
Biocompatibility and Osteogenic Activity of Samarium-Doped Hydroxyapatite—Biomimetic Nanoceramics for Bone Regeneration Applications

 ,
,  ,
,  ,
, 
 ,
, 

Abstract

1. Introduction
2. Materials and Methods
2.1. Samarium-Doped Hydroxyapatite Nanoparticles
2.2. X-ray Diffraction (XRD)
2.3. FTIR Spectroscopy
2.4. X-ray Photoelectron Spectroscopy (XPS)
2.5. Transmission Electron Microscopy (TEM)
2.6. Energy-Dispersive X-ray Spectroscopy (EDS)
2.7. Cell Culture and Treatment
2.8. MTT Assay
2.9. Trypan Blue Cell Viability Assay
2.10. Lactate Dehydrogenase (LDH) Release Assay
2.11. Measurement of NO Production
2.12. Fluorescent Staining of F-Actin Cytoskeleton
2.13. Alkaline Phosphatase Activity
2.14. Western Blot Analysis
2.15. Wound Healing Assay
2.16. Statistical Analysis
3. Results
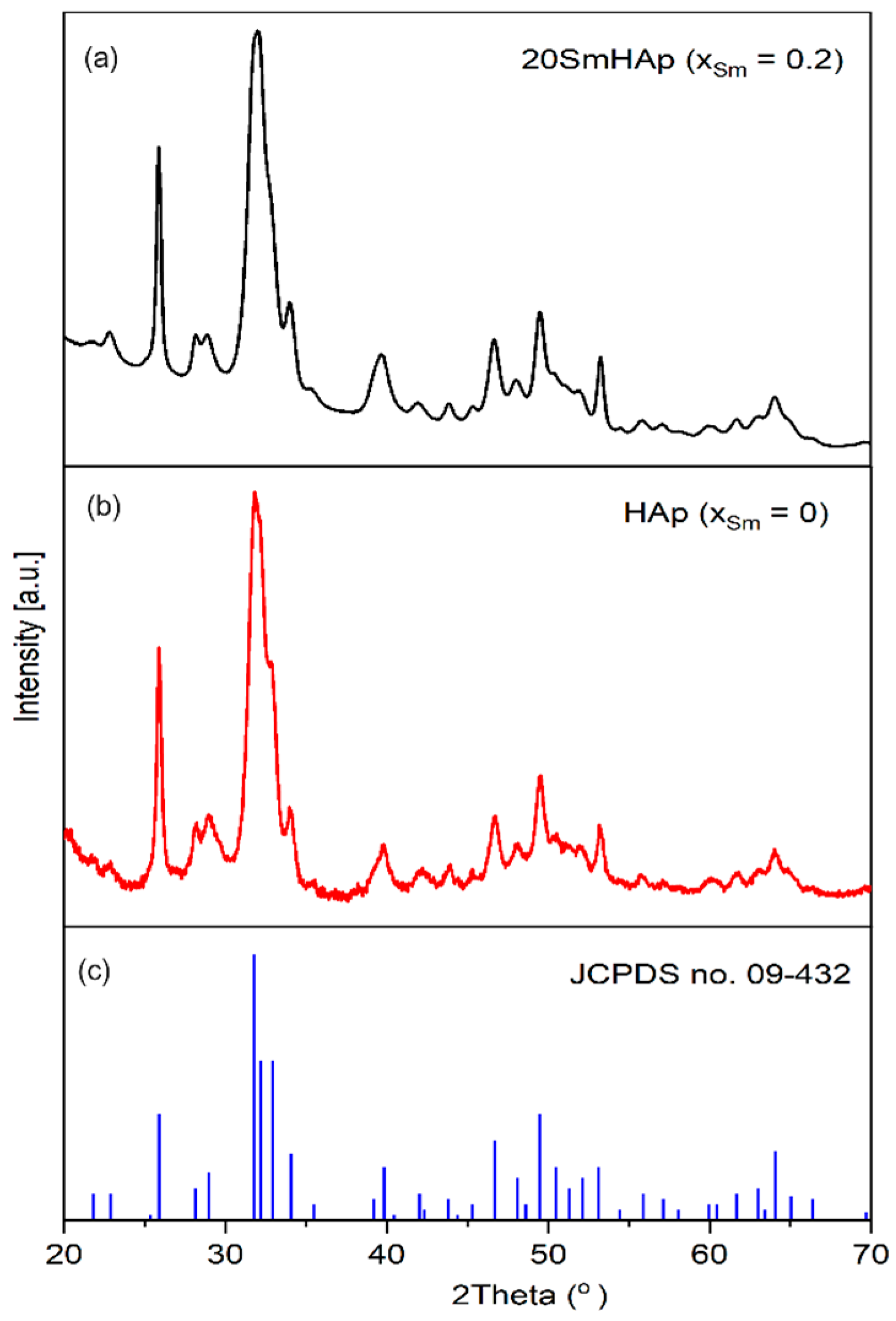
3.1. X-ray Diffraction (XRD)
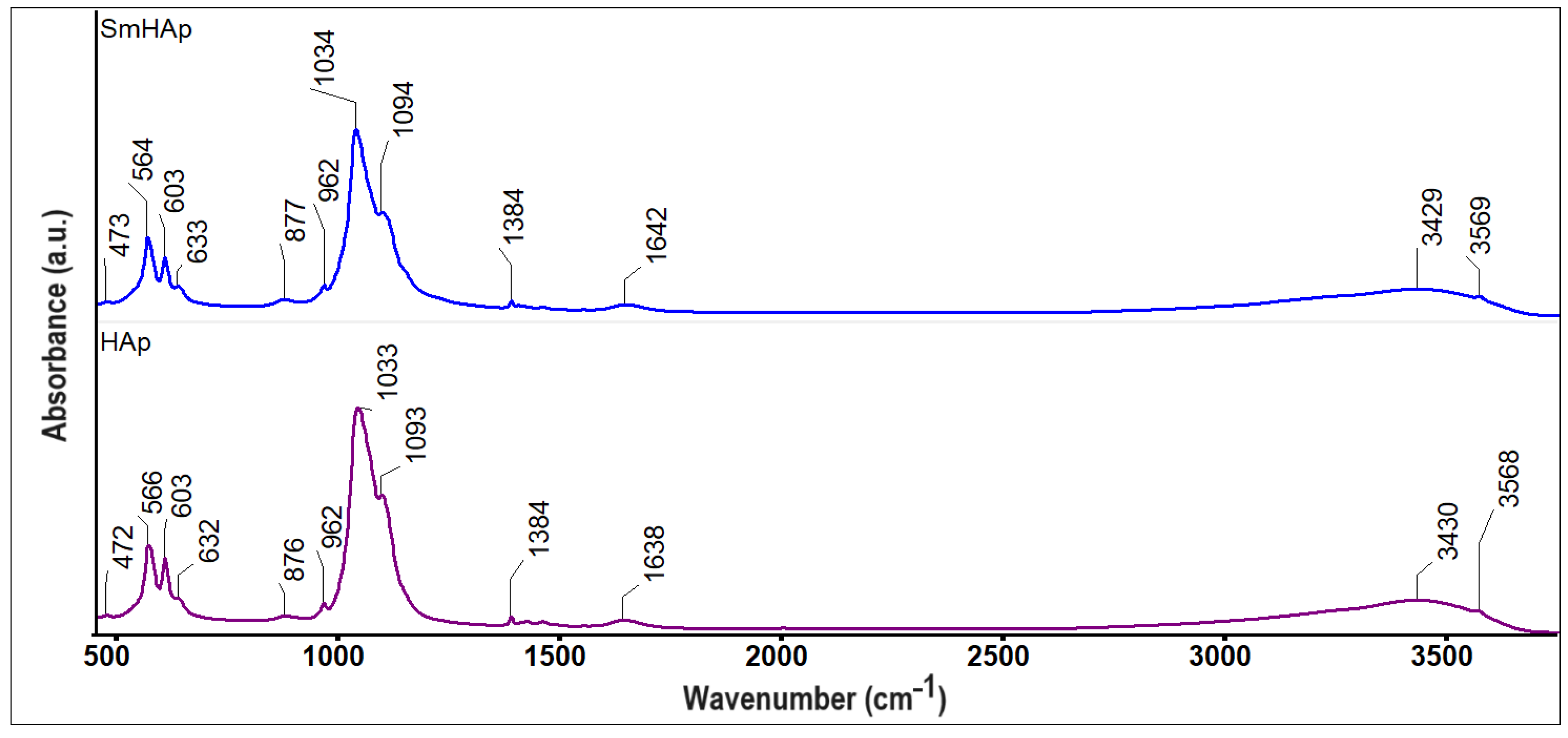
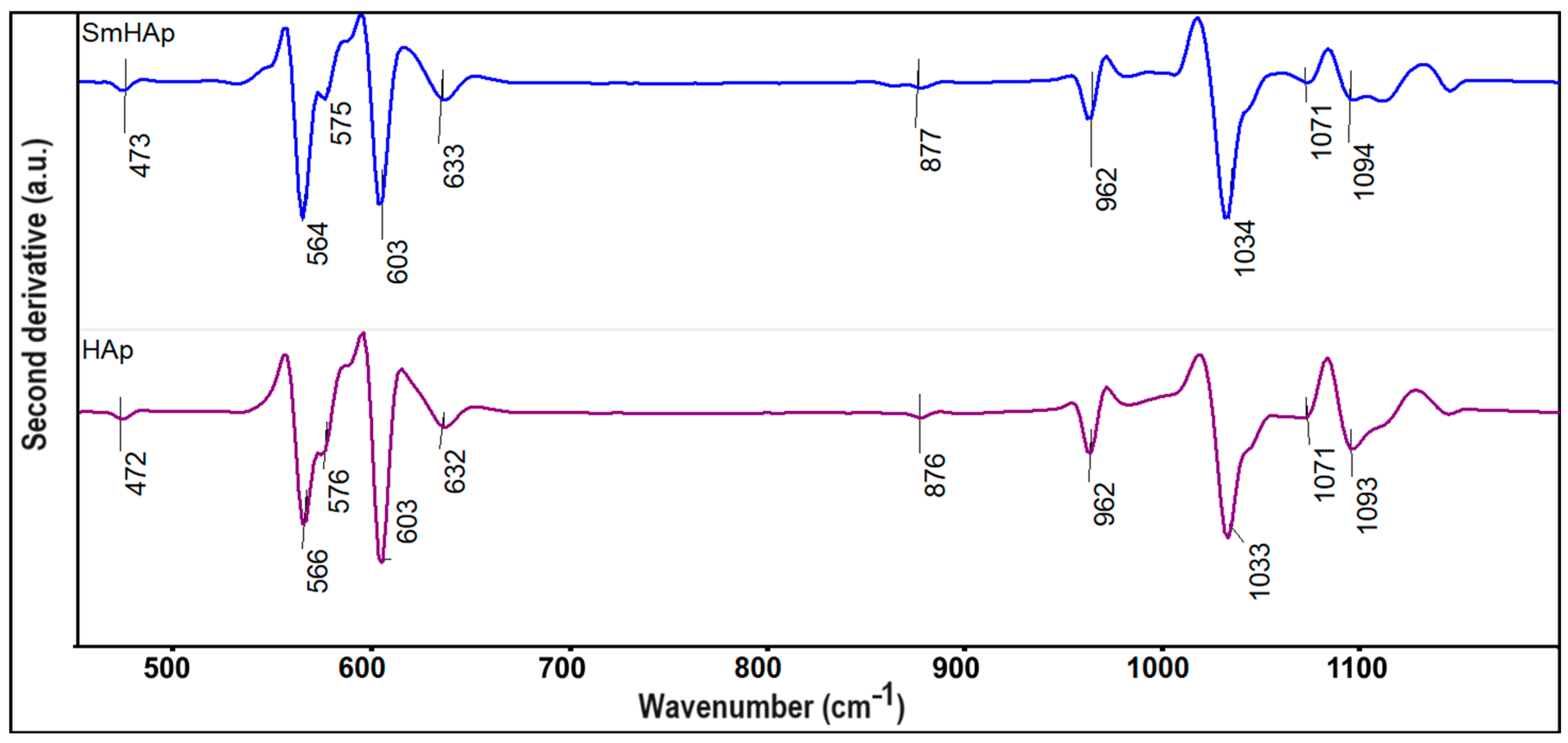
3.2. FTIR Spectroscopy
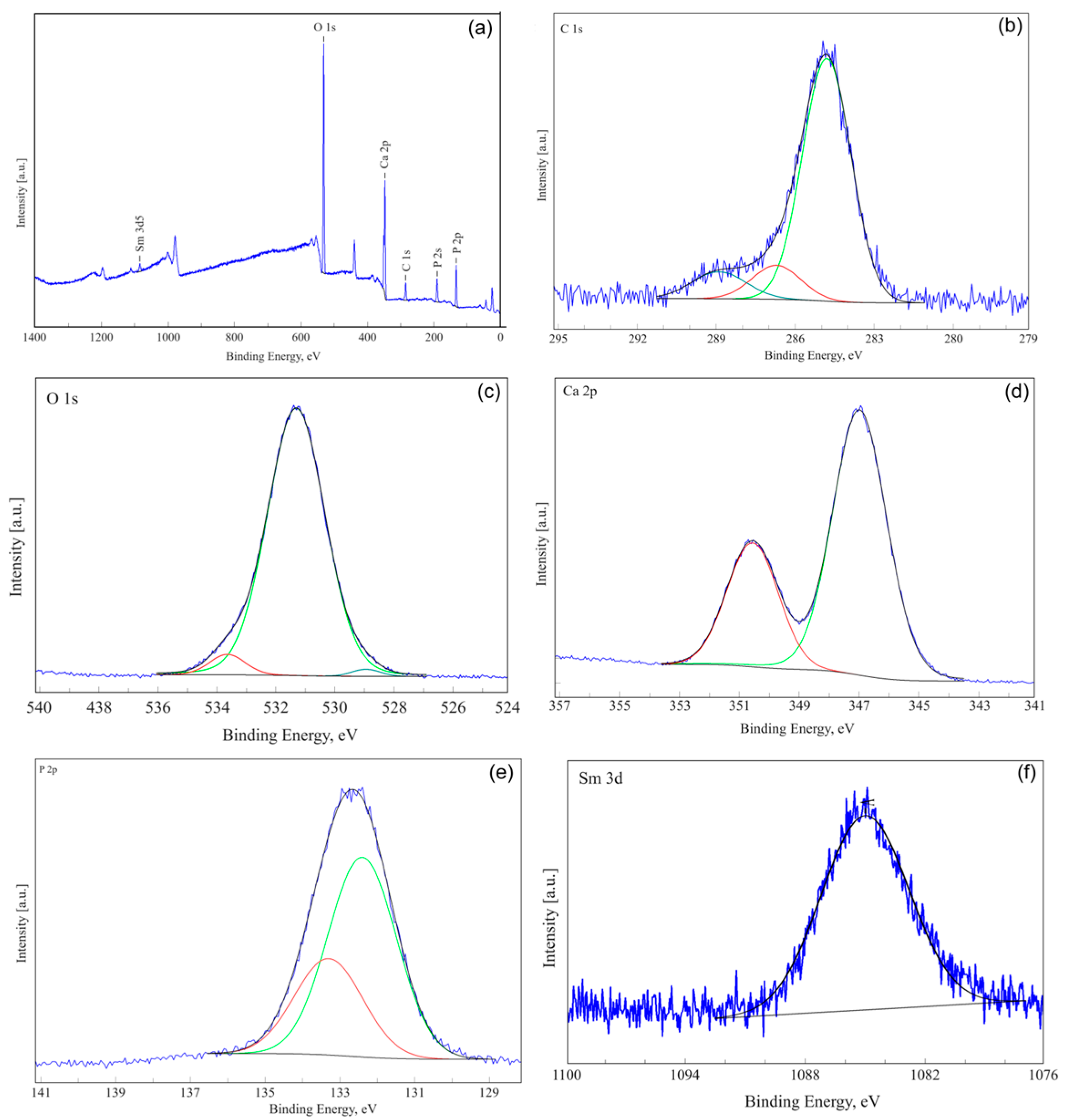
3.3. X-ray Photoelectron Spectroscopy (XPS)
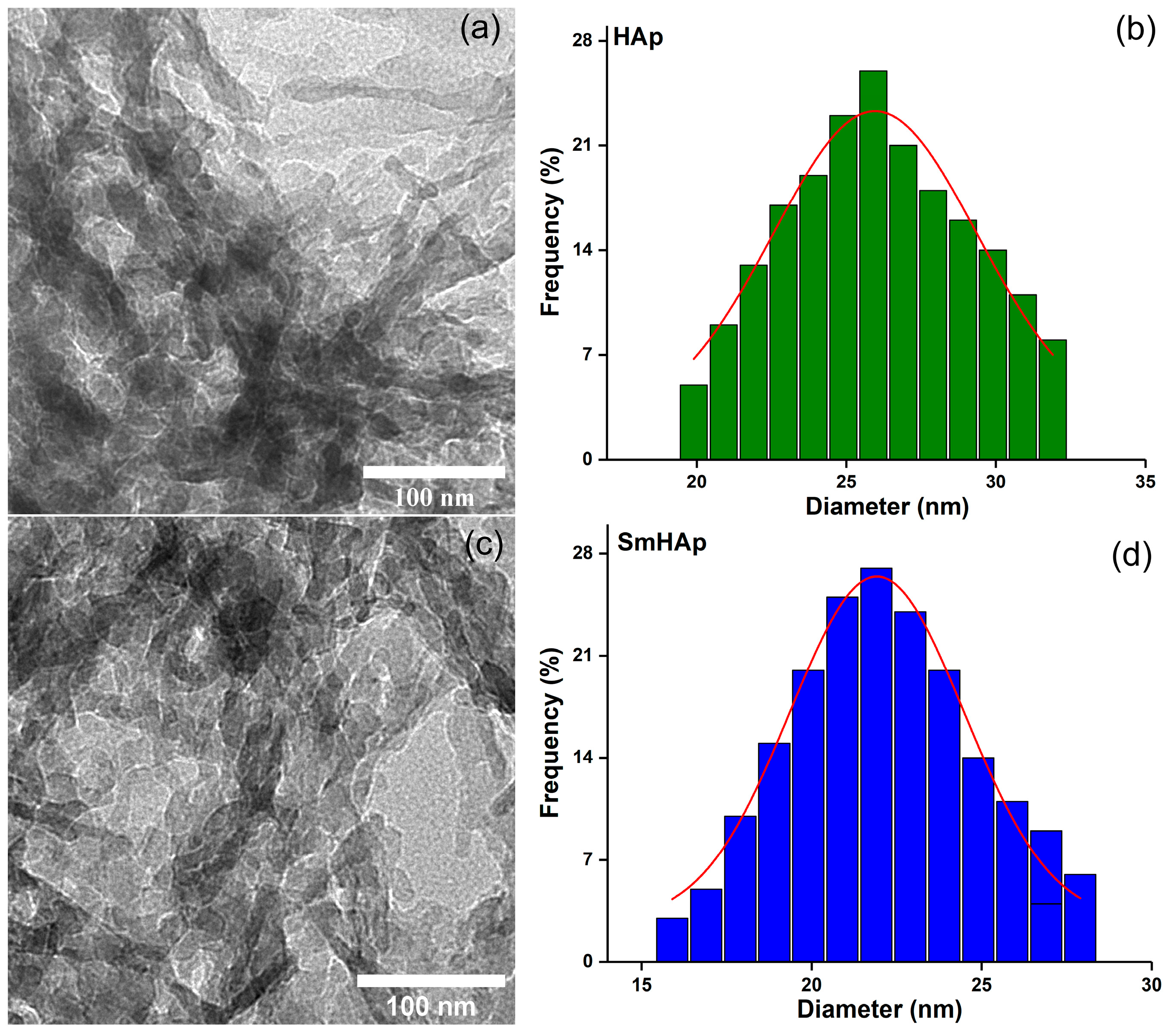
3.4. Transmission Electron Microscopy (TEM)
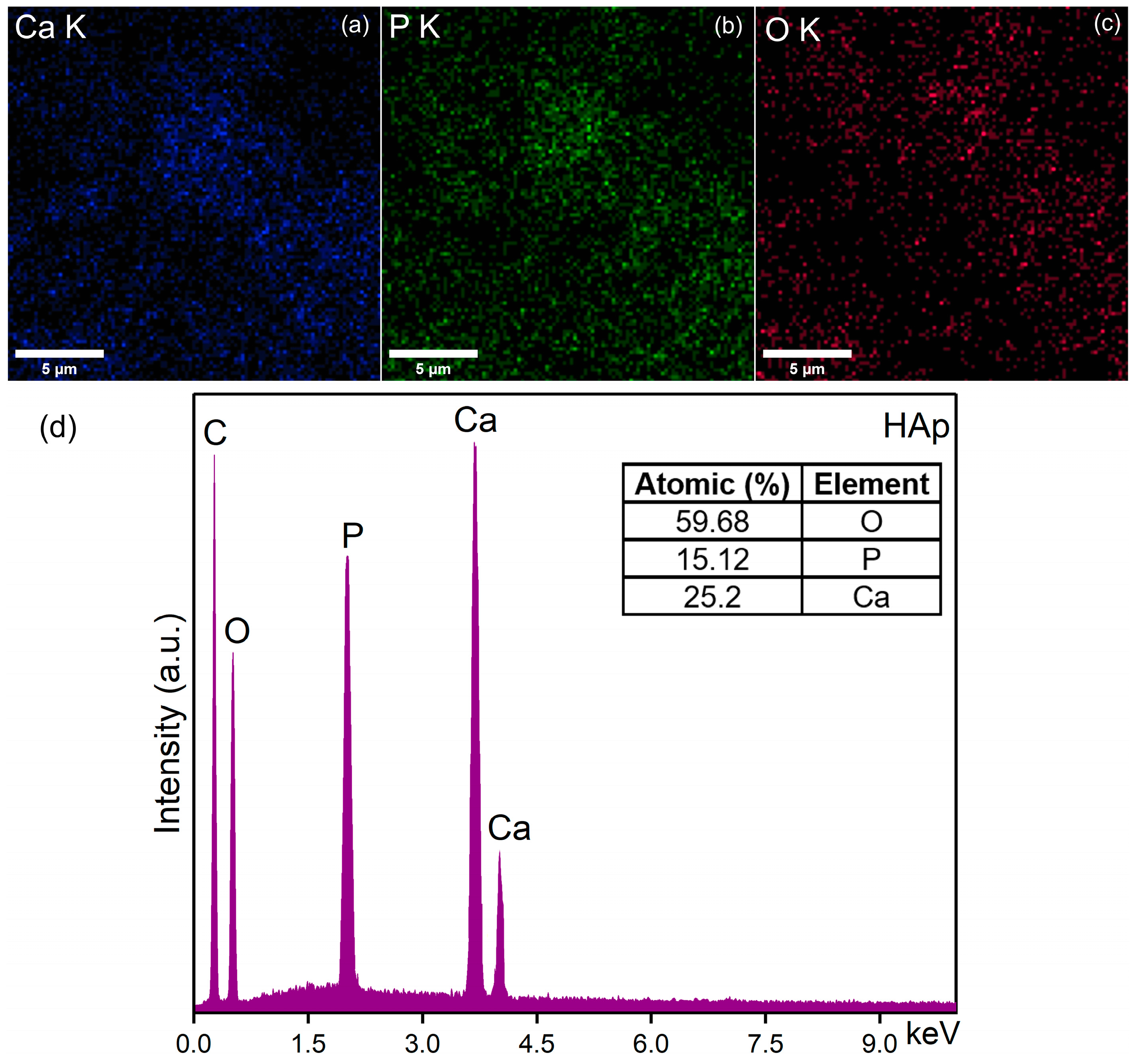
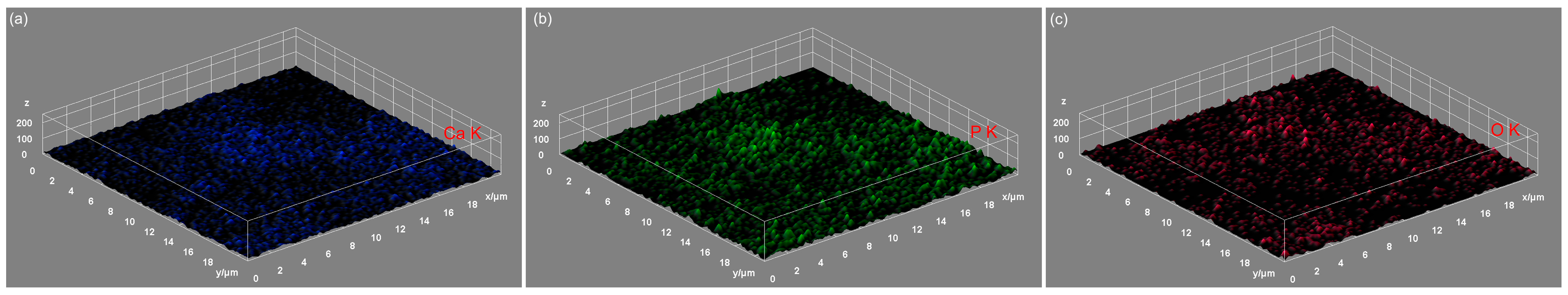
3.5. Energy-Dispersive X-ray Spectroscopy (EDS)
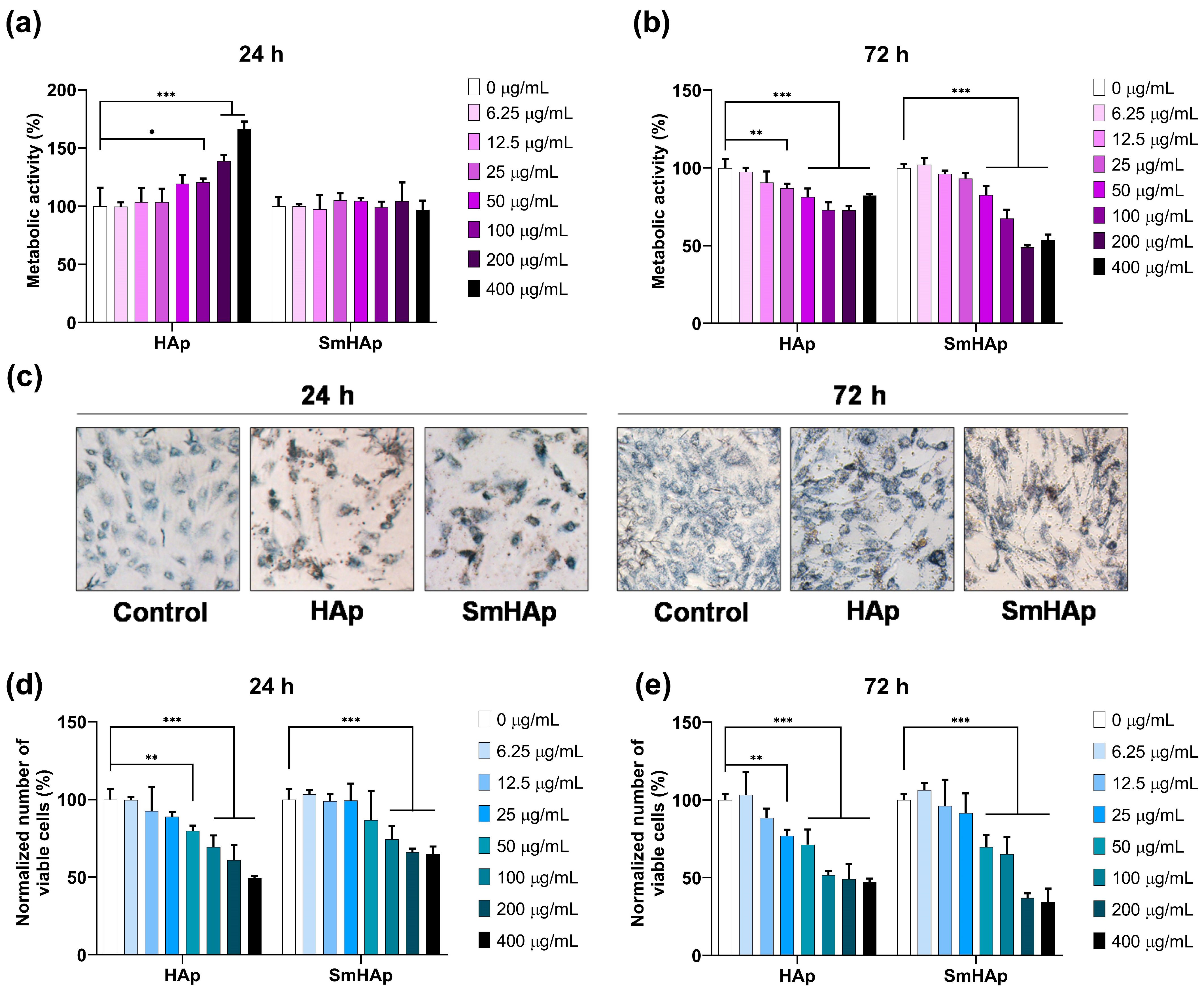
3.6. Osteo-Biocompatibility
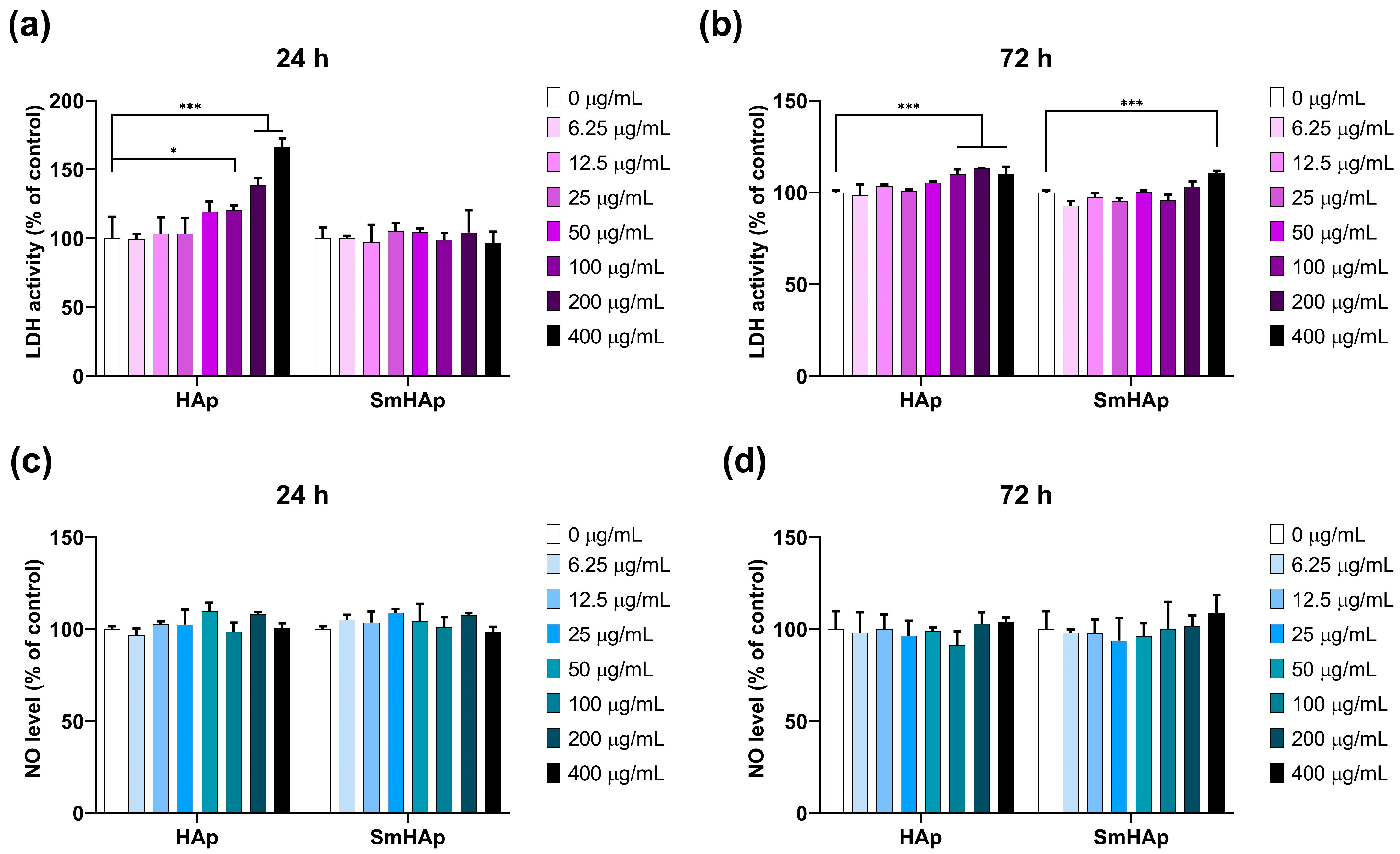
3.7. Effects on Lactate Dehydrogenase (LDH) Activity and Nitric Oxide (NO) Production
3.8. Effects on Organization of F-Actin Cytoskeleton
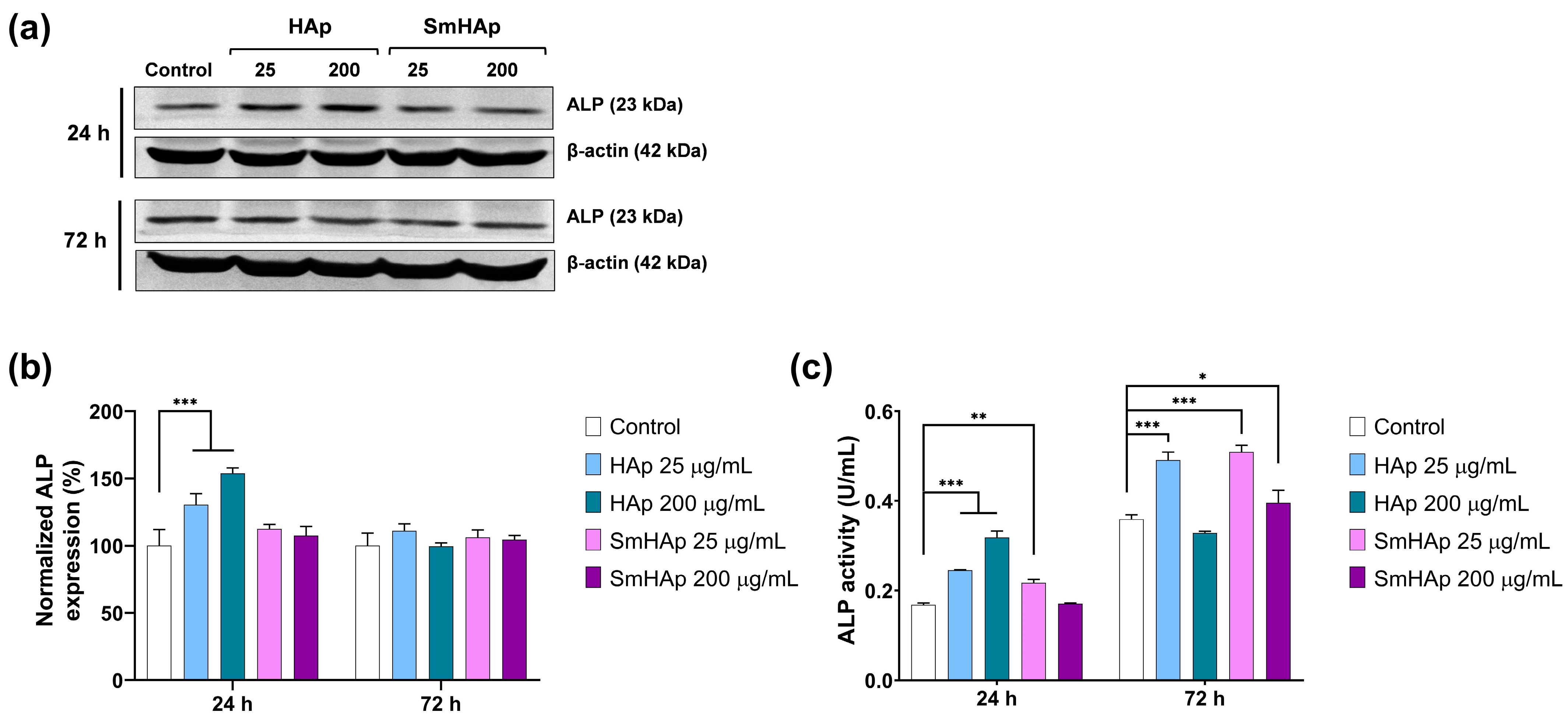
3.9. Osteoinductive Potential
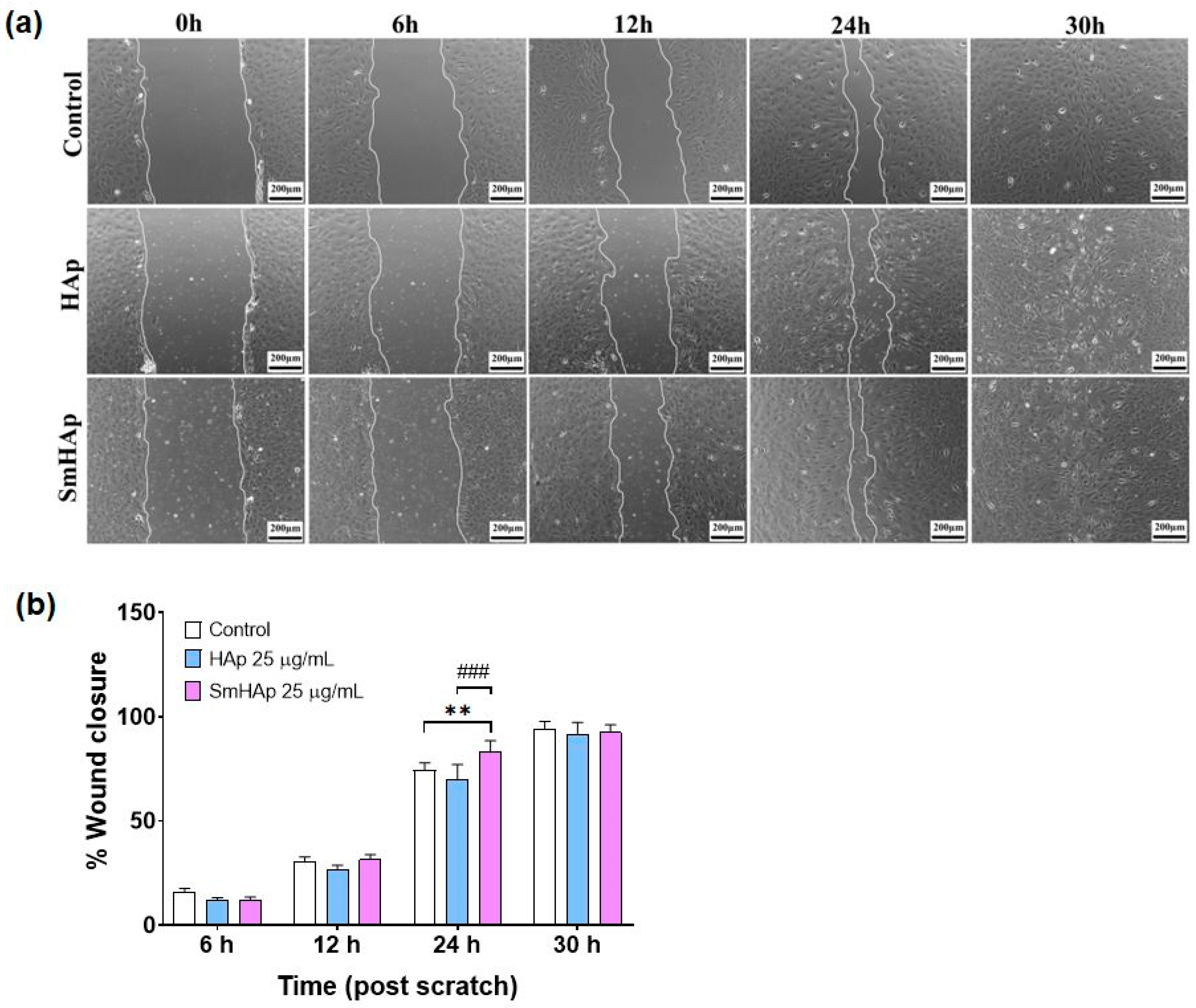
3.10. Effects on Preosteoblast Migration
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, A.-M.; Bisignano, C.; James, S.L.; Abady, G.G.; Abedi, A.; Abu-Gharbieh, E.; Alhassan, R.K.; Alipour, V.; Arabloo, J.; Asaad, M. Global, regional, and national burden of bone fractures in 204 countries and territories, 1990–2019: A systematic analysis from the Global Burden of Disease Study 2019. Lancet Healthy Longev. 2021, 2, e580–e592. [Google Scholar] [CrossRef]
- Shen, Y.; Huang, X.; Wu, J.; Lin, X.; Zhou, X.; Zhu, Z.; Pan, X.; Xu, J.; Qiao, J.; Zhang, T.; et al. The Global Burden of Osteoporosis, Low Bone Mass, and Its Related Fracture in 204 Countries and Territories, 1990–2019. Front. Endocrinol. 2022, 13, 882241. [Google Scholar] [CrossRef]
- Panahi, N.L.; Saeedi Moghaddam, S.; Fahimfar, N.; Rezaei, N.; Sanjari, M.; Rashidi, M.M.; Shobeiri, P.; Larijani, B.; Ostovar, A. Trend in global burden attributable to low bone mineral density in different WHO regions: 2000 and beyond, results from the Global Burden of Disease (GBD) study 2019. Endocr. Connect. 2023, 12, e230160. [Google Scholar] [CrossRef]
- Xue, N.; Ding, X.; Huang, R.; Jiang, R.; Huang, H.; Pan, X.; Min, W.; Chen, J.; Duan, J.A.; Liu, P.; et al. Bone Tissue Engineering in the Treatment of Bone Defects. Pharmaceuticals 2022, 15, 879. [Google Scholar] [CrossRef] [PubMed]
