we review some of the leading NIPT consortiums and programs across various continents, shedding light on their contributions to advancing prenatal screening.
Confirmatory testing following a positive non-invasive prenatal testing (NIPT) result plays a critical role in prenatal diagnosis. While NIPT has revolutionized prenatal screening with its high sensitivity and specificity, it remains a screening tool rather than a diagnostic test. Below is an overview of the importance of confirmatory testing and the challenges associated with NIPT results: accuracy and reliability, informed decision-making, eliminating false positives, preventing unnecessary interventions, and improved pregnancy management.
NIPT is highly effective in detecting common chromosomal abnormalities, such as Down syndrome (Trisomy 21), Trisomy 18, and Trisomy 13. However, as a screening test, it identifies increased risk rather than providing a definitive diagnosis. Confirmatory diagnostic tests, such as amniocentesis or chorionic villus sampling (CVS), are essential to verify the presence of the condition with certainty.
Confirmatory testing empowers parents to make well-informed decisions about their pregnancy. For example, if the confirmatory test results are negative, it can alleviate anxiety and provide reassurance. If the results confirm a chromosomal condition, parents can prepare emotionally, medically, and logistically for the potential challenges ahead.
While NIPT is highly accurate, false positives can occur due to factors such as placental mosaicism, maternal health conditions, or other biological variables. Confirmatory tests analyze fetal DNA directly, reducing uncertainties and ensuring that decisions are based on accurate information [
15,
16,
17].
Negative results from confirmatory diagnostic tests can prevent unnecessary medical interventions or invasive procedures. Additionally, they can reduce the psychological stress and anxiety experienced by parents who may have been concerned about an initial positive NIPT result.
The early confirmation of conditions such as Down syndrome or other chromosomal disorders enables better planning for specialized care and support during pregnancy and after birth. It also allows healthcare providers to offer tailored counseling and resources to help parents navigate their options. In cases of severe chromosomal abnormalities with significant life-long implications, confirmatory testing can guide decisions regarding the continuation of pregnancy.
While NIPT is a powerful tool for early risk assessment in prenatal care, confirmatory diagnostic testing remains indispensable for ensuring accuracy and guiding clinical decisions. By combining the strengths of both screening and diagnostic methods, healthcare providers can offer families comprehensive support and enable informed decision-making throughout pregnancy [
15,
16,
17].
Challenges in Prenatal Diagnosis
Despite the high accuracy of NIPT, several challenges are associated with its results. False positives and false negatives can occur due to factors such as placental mosaicism or maternal DNA contamination, potentially leading to misleading outcomes. A critical issue is the frequent misunderstanding of the distinction between screening (which NIPT provides as a risk assessment) and diagnosis (which requires confirmatory testing for definitive results). This misunderstanding can lead to confusion or inappropriate decision-making among expectant parents and healthcare providers. Furthermore, the emotional impact of a positive NIPT result cannot be underestimated. It can cause significant anxiety for expectant parents, even in cases where subsequent confirmatory testing ultimately rules out the condition. These challenges underscore the importance of comprehensive genetic counseling and clear communication about the capabilities and limitations of NIPT in prenatal care [
11,
12,
15,
16,
17,
18,
19].
The emotional and psychological impact of abnormal NIPT results can be significant, often causing distress and anxiety for expectant parents, even though the test itself is non-invasive. The subsequent need for confirmatory testing may prolong uncertainty, potentially leading to decision fatigue. Access to confirmatory diagnostic procedures can be limited in some regions due to financial, infrastructural, or insurance constraints, potentially creating healthcare disparities. While more accurate than NIPT, invasive confirmatory tests like amniocentesis and CVS carry a small risk of miscarriage, complicating the decision-making process for parents.
Interpreting the results can be challenging, particularly in cases of mosaicism where only some fetal cells carry the chromosomal abnormality. This complexity can lead to inconclusive or ambiguous outcomes, making it difficult for healthcare providers and parents to make informed decisions. The availability of prenatal testing also raises ethical and social questions regarding selective abortion, societal pressures, and genetic privacy.
Confined placental mosaicism or maternal mosaicism are also major causes of false-positive results in NIPTs. Mosaicism is the phenomenon that consists of two or more different cell lines in one individual, originating from one zygote. The word “mosaic”rises from the stone art found in Mesopotamia during the third millennium B.C. and art that was also found in ancient Greece and Rome. Mosaicism can be excluded only by diagnostic tests such as a chorionic villus biopsy (later confirmed by amniocentesis) and the constitutional chromosomal analysis of the mother [
16,
20,
21,
22,
23,
24,
25,
26].
It is important to note that while NIPT is effective for detecting common chromosomal abnormalities, it does not screen for all genetic conditions or structural anomalies. For instance, neural tube defects like spina bifida require additional diagnostic tests such as ultrasound. Lastly, the interpretation of NIPT and confirmatory test results often necessitates careful explanation by genetic counselors or healthcare providers, which is a process that can be challenging, especially when results are unexpected or complex.
