Cationic Nanostructures for Vaccines Design



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
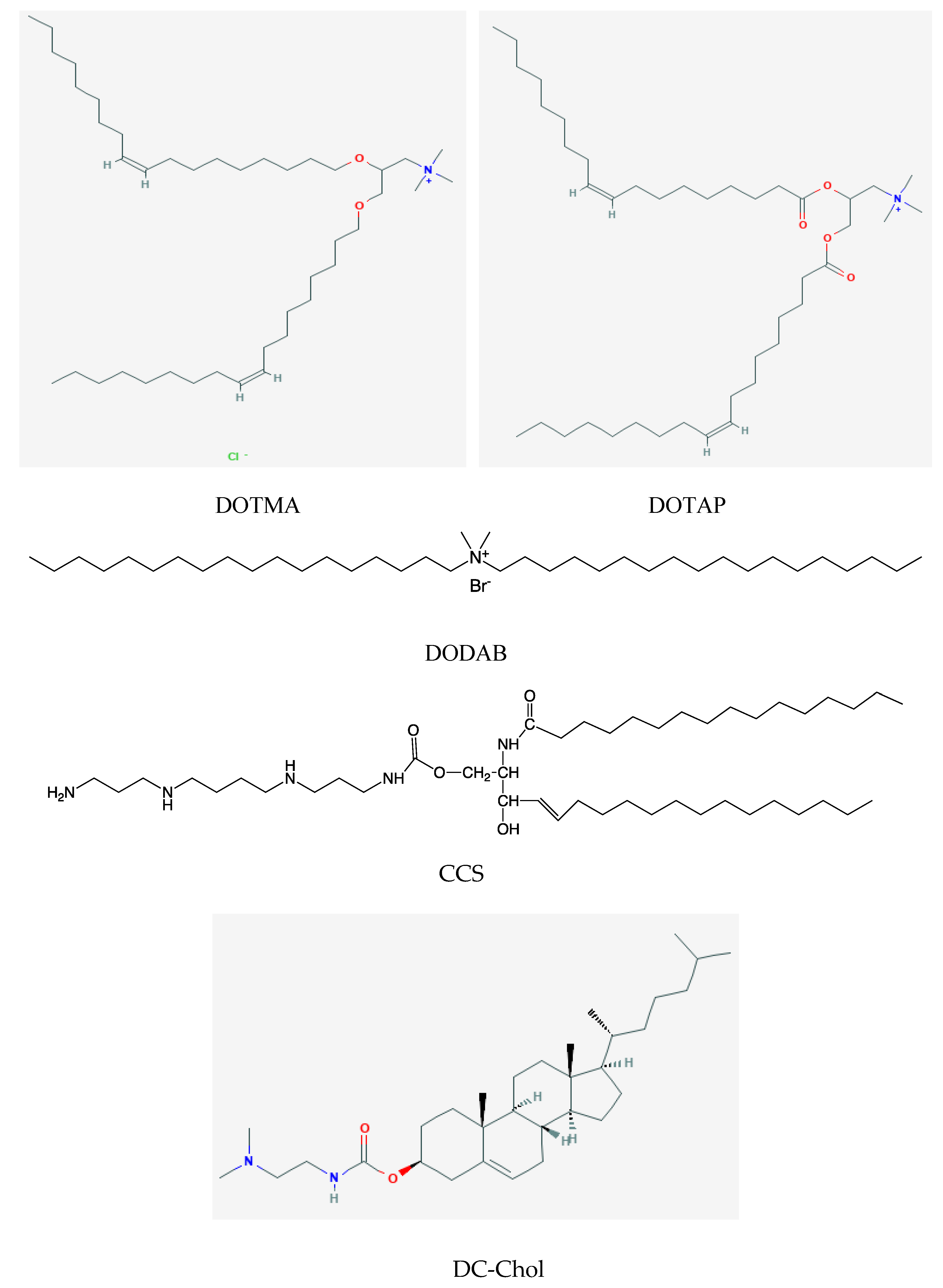
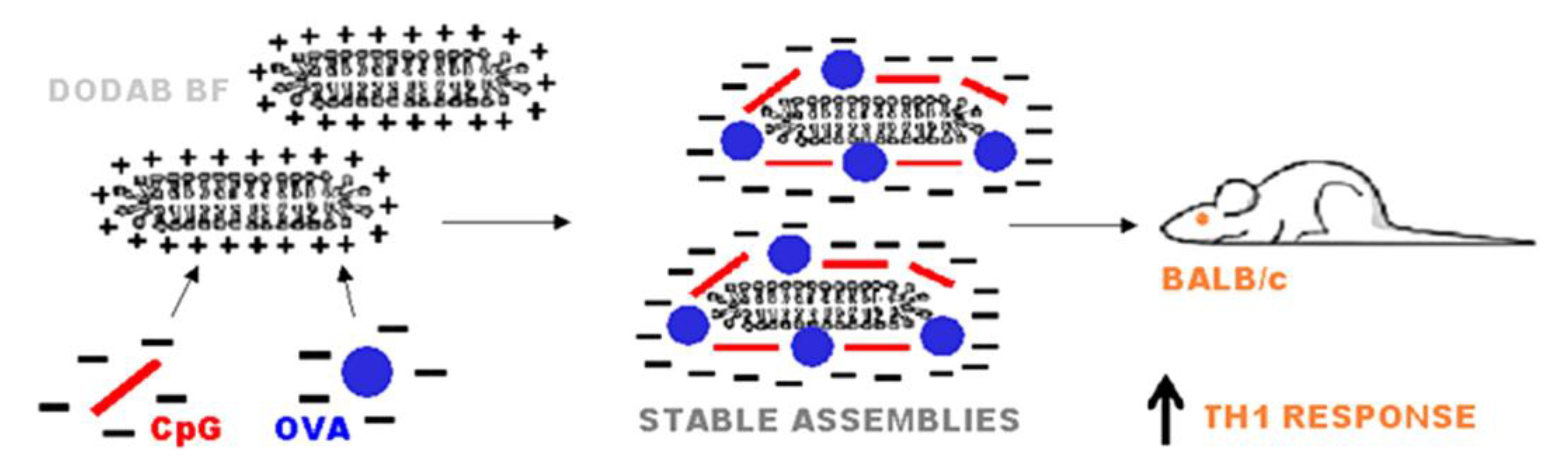
2. Assemblies from Cationic Lipids and Surfactants
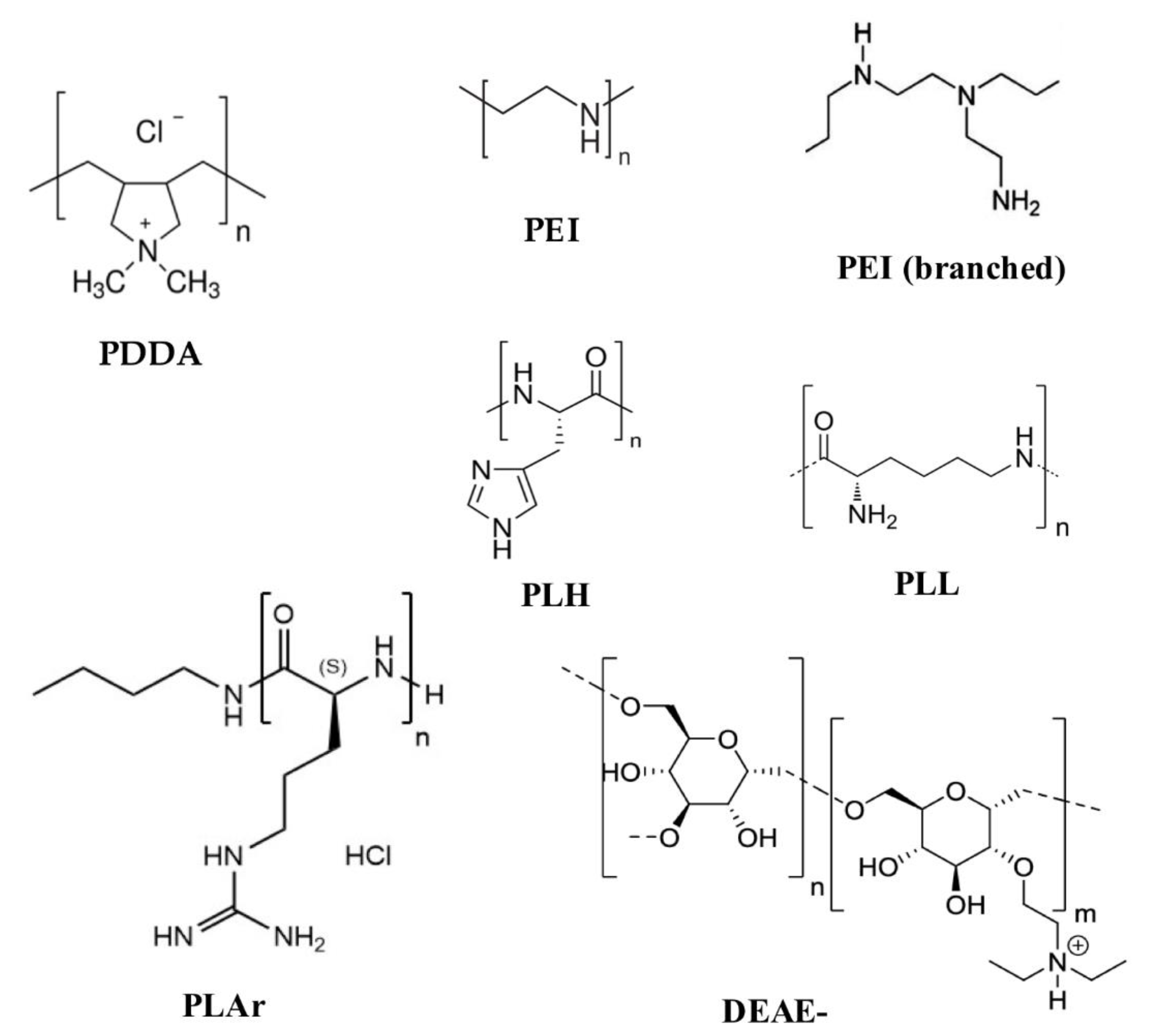
3. Assemblies Based on Cationic Polymers
4. Hybrid Cationic Assemblies of Biocompatible Polymer/Cationic Polymer
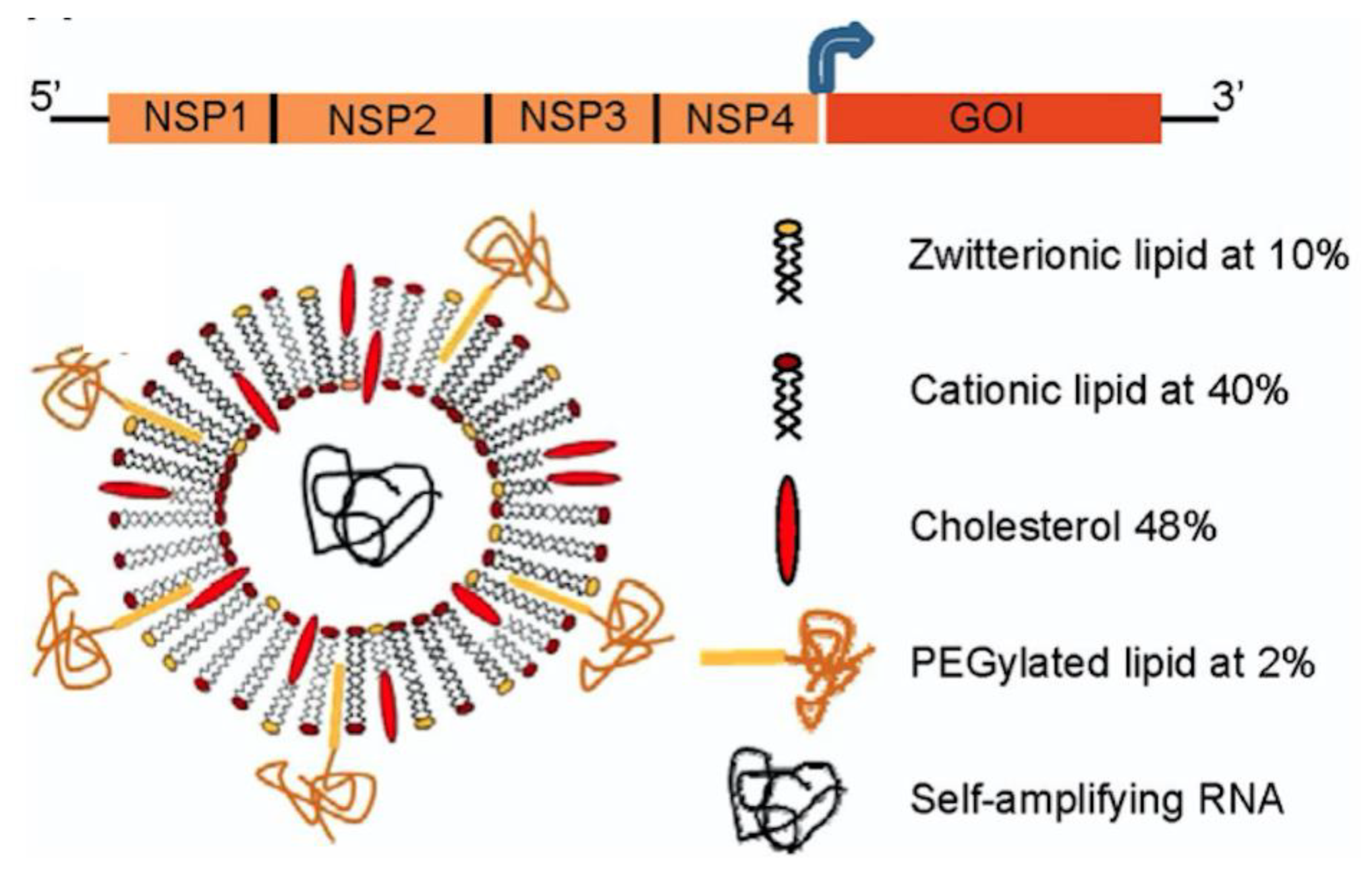
5. Cationic Assemblies of Lipid–Polymer and Polymer–Lipid
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 9th ed.; Elsevier Ltd.: Philadelphia, PA, USA, 2018; ISBN 9780323479783. [Google Scholar]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef]
- Bergmann-Leitner, E.S.; Leitner, W.W. Adjuvants in the driver’s seat: How magnitude, type, fine specificity and longevity of immune responses are driven by distinct classes of immune potentiators. Vaccines 2014, 2, 252–296. [Google Scholar] [CrossRef]
- Pasquale, A.; Preiss, S.; Silva, F.; Garçon, N. Vaccine adjuvants: From 1920 to 2015 and beyond. Vaccines 2015, 3, 320–343. [Google Scholar] [CrossRef]
- Awate, S.; Babiuk, L.A.; Mutwiri, G. Mechanisms of action of adjuvants. Front. Immunol. 2013, 4, 114. [Google Scholar] [CrossRef]
- HogenEsch, H.; O’Hagan, D.T.; Fox, C.B. Optimizing the utilization of aluminum adjuvants in vaccines: You might just get what you want. NPJ Vaccines 2018, 3, 51. [Google Scholar] [CrossRef]
- Martiñón, S.; Cisneros, A.; Villicaña, S.; Hernández-Miramontes, R.; Mixcoha, E.; Calderón-Vargas, P. Chemical and immunological characteristics of aluminum-based, oil-water emulsion, and bacterial-origin adjuvants. J. Immunol. Res. 2019, 2019, 3974127. [Google Scholar] [CrossRef] [PubMed]
- Apostólico, J.D.; Lunardelli, V.A.S.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, modus operandi, and licensing. J. Immunol. Res. 2016, 2016, 1459394. [Google Scholar] [CrossRef]
- Lincopan, N.; Espindola, N.M.; Vaz, A.J.; da Costa, M.H.B.; Faquim-Mauro, E.; Carmona-Ribeiro, A.M. Novel immunoadjuvants based on cationic lipid: Preparation, characterization and activity in vivo. Vaccine 2009, 27, 5760–5771. [Google Scholar] [CrossRef] [PubMed]
- Lincopan, N.; Espindola, N.M.; Vaz, A.J.; Carmona-Ribeiro, A.M. Cationic supported lipid bilayers for antigen presentation. Int. J. Pharm. 2007, 340, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Nawwab Al-Deen, F.M.; Selomulya, C.; Kong, Y.Y.; Xiang, S.D.; Ma, C.; Coppel, R.L.; Plebanski, M. Design of magnetic polyplexes taken up efficiently by dendritic cell for enhanced DNA vaccine delivery. Gene Ther. 2014, 21, 212–218. [Google Scholar] [CrossRef]
- Rose, F.; Wern, J.E.; Gavins, F.; Andersen, P.; Follmann, F.; Foged, C. A strong adjuvant based on glycol-chitosan-coated lipid-polymer hybrid nanoparticles potentiates mucosal immune responses against the recombinant Chlamydia trachomatis fusion antigen CTH522. J. Control. Release 2018, 271, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, L.R.; Quintilio, W.; Costa, M.H.; Carmona-Ribeiro, A.M. Interactions between cationic liposomes and an antigenic protein: The physical chemistry of the immunoadjuvant action. J. Lipid Res. 1997, 38, 2003–2011. [Google Scholar] [PubMed]
- Riehl, M.; Harms, M.; Hanefeld, A.; Baleeiro, R.B.; Walden, P.; Mader, K. Combining R-DOTAP and a particulate antigen delivery platform to trigger dendritic cell activation: Formulation development and in-vitro interaction studies. Int. J. Pharm. 2017, 532, 37–46. [Google Scholar] [CrossRef]