- Battafarano, G.; Rossi, M.; De Martino, V.; Marampon, F.; Borro, L.; Secinaro, A.; Del Fattore, A. Strategies for Bone Regeneration: From Graft to Tissue Engineering. Int. J. Mol. Sci. 2021, 22, 1128. [Google Scholar] [CrossRef]
- Tang, G.; Liu, Z.; Liu, Y.; Yu, J.; Wang, X.; Tan, Z.; Ye, X. Recent Trends in the Development of Bone Regenerative Biomaterials. Front. Cell Dev. Biol. 2021, 9, 665813. [Google Scholar] [CrossRef]
- Epple, M. Review of potential health risks associated with nanoscopic calcium phosphate. Acta Biomater. 2018, 77, 1–14. [Google Scholar] [CrossRef]
- Galindo, T.G.P.; Chai, Y.; Tagaya, M. Hydroxyapatite Nanoparticle Coating on Polymer for Constructing Effective Biointeractive Interfaces. J. Nanomater. 2019, 2019, 6495239. [Google Scholar] [CrossRef]
- Firdaus Hussin, M.S.; Abdullah, H.Z.; Idris, M.I.; Abdul Wahap, M.A. Extraction of natural hydroxyapatite for biomedical applications—A review. Heliyon 2022, 8, e10356. [Google Scholar] [CrossRef] [PubMed]
- Fontana, C.E.; Dos Santos, B.A.; Campos, M.C.; de Lima, S.G.; da Silva, V.C.; Gonçalves, A.D.; de Moura, J.D.; Rocha, D.G.; Pinheiro, S.L.; Bueno, C.S. Evaluation of the apical sealing of an eggshell hydroxyapatite-based sealer. J. Clin. Exp. Dent. 2023, 15, e895–e903. [Google Scholar] [CrossRef]
- Tagliari, I.; Lerner, A.M.; Severo, A.L.; Israel, C.L. Biocomponents Based on Hydroxiapatite: Influence of Sterilization on the Mechanical Resistance. Rev. Bras. Ortop. 2022, 57, 1051–1059. [Google Scholar]
- Wu, X.H.; Wu, Z.Y.; Su, J.C.; Yan, Y.G.; Yu, B.Q.; Wei, J.; Zhao, L.M. Nanohydroxyapatite promotes self-assembly of honeycomb pores in poly(l-lactide) films through breath-figure method and MC3T3-E1 cell functions. RSC Adv. 2015, 5, 6607–6616. [Google Scholar] [CrossRef]
- Shi, X.; Zhou, K.; Huang, F.; Wang, C. Interaction of hydroxyapatite nanoparticles with endothelial cells: Internalization and inhibition of angiogenesis in vitro through the PI3K/Akt pathway. Int. J. Nanomed. 2017, 10, 5781–5795. [Google Scholar] [CrossRef]
- Atak, B.H.; Buyuk, B.; Huysal, M.; Isik, S.; Senel, M.; Metzger, W.; Cetin, G. Preparation and characterization of amine functional nanohydroxyapatite/chitosan bionanocomposite for bone tissue engineering applications. Carbohydr. Polym. 2017, 164, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Huang, X.; Cai, Y.; Tang, R.; Yang, D. Size effect of hydroxyapatite nanoparticles on proliferation and apoptosis of osteoblast-like cells. Acta Biomater. 2009, 5, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Cacciotti, I. Cationic and Anionic Substitutions in Hydroxyapatite. In Handbook of Bioceramics and Biocomposites; Antoniac, I., Ed.; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Bulina, N.V.; Vinokurova, O.B.; Eremina, N.V.; Prosanov, I.Y.; Khusnutdinov, V.R.; Chaikina, M.V. Features of solid-phase mechanochemical synthesis of hydroxyapatite doped by copper and zinc ions. J. Solid State Chem. 2021, 296, 121973. [Google Scholar] [CrossRef]