Higher resolution circulating free (cfDNA) testing is revolutionizing prenatal diagnosis, making it possible to detect a wide range of genetic conditions non-invasively and with greater precision. It holds great promise for improving early diagnosis and the management of genetic disorders, including RhD typing, inherited point mutations, and de novo mutations. However, as these technologies evolve, it is crucial to balance the benefits with the ethical, technical, and clinical challenges they present.
Inferring the inheritance of point mutations from the mother is more complex because cfDNA in maternal plasma consists of a mixture of both fetal and maternal DNA fragments. Since the fetus shares half of its genetic material with the mother, it is impossible to completely separate fetal from maternal DNA. In particular, when the mother is heterozygous at a specific locus, both alleles will be present in the maternal plasma, making it difficult to distinguish fetal DNA. Therefore, methods that rely solely on the presence of an allele in the maternal blood are inadequate, and quantification is required. However, this approach comes with several challenges. First, the amount of cfDNA in plasma is relatively low, complicating accurate quantification. Second, the maternal-derived fragments tend to dominate the cfDNA, which makes the analysis of fetal DNA more difficult. These challenges are even more pronounced in the early stages of pregnancy, which are often the most clinically relevant. For example, the fetal fraction typically averages around 10% at the end of the first trimester. To overcome these issues, either highly accurate counting technologies or DNA amplification methods are necessary.
The first successful approach for determining the inheritance of maternal mutations involved digital PCR (dPCR), a precise quantification technology that can detect small allelic imbalances. This method, referred to as relative mutation dosage (RMD) analysis, examines a genomic locus where the mother is heterozygous within the cfDNA. A shift in the ratio between the two alleles toward one allele suggests that the fetus has inherited homozygosity for that allele. The degree of allelic imbalance correlates with the fetal fraction. This approach has been validated for a range of mutations and diseases, such as chromosomal aneuploidies, monogenic disorders, and chromosomal microdeletions/duplications [
27].
The development of next-generation sequencing (NGS) has greatly advanced the study of monogenic disorders. NGS techniques, including Whole-Exome Sequencing (WES) and whole-genome sequencing (WGS), have facilitated the identification of numerous genetic variants, many of which are pathogenic and linked to monogenic diseases. Invasive prenatal WES/WGS, typically performed via amniocentesis, enables the diagnosis of monogenic disorders in the early stages of life, allowing for early interventions such as treatment or, if necessary, the termination of the pregnancy. There is increasing interest in achieving non-invasive prenatal WES/WGS, but the methodologies to detect point mutations are not feasible for large-scale applications. This is particularly challenging when it comes to maternal inheritance and de novo mutations. For instance, the RMD approach—based on dPCR—is not suitable for genome-wide analyses, as it requires the design of specific primer sets for each mutation. Clearly, the genome-wide detection of de novo mutations is a powerful application of WES/WGS, but it demands highly accurate and deep sequencing. Unlike targeted mutation tests, searching the entire genome for rare mutations is inherently more difficult because there is no prior knowledge of what mutations to expect. As a result, distinguishing true de novo mutations from sequencing noise can be challenging [
18,
28,
29].
The progress made in NIPD and the growing availability of NGS technologies eventually led to the first successful attempts for non-invasive prenatal WGS, enabling the genome-wide NIPD of monogenic diseases. However, despite these early successes, genome-wide non-invasive prenatal WGS has not yet become clinically available. Studies have shown that accurate non-invasive genome-wide genotyping relies heavily on both the fetal fraction and sequencing depth [
30,
31,
32]. These factors pose significant limitations, even with NGS-based methods, primarily due to the low availability and high costs of the necessary technologies. Improving computational and algorithmic techniques is crucial to overcoming these challenges.
Although much of the literature focuses on the clinical implementation of NIPD (Non-Invasive Prenatal Diagnosis), few studies have proposed novel algorithmic approaches. This review focuses on the methods developed for the genome-wide NIPD of monogenic diseases and provides an overview of various approaches that have emerged over the past decade. We also discuss the future of this field, identifying the remaining research and clinical gaps that must be addressed to make these advanced diagnostic methods clinically accessible. New management plans, such as in utero therapy for fetal genetic conditions, are already being implemented worldwide alongside new emerging techniques such as WES/WGS NIPT [
33,
34].
5. Conclusions
We concluded that the female fetus did not inherit either of the parent’s variants; the prenatal WES did not yield any pathogenic or likely pathogenic variants. The chromosomal analysis was that of a normal female fetus. We selected this case to highlight the importance of a prenatal diagnosis versus an NIPT that yields a very high-risk result and the impact that screening followed by a diagnostic procedure has on the respective family.
Prenatal WES was, therefore, the perfect tool in this case due to its comprehensive and time-efficient qualities, which helped us—the multidisciplinary team involved in the case—and also the parents by providing a diagnostic in less than three weeks. This whole assessment, starting with a screening test, allowed us to know and counsel the family according to their variants. Genetic advice is mandatory in such cases, and it allows the expecting parents to know their risks for each pregnancy to come.



 ,
, 

{kind=link}
{kind=link}
{kind=link}