- Davidsen, J.; Rosenkrands, I.; Christensen, D.; Vangala, A.; Kirby, D.; Perrie, Y.; Agger, E.M.; Andersen, P. Characterization of cationic liposomes based on dimethyldioctadecylammonium and synthetic cord factor from M. tuberculosis (trehalose 6,6′-dibehenate)-a novel adjuvant inducing both strong CMI and antibody responses. Biochim. Biophys. Acta 2005, 1718, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, J.H.K.; Silva, S.R.; Raneia, P.A.; Faquim-Mauro, E.; Carmona-Ribeiro, A.M. Stable assemblies of cationic bilayer fragments and CpG oligonucleotide with enhanced immunoadjuvant activity in vivo. J. Control. Release 2012, 160, 367–373. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.F.; De Gaspari, E. Dioctadecyldimethylammonium bromide (DODAB-BF) as a new adjuvant for maternal-fetal immunization in mice against Neisseria meningitidis: Evaluation of humoral response. Pathog. Dis. 2018, 76. [Google Scholar] [CrossRef]
- Lincopan, N.; Santana, M.R.; Faquim-Mauro, E.; da Costa, M.H.B.; Carmona-Ribeiro, A.M. Silica-based cationic bilayers as immunoadjuvants. BMC Biotechnol. 2009, 9, 5. [Google Scholar] [CrossRef]
- Nawwab Al-Deen, F.; Ma, C.; Xiang, S.D.; Selomulya, C.; Plebanski, M.; Coppel, R.L. On the efficacy of malaria DNA vaccination with magnetic gene vectors. J. Control. Release 2013, 168, 10–17. [Google Scholar] [CrossRef]
- Xiang, S.D.; Scholzen, A.; Minigo, G.; David, C.; Apostolopoulos, V.; Mottram, P.L.; Plebanski, M. Pathogen recognition and development of particulate vaccines: Does size matter? Methods 2006, 40, 1–9. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Cationic Nanostructures for Vaccines. In Immune Response Activation; Duc, G.H.T., Ed.; IntechOpen: Rijeka, Croatia, 2014; pp. 1–45. ISBN 978-953-51-1374-4. [Google Scholar]
- Pelkmans, L. Secrets of caveolae- and lipid raft-mediated endocytosis revealed by mammalian viruses. Biochim. Biophys. Acta 2005, 1746, 295–304. [Google Scholar] [CrossRef]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.C.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.T.; MacKichan, M.L.; Singh, M. Recent developments in adjuvants for vaccines against infectious diseases. Biomol. Eng. 2001, 18, 69–85. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Nanomaterials based on lipids for vaccine development. In Micro and Nano Technologies; Skwarczynski, M., Toth, I.B.T.M., Eds.; Elsevier: Oxford, UK, 2017; pp. 241–257. ISBN 978-0-323-39981-4. [Google Scholar]
- Holten-Andersen, L.; Doherty, T.M.; Korsholm, K.S.; Andersen, P. Combination of the cationic surfactant dimethyl dioctadecyl ammonium bromide and synthetic mycobacterial cord factor as an efficient adjuvant for tuberculosis subunit vaccines. Infect. Immun. 2004, 72, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Santos, F.A.; Lincopan, N.; De Gaspari, E. Evaluation of intranasal and subcutaneous route of immunization in neonatal mice using DODAB-BF as adjuvant with outer membrane vesicles of Neisseria meningitis B. Immunobiology 2018, 223, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M. The versatile dioctadecyldimethylammonium bromide. In Application and Characterization of Surfactants; Najjar, R., Ed.; IntechOpen: Rijeka, Croatia, 2017; pp. 157–182. ISBN 9789535133261. [Google Scholar]
- Ju, J.; Huan, M.L.; Wan, N.; Qiu, H.; Zhou, S.Y.; Zhang, B.L. Novel cholesterol-based cationic lipids as transfecting agents of DNA for efficient gene delivery. Int. J. Mol. Sci. 2015, 16, 5666–5681. [Google Scholar] [CrossRef] [PubMed]
- Tada, R.; Suzuki, H.; Takahashi, S.; Negishi, Y.; Kiyono, H.; Kunisawa, J.; Aramaki, Y. Nasal vaccination with pneumococcal surface protein A in combination with cationic liposomes consisting of DOTAP and DC-chol confers antigen-mediated protective immunity against Streptococcus pneumoniae infections in mice. Int. Immunopharmacol. 2018, 61, 385–393. [Google Scholar] [CrossRef]
- Milicic, A.; Kaur, R.; Reyes-Sandoval, A.; Tang, C.K.; Honeycutt, J.; Perrie, Y.; Hill, A.V.S. Small cationic DDA:TDB liposomes as protein vaccine adjuvants obviate the need for TLR agonists in inducing cellular and humoral responses. PLoS ONE 2012, 7, e34255. [Google Scholar] [CrossRef]