- Basirun, W.J.; Nasiri-Tabrizi, B.; Baradaran, S. Overview of hydroxyapatite–graphene nanoplatelets composite as bone graft substitute: Mechanical behavior and in-vitro biofunctionality. Crit. Rev. Solid State Mater. Sci. 2018, 43, 177–212. [Google Scholar] [CrossRef]
- Kurzyk, A.; Szwed-Georgiou, A.; Pagacz, J.; Antosik, A.; Tymowicz-Grzyb, P.; Gerle, A.; Szterner, P.; Włodarczyk, M.; Płociński, P.; Urbaniak, M.M.; et al. Calcination and ion substitution improve physicochemical and biological properties of nanohydroxyapatite for bone tissue engineering applications. Sci. Rep. 2023, 13, 15384. [Google Scholar] [CrossRef] [PubMed]
- Badea, M.A.; Balas, M.; Popa, M.; Borcan, T.; Bunea, A.-C.; Predoi, D.; Dinischiotu, A. Biological Response of Human Gingival Fibroblasts to Zinc-Doped Hydroxyapatite Designed for Dental Applications—An In Vitro Study. Materials 2023, 16, 4145. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yuan, Z.; Huang, J. Substituted hydroxyapatite: A recent development. Mater. Technol. 2020, 35, 785–796. [Google Scholar] [CrossRef]
- Tite, T.; Popa, A.-C.; Balescu, L.M.; Bogdan, I.M.; Pasuk, I.; Ferreira, J.M.F.; Stan, G.E. Cationic Substitutions in Hydroxyapatite: Current Status of the Derived Biofunctional Effects and Their In Vitro Interrogation Methods. Materials 2018, 11, 2081. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Das, S.; Mallick, S.P.; Beyene, Z. A review on the antimicrobial and antibiofilm activity of doped hydroxyapatite and its composites for biomedical applications. Mater. Today Commun. 2022, 31, 103311. [Google Scholar] [CrossRef]
- Gu, M.; Li, W.; Jiang, L.; Li, X. Recent progress of rare earth doped hydroxyapatite nanoparticles: Luminescence properties, synthesis and biomedical applications. Acta Biomater. 2022, 148, 22–43. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Dorozhkin, S.V.; Pal, U. Recent progress on fabrication and drug delivery applications of nanostructured hydroxyapatite. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1504. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhou, Z.; Li, W.; Fan, Y.; Li, Z.; Wei, J. Hydroxyapatite based materials for bone tissue engineering: A brief and comprehensive introduction. Crystals 2021, 11, 149. [Google Scholar] [CrossRef]
- Kazin, P.E.; Gazizova, O.R.; Karpov, A.S.; Jansen, M.; Tretyakov, Y.D. Incorporation of 3d-metal ions in the hexagonal channels of the Sr5(PO4)3OH apatite. Solid State Sci. 2007, 9, 82–87. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, T.; Xu, S.; Wang, K.; Yu, S.; Yang, M. Effects of lanthanum on formation and bone-resorbing activity of osteoclast-like cells. J. Rare Earths 2004, 22, 891–895. [Google Scholar]
- Wieszczycka, K.; Staszak, K.; Woźniak-Budych, M.J.; Jurga, S. Lanthanides and tissue engineering strategies for bone regeneration. Coord. Chem. Rev. 2019, 388, 248–267. [Google Scholar] [CrossRef]
- Etchebehere, E.C.; Pereira Neto, C.A.; Lima, M.C.; Santos Ade, O.; Ramos, C.D.; Silva, C.M.; Camargo, E.E. Treatment of bone pain secondary to metastases using samarium-153-EDTMP. Sao Paulo Med. J. 2004, 122, 208–212. [Google Scholar] [CrossRef]
- Herath, H.M.; Di Silvio, L.; Evans, J.R. In vitro evaluation of samarium (III) oxide as a bone substituting material. J. Biomed. Mater. Res. Part A 2010, 94, 130. [Google Scholar] [CrossRef]
- Wilky, B.A.; Loeb, D.M. Beyond palliation: Therapeutic applications of 53Samarium-EDTMP. J. Clin. Exp. Pharmacol. 2013, 3, 1000131. [Google Scholar] [CrossRef] [PubMed]
- Ciobanu, C.S.; Iconaru, S.L.; Popa, C.L.; Motelica-Heino, M.; Predoi, D. Evaluation of Samarium Doped Hydroxyapatite, Ceramics for Medical Application: Antimicrobial Activity. J. Nanomater. 2015, 2015, 849216. [Google Scholar] [CrossRef]
- Iconaru, S.L.; Groza, A.; Gaiaschi, S.; Rokosz, K.; Raaen, S.; Ciobanu, S.C.; Chapon, P.; Predoi, D. Antimicrobial Properties of Samarium Doped Hydroxyapatite Suspensions and Coatings. Coatings 2020, 10, 1124. [Google Scholar] [CrossRef]
- Morais, D.S.; Coelho, J.; Ferraz, M.P.; Gomes, P.S.; Fernandes, M.H.; Hussain, N.S.; Santos, J.D.; Lopes, M.A. Samarium doped glass-reinforced hydroxyapatite with enhanced osteoblastic performance and antibacterial properties for bone tissue regeneration. J. Mater. Chem. B 2014, 2, 5872–5881. [Google Scholar] [CrossRef] [PubMed]
- Clunie, G.; Lui, D.; Cullum, I.; Edwards, J.C.W.; Peter, J.E. Samarium-153-particulate hydroxyapatite radiation synovectomy: Biodistribution data for chronic knee synovitis. J. Nucl. Med. 1995, 36, 51–57. [Google Scholar] [PubMed]
- Zantye, P.; Fernandes, F.; Ramanan, S.R.; Kowshik, M. Rare earth doped hydroxyapatite nanoparticles for in vitro bioimaging applications. Curr. Phys. Chem. 2019, 9, 94–109. [Google Scholar] [CrossRef]
- Nabeel, A.I. Samarium enriches antitumor activity of ZnO nanoparticles via downregulation of CXCR4 receptor and cytochrome P450. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2020, 42, 1010428320909999. [Google Scholar] [CrossRef]
- Costescu, A.; Ciobanu, C.S.; Iconaru, S.L.; Ghita, R.V.; Chifiriuc, C.M.; Marutescu, L.G.; Predoi, D. Fabrication, characterization, and antimicrobial activity, evaluation of low silver concentrations in silver-doped hydroxyapatite nanoparticles. J. Nanomater. 2013, 2013, 5. [Google Scholar] [CrossRef]
- Rodríguez-Lugo, V.; Karthik, T.V.K.; Mendoza-Anaya, D.; Rubio-Rosas, E.; Villaseñor Cerón, L.S.; Reyes-Valderrama, M.I.; Salinas-Rodríguez, E. Wet chemical synthesis of nanocrystalline hydroxyapatite flakes: Effect of pH and sintering temperature on structural and morphological properties. R. Soc. Open Sci. 2018, 5, 180962. [Google Scholar] [CrossRef]
- de Araujo, T.S.; Macedo, Z.S.; de Oliveira, P.A.; Valerio, M.E. Production and characterization of pure and Cr 3+-doped hydroxyapatite for biomedical applications as fluorescent probes. J. Mater. Sci. 2007, 42, 2236–2243. [Google Scholar] [CrossRef]