- De Serrano, L.O.; Burkhart, D.J. Liposomal vaccine formulations as prophylactic agents: Design considerations for modern vaccines. J. Nanobiotechnol. 2017, 15, 83. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.H.; Gringhuis, S.I. C-type lectin receptors in the control of T helper cell differentiation. Nat. Rev. Immunol. 2016, 16, 433–448. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Biomimetic nanoparticles: Preparation, characterization and biomedical applications. Int. J. Nanomed. 2010, 5, 249–259. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Biomimetic particles in drug and vaccine delivery. J. Liposome Res. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.M.; Chaimovich, H. Preparation and characterization of large dioctadecyldimethylammonium chloride liposomes and comparison with small sonicated vesicles. Biochim. Biophys. Acta 1983, 733, 172–179. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Synthetic amphiphile vesicles. Chem. Soc. Rev. 1992, 21, 209–214. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Bilayer-forming synthetic lipids: Drugs or carriers? Curr. Med. Chem. 2003, 10, 2425–2446. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M. Lipid bilayer fragments and disks in drug delivery. Curr. Med. Chem. 2006, 13, 1359–1370. [Google Scholar] [CrossRef]
- Carvalho, L.A.; Carmona-Ribeiro, A.M. Interactions between cationic vesicles and serum proteins. Langmuir 1998, 14, 6077–6081. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Interactions between cationic liposomes and drugs or biomolecules. An. Acad. Bras. Cienc. 2000, 72, 39–43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rosa, H.; Petri, D.F.S.; Carmona-Ribeiro, A.M. Interactions between bacteriophage DNA and cationic biomimetic particles. J. Phys. Chem. B 2008, 112, 16422–16430. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, I.S.; Carmona-Ribeiro, A.M. Interactions between DNA and Synthetic Cationic Liposomes. J. Phys. Chem. B 2000, 104, 2829–2835. [Google Scholar] [CrossRef]
- Kikuchi, I.S.; Viviani, W.; Carmona-Ribeiro, A.M. Nucleotide Insertion in Cationic Bilayers. J. Phys. Chem. A 1999, 103, 8050–8055. [Google Scholar] [CrossRef]
- Nantes, I.L.; Correia, F.M.; Faljoni-Alario, A.; Kawanami, A.E.; Ishiki, H.M.; Amaral, A.T.; Carmona-Ribeiro, A.M. Nucleotide conformational change induced by cationic bilayers. Arch. Biochem. Biophys. 2003, 416, 25–30. [Google Scholar] [CrossRef]
- Rozenfeld, J.H.K.; Oliveira, T.R.; Lamy, M.T.; Carmona-Ribeiro, A.M. Interaction of cationic bilayer fragments with a model oligonucleotide. Biochim. Biophys. Acta 2011, 1808, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Azmi, F.; Ahmad Fuaad, A.A.H.; Skwarczynski, M.; Toth, I. Recent progress in adjuvant discovery for peptide-based subunit vaccines. Hum. Vaccines Immunother. 2014, 10, 778–796. [Google Scholar] [CrossRef] [PubMed]
- Amorij, J.P.; Kersten, G.F.A.; Saluja, V.; Tonnis, W.F.; Hinrichs, W.L.J.; Slutter, B.; Bal, S.M.; Bouwstra, J.A.; Huckriede, A.; Jiskoot, W. Towards tailored vaccine delivery: Needs, challenges and perspectives. J. Control. Release 2012, 161, 363–376. [Google Scholar] [CrossRef]
- Christensen, D.; Korsholm, K.S.; Andersen, P.; Agger, E.M. Cationic liposomes as vaccine adjuvants. Expert Rev. Vaccines 2011, 10, 513–521. [Google Scholar] [CrossRef]
- Joseph, A.; Itskovitz-Cooper, N.; Samira, S.; Flasterstein, O.; Eliyahu, H.; Simberg, D.; Goldwaser, I.; Barenholz, Y.; Kedar, E. A new intranasal influenza vaccine based on a novel polycationic lipid—Ceramide carbamoyl-spermine (CCS): I. Immunogenicity and efficacy studies in mice. Vaccine 2006, 24, 3990–4006. [Google Scholar] [CrossRef]
- Even-Or, O.; Samira, S.; Rochlin, E.; Balasingam, S.; Mann, A.J.; Lambkin-Williams, R.; Spira, J.; Goldwaser, I.; Ellis, R.; Barenholz, Y. Immunogenicity, protective efficacy and mechanism of novel CCS adjuvanted influenza vaccine. Vaccine 2010, 28, 6527–6541. [Google Scholar] [CrossRef]
- Hilgers, L.A.; Snippe, H. DDA as an immunological adjuvant. Res. Immunol. 1992, 143, 494–496. [Google Scholar] [CrossRef]
- Calzas, C.; Chevalier, C. Innovative mucosal vaccine formulations against influenza A virus infections. Front. Immunol. 2019, 10, 1605. [Google Scholar] [CrossRef]
- Bernocchi, B.; Carpentier, R.; Betbeder, D. Nasal nanovaccines. Int. J. Pharm. 2017, 530, 128–138. [Google Scholar] [CrossRef]
- Shen, H.; Ackerman, A.L.; Cody, V.; Giodini, A.; Hinson, E.R.; Cresswell, P.; Edelson, R.L.; Saltzman, W.M.; Hanlon, D.J. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 2006, 117, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, P.; Liu, L.; Pan, H.; Li, H.; Cai, L.; Ma, Y. Self-adjuvanted nanovaccine for cancer immunotherapy: Role of lysosomal rupture-induced ROS in MHC class I antigen presentation. Biomaterials 2016, 79, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Korsholm, K.S.; Agger, E.M.; Foged, C.; Christensen, D.; Dietrich, J.; Andersen, C.S.; Geisler, C.; Andersen, P. The adjuvant mechanism of cationic dimethyldioctadecylammonium liposomes. Immunology 2007, 121, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Italiani, P. From antigen delivery system to adjuvanticy: The board application of nanoparticles in vaccinology. Vaccines 2015, 3, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Nature and function of gastrointestinal antigen-presenting cells. Allergy 2001, 56, 16–20. [Google Scholar] [CrossRef]
- Bungener, L.; Huckriede, A.; Wilschut, J.; Daemen, T. Delivery of protein antigens to the immune system by fusion-active virosomes: A comparison with liposomes and ISCOMs. Biosci. Rep. 2002, 22, 323–338. [Google Scholar] [CrossRef]
- Diebold, S.S.; Cotten, M.; Koch, N.; Zenke, M. MHC class II presentation of endogenously expressed antigens by transfected dendritic cells. Gene Ther. 2001, 8, 487–493. [Google Scholar] [CrossRef]
- Heath, W.R.; Belz, G.T.; Behrens, G.M.N.; Smith, C.M.; Forehan, S.P.; Parish, I.A.; Davey, G.M.; Wilson, N.S.; Carbone, F.R.; Villadangos, J.A. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol. Rev. 2004, 199, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Sigal, L.J.; Crotty, S.; Andino, R.; Rock, K.L. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature 1999, 402, 25–29. [Google Scholar] [CrossRef]
- Sun, H.X.; Xie, Y.; Ye, Y.P. ISCOMs and ISCOMATRIX. Vaccine 2009, 27, 4388–4401. [Google Scholar] [CrossRef] [PubMed]
- Perez-Betancourt, Y.; Tavora, B.C.; Colombini, M.; Faquim-Mauro, E.L.; Carmona-Ribeiro, A.M. Simple nanoparticles from the assembly of cationic polymer and antigen as immunoadjuvants. Vaccines 2020, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Zhi, D.; Bai, Y.; Yang, J.; Cui, S.; Zhao, Y.; Chen, H.; Zhang, S. A review on cationic lipids with different linkers for gene delivery. Adv. Colloid Interface Sci. 2018, 253, 117–140. [Google Scholar] [CrossRef] [PubMed]
- Chernikov, I.V.; Vlassov, V.V.; Chernolovskaya, E.L. Current development of siRNA bioconjugates: From research to the clinic. Front. Pharmacol. 2019, 10, 444. [Google Scholar] [CrossRef]
- Scherman, D.; Rousseau, A.; Bigey, P.; Escriou, V. Genetic pharmacology: Progresses in siRNA delivery and therapeutic applications. Gene Ther. 2017, 24, 151–156. [Google Scholar] [CrossRef]
- Byk, G.; Dubertret, C.; Escriou, V.; Frederic, M.; Jaslin, G.; Rangara, R.; Pitard, B.; Crouzet, J.; Wils, P.; Schwartz, B.; et al. Synthesis, activity, and structure--activity relationship studies of novel cationic lipids for DNA transfer. J. Med. Chem. 1998, 41, 229–235. [Google Scholar] [CrossRef]
- Pizzuto, M.; Bigey, P.; Lachages, A.M.; Hoffmann, C.; Ruysschaert, J.M.; Escriou, V.; Lonez, C. Cationic lipids as one-component vaccine adjuvants: A promising alternative to alum. J. Control. Release 2018, 287, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Puapatanakul, P.; Chansritrakul, S.; Susantitaphong, P.; Ueaphongsukkit, T.; Eiam-Ong, S.; Praditpornsilpa, K.; Kittanamongkolchai, W.; Avihingsanon, Y. Interferon-inducible protein 10 and disease activity in systemic lupus erythematosus and lupus nephritis: A systematic review and meta-analysis. Int. J. Mol. Sci. 2019, 20, 4954. [Google Scholar] [CrossRef]