- Guzman Vazquez, C.; Piña Barba, C.; Munguia, N. Stoichiometric hydroxyapatite obtained by precipitation and sol gel processes. Rev. Mex. Fis. 2005, 51, 284–293. [Google Scholar]
- Ciobanu, C.S.; Iconaru, S.L.; Pasuk, I.; Vasile, B.S.; Lupu, A.R.; Hermenean, A.; Dinischiotu, A.; Predoi, D. Structural properties of silver doped hydroxyapatite and their biocompatibility. Mater. Sci. Eng. C 2013, 33, 1395–1402. [Google Scholar] [CrossRef]
- Ciobanu, C.S.; Massuyeau, F.; Constantin, L.V.; Predoi, D. Structural and physical properties of antibacterial Ag-doped nano-hydroxyapatite synthesized at 100 C. Nanoscale Res. Lett. 2011, 6, 613. [Google Scholar] [CrossRef]
- Predoi, D.; Iconaru, S.L.; Ciobanu, S.C.; Predoi, S.-A.; Buton, N.; Megier, C.; Beuran, M. Development of Iron-Doped Hydroxyapatite Coatings. Coatings 2021, 11, 186. [Google Scholar] [CrossRef]
- Alshemary, A.Z.; Akram, M.; Goh, Y.F.; Kadir, M.R.A.; Abdolahi, A.; Hussain, R. Structural characterization, optical properties and in vitro bioactivity of mesoporous erbium-doped hydroxyapatite. J. Alloys Compd. 2015, 645, 478–486. [Google Scholar] [CrossRef]
- Demirel, B.; Saban, E.; Yaras, A.; Akkurt, F. Synthesis of Gd+3 doped hydroxyapatite ceramics: Optical, thermal and electrical properties. J. Asian Ceram. Soc. 2021, 9, 865–873. [Google Scholar] [CrossRef]
- Ignjatović, N.L.; Mančić, L.; Vuković, M.; Stojanović, Z.; Nikolić, M.G.; Škapin, S.; Jovanović, S.; Veselinović, L.; Uskoković, V.; Lazić, S.; et al. Rare-earth (Gd3+, Yb3+/Tm3+, Eu3+) co-doped hydroxyapatite as magnetic, up-conversion and down-conversion materials for multimodal imaging. Sci. Rep. 2019, 9, 16305. [Google Scholar] [CrossRef]
- Iconaru, S.L.; Motelica-Heino, M.; Predoi, D. Study on europium-doped hydroxyapatite nanoparticles by fourier transform infrared spectroscopy and their antimicrobial properties. J. Spectrosc. 2013, 2013, 284285. [Google Scholar] [CrossRef]
- Tian, M.; Hu, X.; Qu, L.; Zhu, S.; Sun, Y.; Han, G. Versatile and ductile cotton fabric achieved via layer-by-layer self-assembly by consecutive adsorption of graphene doped PEDOT: PSS and chitosan. Carbon 2016, 96, 166–1174. [Google Scholar] [CrossRef]
- ImageJ. Available online: http://imagej.nih.gov/ij (accessed on 10 January 2024).
- Predoi, S.A.; Ciobanu, S.C.; Chifiriuc, C.M.; Iconaru, S.L.; Predoi, D.; Negrila, C.C.; Marinas, I.C.; Raaen, S.; Rokosz, K.; Motelica-Heino, M. Sodium bicarbonate-hydroxyapatite used for removal of lead ions from aqueous solution. Ceram. Int. 2024, 50, 1742–1755. [Google Scholar] [CrossRef]
- Habib, M.L.; Disha, S.A.; Hossain, M.S.; Uddin, M.N.; Ahmed, S. Enhancement of antimicrobial properties by metals doping in nano-crystalline hydroxyapatite for efficient biomedical applications. Heliyon 2024, 10, e23845. [Google Scholar] [CrossRef] [PubMed]