- Eisenbarth, S.C.; Colegio, O.R.; O’Connor, W.; Sutterwala, F.S.; Flavell, R.A. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 2008, 453, 1122–1126. [Google Scholar] [CrossRef]
- Schweneker, K.; Gorka, O.; Schweneker, M.; Poeck, H.; Tschopp, J.; Peschel, C.; Ruland, J.; Gross, O. The mycobacterial cord factor adjuvant analogue trehalose-6,6′-dibehenate (TDB) activates the Nlrp3 inflammasome. Immunobiology 2013, 218, 664–673. [Google Scholar] [CrossRef]
- Nguyen, T.K.T.; D’Aigle, J.; Chinea, L.; Niaz, Z.; Hunter, R.L.; Hwang, S.A.; Actor, J.K. Mycobacterial trehalose 6,6′dimycolate-induced M1-type inflammation. Am. J. Pathol. 2020, 190, 286–294. [Google Scholar] [CrossRef]
- Nascimento, D.B.; Rapuano, R.; Lessa, M.M.; Carmona-Ribeiro, A.M. Counterion effects on properties of cationic vesicles. Langmuir 1998, 14, 7387–7391. [Google Scholar] [CrossRef]
- Henriksen-Lacey, M.; Bramwell, V.W.; Christensen, D.; Agger, E.M.; Andersen, P.; Perrie, Y. Liposomes based on dimethyldioctadecylammonium promote a depot effect and enhance immunogenicity of soluble antigen. J. Control. Release 2010, 142, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, C.; Correia, A.; Collins, T.; Vilanova, M.; Pais, C.; Gomes, A.C.; Real Oliveira, M.E.C.D.; Sampaio, P. DODAB:monoolein liposomes containing Candida albicans cell wall surface proteins: A novel adjuvant and delivery system. Eur. J. Pharm. Biopharm. 2015, 89, 190–200. [Google Scholar] [CrossRef]
- Martins, L.S.; Nomura, D.A.; Duarte, E.L.; Riske, K.A.; Lamy, M.T.; Rozenfeld, J.H.K. Structural characterization of cationic DODAB bilayers containing C24:1 beta-glucosylceramide. Biochim. Biophys. Acta Biomembr. 2019, 1861, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Wui, S.R.; Ryu, J.I.; Lee, Y.J.; Hien, D.T.T.; Rhee, I.; Shin, S.J.; Park, S.A.; Kim, K.S.; Cho, Y.J.; et al. Potentiation of Th1-Type Immune Responses to Mycobacterium tuberculosis Antigens in Mice by Cationic Liposomes Combined with De-O-Acylated Lipooligosaccharide. J. Microbiol. Biotechnol. 2018, 28, 136–144. [Google Scholar] [CrossRef]
- Qu, W.; Li, N.; Yu, R.; Zuo, W.; Fu, T.; Fei, W.; Hou, Y.; Liu, Y.; Yang, J. Cationic DDA/TDB liposome as a mucosal vaccine adjuvant for uptake by dendritic cells in vitro induces potent humoural immunity. Artif. Cells Nanomed. Botechnol. 2018, 46, 852–860. [Google Scholar] [CrossRef]
- Silva, J.P.N.; Oliveira, A.C.N.; Casal, M.P.P.A.; Gomes, A.C.; Coutinho, P.J.G.; Coutinho, O.P.; Oliveira, M.E. DODAB:monoolein-based lipoplexes as non-viral vectors for transfection of mammalian cells. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2440–2449. [Google Scholar] [CrossRef]
- Oliveira, A.C.N.; Martens, T.F.; Raemdonck, K.; Adati, R.D.; Feitosa, E.; Botelho, C.; Gomes, A.C.; Braeckmans, K.; Real Oliveira, M.E. Dioctadecyldimethylammonium:monoolein nanocarriers for efficient in vitro gene silencing. ACS Appl. Mater. Interfaces 2014, 6, 6977–6989. [Google Scholar] [CrossRef]
- Carneiro, C.; Correia, A.; Lima, T.; Vilanova, M.; Pais, C.; Gomes, A.C.; Real Oliveira, M.E.; Sampaio, P. Protective effect of antigen delivery using monoolein-based liposomes in experimental hematogenously disseminated candidiasis. Acta Biomater. 2016, 39, 133–145. [Google Scholar] [CrossRef]
- Tada, R.; Hidaka, A.; Iwase, N.; Takahashi, S.; Yamakita, Y.; Iwata, T.; Muto, S.; Sato, E.; Takayama, N.; Honjo, E.; et al. Intranasal immunization with DOTAP cationic liposomes combined with DC-cholesterol induces potent antigen-specific mucosal and systemic immune responses in mice. PLoS ONE 2015, 10, e0139785. [Google Scholar] [CrossRef]
- Jiang, L.; Schinkel, M.; van Essen, M.; Schiffelers, R.M. Bacterial membrane vesicles as promising vaccine candidates. Eur. J. Pharm. Biopharm. 2019, 145, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gnopo, Y.M.D.; Mirsa, A.; Hsu, H.L.; DeLisa, M.P.; Daniel, S.; Putnam, D. Induced fusion and aggregation of bacterial outer membrane vesicles: Experimental and theoretical analysis. J. Colloid Interface Sci. 2020, 578, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Daleke-Schermerhorn, M.H.; Felix, T.; Soprova, Z.; Ten Hagen-Jongman, C.M.; Vikstrom, D.; Majlessi, L.; Beskers, J.; Follmann, F.; de Punder, K.; van der Wel, N.N.; et al. Decoration of outer membrane vesicles with multiple antigens by using an autotransporter approach. Appl. Environ. Microbiol. 2014, 80, 5854–5865. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.T.; Wang, S.H.; Ciotti, S.; Makidon, P.E.; Smith, D.M.; Fan, Y.; Schuler, C.F.; Baker, J.R.J. Formulation and characterization of nanoemulsion intranasal adjuvants: Effects of surfactant composition on mucoadhesion and immunogenicity. Mol. Pharm. 2014, 11, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Barnier Quer, C.; Elsharkawy, A.; Romeijn, S.; Kros, A.; Jiskoot, W. Cationic liposomes as adjuvants for influenza hemagglutinin: More than charge alone. Eur. J. Pharm. Biopharm. 2012, 81, 294–302. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef]
- Kang, T.; Huang, Y.; Zhu, Q.; Cheng, H.; Pei, Y.; Feng, J.; Xu, M.; Jiang, G.; Song, Q.; Jiang, T.; et al. Necroptotic cancer cells-mimicry nanovaccine boosts anti-tumor immunity with tailored immune-stimulatory modality. Biomaterials 2018, 164, 80–97. [Google Scholar] [CrossRef]
- Zhuang, X.; Qi, Y.; Wang, M.; Yu, N.; Nan, F.; Zhang, H.; Tian, M.; Li, C.; Lu, H.; Jin, N. mRNA vaccines encoding the HA protein of influenza A H1N1 virus delivered by cationic lipid nanoparticles induce protective immune responses in mice. Vaccines 2020, 8, 123. [Google Scholar] [CrossRef]
- Guevara, M.L.; Persano, S.; Persano, F. Lipid-based vectors for therapeutic mRNA-based anti-cancer vaccines. Curr. Pharm. Des. 2019, 25, 1443–1454. [Google Scholar] [CrossRef]
- Wang, C.; Liu, P.; Zhuang, Y.; Li, P.; Jiang, B.; Pan, H.; Liu, L.; Cai, L.; Ma, Y. Lymphatic-targeted cationic liposomes: A robust vaccine adjuvant for promoting long-term immunological memory. Vaccine 2014, 32, 5475–5483. [Google Scholar] [CrossRef]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-targeted therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Heyes, J.; Palmer, L.; Bremner, K.; MacLachlan, I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release 2005, 107, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef]
- Shetab Boushehri, M.A.; Lamprecht, A. TLR4-based immunotherapeutics in cancer: A review of the achievements and shortcomings. Mol. Pharm. 2018, 15, 4777–4800. [Google Scholar] [CrossRef]
- Meraz, I.M.; Savage, D.J.; Segura-Ibarra, V.; Li, J.; Rhudy, J.; Gu, J.; Serda, R.E. Adjuvant cationic liposomes presenting MPL and IL-12 induce cell death, suppress tumor growth, and alter the cellular phenotype of tumors in a murine model of breast cancer. Mol. Pharm. 2014, 11, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Kuai, R.; Ochyl, L.J.; Bahjat, K.S.; Schwendeman, A.; Moon, J.J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16, 489–496. [Google Scholar] [CrossRef]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Tada, R.; Muto, S.; Iwata, T.; Hidaka, A.; Kiyono, H.; Kunisawa, J.; Aramaki, Y. Attachment of class B CpG ODN onto DOTAP/DC-chol liposome in nasal vaccine formulations augments antigen-specific immune responses in mice. BMC Res. Notes 2017, 10, 68. [Google Scholar] [CrossRef]
- de Oliveira, C.M.; Fregnani, J.H.; Villa, L.L. HPV vaccine: Updates and highlights. Acta Cytol. 2019, 63, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yan, W.; Huang, L. A simple but effective cancer vaccine consisting of an antigen and a cationic lipid. Cancer Immunol. Immunother. 2008, 57, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Varypataki, E.M.; Benne, N.; Bouwstra, J.; Jiskoot, W.; Ossendorp, F. Efficient eradication of established tumors in mice with cationic liposome-based synthetic long-peptide vaccines. Cancer Immunol. Res. 2017, 5, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Brunel, F.; Darbouret, A.; Ronco, J. Cationic lipid DC-Chol induces an improved and balanced immunity able to overcome the unresponsiveness to the hepatitis B vaccine. Vaccine 1999, 17, 2192–2203. [Google Scholar] [CrossRef]