- Patty, D.J.; Nugraheni, A.D.; Ana, I.D.; Yusuf, Y. In vitro bioactivity of 3D microstructure hydroxyapatite/collagen based-egg white as an antibacterial agent. J. Biomed. Mater. Res. 2022, 110, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Debye, P.; Scherrer, P. Interference of irregularly oriented particles in X-rays. Physik. Z. 1916, 17, 277–283. [Google Scholar]
- Yilmaz, B.; Alshemary, A.Z.; Evis, Z. Co-doped hydroxyapatites as potential materials for biomedical applications. Microchem. J. 2019, 144, 443–453. [Google Scholar] [CrossRef]
- Predoi, D.; Iconaru, S.L.; Ciobanu, S.C.; Buton, N.; Predoi, M.V. Complex Evaluation of Nanocomposite-Based Hydroxyapatite for Biomedical Applications. Biomimetics 2023, 8, 528. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Zhang, H.; Wang, S.; Liu, L.; Tan, X.; Liu, S. Atomic Level Design of CoOH+−Hydroxyapatite@C Catalysts for Superfast Degradation of Organics via Peroxymonosulfate Activation. Chem. Commun. 2018, 54, 4919–4922. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yu, H.; Dong, F.; Zhu, B.; Huang, W.; Zhang, S. High efficiency and stability of Au-Cu/hydroxyapatite catalyst for the oxidation of carbon monoxide. RSC Adv. 2017, 7, 45420–45431. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Physical Electronics Inc.: Chanhassen, MN, USA, 1995. [Google Scholar]
- Popa, C.L.; Ciobanu, C.S.; Iconaru, S.L.; Stan, M.; Dinischiotu, A.; Negrila, C.C.; Motelica-Heino, M.; Guegan, R.; Predoi, D. Systematic Investigation and in Vitro Biocompatibility Studies on Mesoporous Europium Doped Hydroxyapatite. Cent. Eur. J. Chem. 2014, 12, 1032–1046. [Google Scholar] [CrossRef]
- Bee, S.-L.; Bustami, Y.; Ul-Hamid, A.; Lim, K.; Abdul Hamid, Z.A. Synthesis of Silver Nanoparticle-Decorated Hydroxyapatite Nanocomposite with Combined Bioactivity and Antibacterial Properties. J. Mater. Sci. Mater. Med. 2021, 32, 106. [Google Scholar] [CrossRef]
- Gomes, G.C.; Borghi, F.F.; Ospina, R.O.; Lopez, E.O.; Borges, F.O.; Mello, A. Nd:YAG (532 nm) pulsed laser deposition produces crystalline hydroxyapatite thin coatings at room temperature. Surf. Coat. Technol. 2017, 329, 174–183. [Google Scholar] [CrossRef]
- Sinulingga, K.; Sirait, M.; Siregar, N.; Abdullah, H. Synthesis and characterizations of natural limestone-derived nano-hydroxyapatite (HAp): A comparison study of different metals doped Haps on antibacterial activity. RSC Adv. 2021, 11, 15896–15904. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Schleg, H.; Kim, K.; Irvine, J.T.S.; Kim, J.H. X-ray photoelectron spectroscopy of Sm-doped layered perovskite for intermediate temperature-operating solid oxide fuel cell. Appl. Surf. Sci. 2014, 288, 695–701. [Google Scholar] [CrossRef]