- Friend, D.S.; Papahadjopoulos, D.; Debs, R.J. Endocytosis and intracellular processing accompanying transfection mediated by cationic liposomes. Biochim. Biophys. Acta 1996, 1278, 41–50. [Google Scholar] [CrossRef]
- Hobernik, D.; Bros, M. DNA Vaccines-How Far From Clinical Use? Int. J. Mol. Sci. 2018, 19, 3605. [Google Scholar] [CrossRef]
- Hermanson, G.; Whitlow, V.; Parker, S.; Tonsky, K.; Rusalov, D.; Ferrari, M.; Lalor, P.; Komai, M.; Mere, R.; Bell, M.; et al. A cationic lipid-formulated plasmid DNA vaccine confers sustained antibody-mediated protection against aerosolized anthrax spores. Proc. Natl. Acad. Sci. USA 2004, 101, 13601–13606. [Google Scholar] [CrossRef]
- Perrie, Y.; McNeil, S.; Vangala, A. Liposome-mediated DNA immunisation via the subcutaneous route. J. Drug Target. 2003, 11, 555–563. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Bacon, A.; Caparros-Wanderley, W.; McCormack, B. A role for liposomes in genetic vaccination. Vaccine 2002, 20. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Saffie, R.; de Souza, J.B. Liposome-mediated DNA vaccination. FEBS Lett. 1997, 402, 107–110. [Google Scholar] [CrossRef]
- Perrie, Y.; Barralet, J.E.; McNeil, S.; Vangala, A. Surfactant vesicle-mediated delivery of DNA vaccines via the subcutaneous route. Int. J. Pharm. 2004, 284, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W.; Gregoriadis, G.; Black, C.D. Liposomes as vehicles for the local release of drugs. Clin. Sci. Mol. Med. 1975, 49, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Szoka, F.C.; Xu, Y.; Zelphati, O. How are nucleic acids released in cells from cationic lipid-nucleic acid complexes? J. Liposome Res. 1996, 6, 567–587. [Google Scholar] [CrossRef]
- Zelphati, O.; Szoka, F.C.J. Intracellular distribution and mechanism of delivery of oligonucleotides mediated by cationic lipids. Pharm. Res. 1996, 13, 1367–1372. [Google Scholar] [CrossRef]
- Velinova, M.; Read, N.; Kirby, C.; Gregoriadis, G. Morphological observations on the fate of liposomes in the regional lymph nodes after footpad injection into rats. Biochim. Biophys. Acta 1996, 1299, 207–215. [Google Scholar] [CrossRef]
- Smith, D.M.; Simon, J.K.; Baker, J.R.J. Applications of nanotechnology for immunology. Nat. Rev. Immunol. 2013, 13, 592–605. [Google Scholar] [CrossRef]
- Wade, A.; Weller, P. Pharmaceutical Excipients, 2nd ed.; American Pharmaceutical Association: Washington, DC, USA; The Pharmaceutical Press: London, UK, 1994; ISBN 0 85369 305 6. [Google Scholar]
- Cui, Z.; Qiu, F.; Sloat, B.R. Lecithin-based cationic nanoparticles as a potential DNA delivery system. Int. J. Pharm. 2006, 313, 206–213. [Google Scholar] [CrossRef]
- Lonez, C.; Vandenbranden, M.; Ruysschaert, J.M. Cationic lipids activate intracellular signaling pathways. Adv. Drug Deliv. Rev. 2012, 64, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, P.; Bhatia, E.; Sharma, S.; Ahamad, N.; Banerjee, R. Advancements in prophylactic and therapeutic nanovaccines. Acta Biomater. 2020, 108, 1–21. [Google Scholar] [CrossRef]
- Mikelez-Alonso, I.; Aires, A.; Cortajarena, A.L. Cancer nano-immunotherapy from the injection to the target: The role of protein corona. Int. J. Mol. Sci. 2020, 21, 519. [Google Scholar] [CrossRef]
- Fadeel, B. Hide and seek: Nanomaterial interactions with the immune system. Front. Immunol. 2019, 10, 133. [Google Scholar] [CrossRef]
- Gilabert-Oriol, R.; Ryan, G.M.; Leung, A.W.Y.; Firmino, N.S.; Bennewith, K.L.; Bally, M.B. Liposomal formulations to modulate the tumour microenvironment and antitumour immune response. Int. J. Mol. Sci. 2018, 19, 2922. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wijewardhana, C.; Mann, J.F.S. Virus-like particle, liposome, and polymeric particle-based vaccines against HIV-1. Front. Immunol. 2018, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Monnery, B.D.; Wright, M.; Cavill, R.; Hoogenboom, R.; Shaunak, S.; Steinke, J.H.G.; Thanou, M. Cytotoxicity of polycations: Relationship of molecular weight and the hydrolytic theory of the mechanism of toxicity. Int. J. Pharm. 2017, 521, 249–258. [Google Scholar] [CrossRef]