- Westhauser, F.; Karadjian, M.; Essers, C.; Senger, A.-S.; Hagmann, S.; Schmidmaier, G.; Moghaddam, A. Osteogenic differentiation of mesenchymal stem cells is enhanced in a 45S5-supplemented β-TCP composite scaffold: An in-vitro comparison of Vitoss and Vitoss BA. PLoS ONE 2019, 14, e0212799. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.J.; Cho, Y.E.; Kim, T.; Shin, H.I.; Kwun, I.S. Zinc may increase bone formation through stimulating cell proliferation, alkaline phosphatase activity and collagen synthesis in osteoblastic MC3T3-E1 cells. Nutr. Res. Pract. 2010, 4, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Xu, G.; Cai, S.; Dou, Y.; Zhu, Y.; Hu, H. Preparation, surface properties, and MC3T3-E1 cell response of mesoporous hydroxyapatite thin films. J. Mater. Sci. 2012, 47, 3763–3769. [Google Scholar] [CrossRef]
- Pendleton, E.; Chandar, N. In Vitro Differentiation of Preosteoblast-Like Cells, MC3T3-E1, to Adipocytes Is Enhanced by 1,25(OH)2 Vitamin D3. Front. Endocrinol. 2017, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Hadjidakis, D.J.; Androulakis, I.I. Bone remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef]
- Vimalraj, S. Alkaline phosphatase: Structure, expression and its function in bone mineralization. Gene 2020, 5, 144855. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Li, S.; Han, Y.; Yuan, L.; Yin, M. Internalization of hydroxyapatite nanoparticles in liver cancer cells. J. Mater. Sci. Mater. Med. 2008, 19, 1091–1095. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, C.; Gao, D.; Chen, S.; Zhu, Y.; Sun, J.; Luo, H.; Yu, K.; Fan, H.; Zhang, X. Antitumor Effect by Hydroxyapatite Nanospheres: Activation of Mitochondria-Dependent Apoptosis and Negative Regulation of Phosphatidylinositol-3-Kinase/Protein Kinase B Pathway. ACS Nano 2018, 12, 7838–7854. [Google Scholar] [CrossRef]
- Boanini, E.; Torricelli, P.; Gazzano, M.; Giardino, R.; Bigi, A. Nanocomposites of hydroxyapatite with aspartic acid and glutamic acid and their interaction with osteoblast-like cells. Biomaterials 2006, 27, 4428–4433. [Google Scholar] [CrossRef]
- Ha, S.; Jang, H.; Nam, K.; Beck, G. Nano-hydroxyapatite modulates osteoblast lineage commitment by stimulation of DNA methylation and regulation of gene expression. Biomaterials 2015, 65, 32–42. [Google Scholar] [CrossRef]
- Cui, X.; Liang, T.; Liu, C.; Yuan, Y.; Qian, J. Correlation of particle properties with cytotoxicity and cellular uptake of hydroxyapatite nanoparticles in human gastric cancer cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 67, 453–460. [Google Scholar] [CrossRef]
- Rai, Y.; Pathak, R.; Kumari, N.; Sah, D.K.; Pandey, S.; Kalra, N.; Soni, R.; Dwarakanath, B.S.; Bhatt, A.N. Mitochondrial biogenesis and metabolic hyperactivation limits the application of MTT assay in the estimation of radiation induced growth inhibition. Sci. Rep. 2018, 8, 1531. [Google Scholar] [CrossRef]
- Theiszová, M.; Jantová, S.; Dragúňová, J.; Grznárová, P.; Palou, M. Comparison the cytotoxicity of hydroxyapatite measured by direct cell counting and MTT test in murine fibroblast NIH-3T3 cells. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2005, 149, 393–396. [Google Scholar] [CrossRef]
- Qiao, Y. Tunable luminescence and cytotoxicity of Samarium-doped calcium–strontium–hydroxyapatite for cell imaging. Funct. Mater. Lett. 2022, 15, 2250005. [Google Scholar] [CrossRef]
- Nica, I.C.; Popa, M.; Marutescu, L.; Dinischiotu, A.; Iconaru, S.L.; Ciobanu, S.C.; Predoi, D. Biocompatibility and Antibiofilm Properties of Samarium Doped Hydroxyapatite Coatings: An In Vitro Study. Coatings 2021, 11, 1185. [Google Scholar] [CrossRef]
- Kanematsu, M.; Ikeda, K.; Yamada, Y. Interaction between nitric oxide synthase and cyclooxygenase pathways in osteoblastic MC3T3-E1 cells. J. Bone Miner. Res. 1997, 12, 1789–1796. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta. 2004, 1658, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Salido, M.; Vilches, J.I.; Gutiérrez, J.L.; Vilches, J. Actin cytoskeletal organization in human osteoblasts grown on different dental titanium implant surfaces. Histol. Histopathol. 2007, 22, 1355–1364. [Google Scholar] [CrossRef]
- Suzuki, H.; Tatei, K.; Ohshima, N.; Sato, S.; Izumi, T. Regulation of MC3T3-E1 differentiation by actin cytoskeleton through lipid mediators reflecting the cell differentiation stage. Biochem. Biophys. Res. Commun. 2019, 514, 393–400. [Google Scholar] [CrossRef]
- Gonnerman, K.N.; Brown, L.S.; Chu, T.M. Effects of growth factors on cell migration and alkaline phosphatase release. Biomed. Sci. Instrum. 2006, 42, 60–65. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Lattice Parameter (Å) | c/a Ratio | Volume of Unit Cell (Å)3 | Crystallite Size (nm) | |
|---|---|---|---|---|---|
| a | c | ||||
| HAp | 9.414 | 6.882 | 0.731 | 528.18 | 25.4 |
| SmHAp | 9.442 | 6.892 | 0.729 | 532.10 | 21.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balas, M.; Badea, M.A.; Ciobanu, S.C.; Piciu, F.; Iconaru, S.L.; Dinischiotu, A.; Predoi, D. Biocompatibility and Osteogenic Activity of Samarium-Doped Hydroxyapatite—Biomimetic Nanoceramics for Bone Regeneration Applications. Biomimetics 2024, 9, 309. https://doi.org/10.3390/biomimetics9060309
Balas M, Badea MA, Ciobanu SC, Piciu F, Iconaru SL, Dinischiotu A, Predoi D. Biocompatibility and Osteogenic Activity of Samarium-Doped Hydroxyapatite—Biomimetic Nanoceramics for Bone Regeneration Applications. Biomimetics. 2024; 9(6):309. https://doi.org/10.3390/biomimetics9060309
Chicago/Turabian StyleBalas, Mihaela, Madalina Andreea Badea, Steluta Carmen Ciobanu, Florentina Piciu, Simona Liliana Iconaru, Anca Dinischiotu, and Daniela Predoi. 2024. "Biocompatibility and Osteogenic Activity of Samarium-Doped Hydroxyapatite—Biomimetic Nanoceramics for Bone Regeneration Applications" Biomimetics 9, no. 6: 309. https://doi.org/10.3390/biomimetics9060309
APA StyleBalas, M., Badea, M. A., Ciobanu, S. C., Piciu, F., Iconaru, S. L., Dinischiotu, A., & Predoi, D. (2024). Biocompatibility and Osteogenic Activity of Samarium-Doped Hydroxyapatite—Biomimetic Nanoceramics for Bone Regeneration Applications. Biomimetics, 9(6), 309. https://doi.org/10.3390/biomimetics9060309