- Carrasco, L.D.; Sampaio, J.L.M.; Carmona-Ribeiro, A.M. Supramolecular cationic assemblies against multidrug-resistant microorganisms: Activity and mechanism of action. Int. J. Mol. Sci. 2015, 16, 6337–6352. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Hwang, J.; Lee, H.; Hammond, P.T.; Choi, J.; Hong, J. In vitro blood cell viability profiling of polymers used in molecular assembly. Sci. Rep. 2017, 7, 9481. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Ortis, F.; Schumacher, R.I.; Armelin, M.C.S. Interactions between cationic vesicles and cultured mammalian cells. Langmuir 1997, 13, 2215–2218. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, K.; Si, Y.; Guo, X.; Xu, Y. Protein-polyelectrolyte interaction: Thermodynamic analysis based on the titration method (†). Polymers 2019, 11, 82. [Google Scholar] [CrossRef]
- Singh, M.; Briones, M.; Ott, G.; O’Hagan, D. Cationic microparticles: A potent delivery system for DNA vaccines. Proc. Natl. Acad. Sci. USA 2000, 97, 811–816. [Google Scholar] [CrossRef]
- Wusiman, A.; Gu, P.; Liu, Z.; Xu, S.; Zhang, Y.; Hu, Y.; Liu, J.; Wang, D.; Huang, X. Cationic polymer modified PLGA nanoparticles encapsulating Alhagi honey polysaccharides as a vaccine delivery system for ovalbumin to improve immune responses. Int. J. Nanomed. 2019, 14, 3221–3234. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, F.; Gartlan, K.H.; Harandi, A.M.; Brinckmann, S.A.; Coccia, M.; Hillson, W.R.; Kok, W.L.; Cole, S.; Ho, L.P.; Lambe, T.; et al. Polyethyleneimine is a potent mucosal adjuvant for viral glycoprotein antigens. Nat. Biotechnol. 2012, 30, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, N.C.; Brinckmann, S.A.; Gartlan, K.H.; Puthia, M.; Svanborg, C.; Krashias, G.; Eisenbarth, S.C.; Flavell, R.A.; Sattentau, Q.J.; Wegmann, F. Polyethyleneimine is a potent systemic adjuvant for glycoprotein antigens. Int. Immunol. 2014, 26, 531–538. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; de Melo Carrasco, L.D. Cationic antimicrobial polymers and their assemblies. Int. J. Mol. Sci. 2013, 14, 9906–9946. [Google Scholar] [CrossRef] [PubMed]
- Vieira, D.B.; Carmona-Ribeiro, A.M. Cationic nanoparticles for delivery of amphotericin B: Preparation, characterization and activity in vitro. J. Nanobiotechnol. 2008, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.D.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Antimicrobial particles from cationic lipid and polyelectrolytes. Langmuir 2010, 26, 12300–12306. [Google Scholar] [CrossRef]
- Bohidar, H.; Dubin, P.L.; Majhi, P.R.; Tribet, C.; Jaeger, W. Effects of protein-polyelectrolyte affinity and polyelectrolyte molecular weight on dynamic properties of bovine serum albumin-poly(diallyldimethylammonium chloride) coacervates. Biomacromolecules 2005, 6, 1573–1585. [Google Scholar] [CrossRef]
- Kayitmazer, A.B.; Strand, S.P.; Tribet, C.; Jaeger, W.; Dubin, P.L. Effect of polyelectrolyte structure on protein-polyelectrolyte coacervates: Coacervates of bovine serum albumin with poly(diallyldimethylammonium chloride) versus chitosan. Biomacromolecules 2007, 8, 3568–3577. [Google Scholar] [CrossRef]
- Xu, L.; Liu, Y.; Chen, Z.; Li, W.; Liu, Y.; Wang, L.; Liu, Y.; Wu, X.; Ji, Y.; Zhao, Y.; et al. Surface-engineered gold nanorods: Promising DNA vaccine adjuvant for HIV-1 treatment. Nano Lett. 2012, 12, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Chen, W.C.W.; Heo, Y.; Wang, Y. Polycations and their biomedical applications. Prog. Polym. Sci. 2016, 60, 18–50. [Google Scholar] [CrossRef]
- Shen, C.; Li, J.; Zhang, Y.; Li, Y.; Shen, G.; Zhu, J.; Tao, J. Polyethylenimine-based micro/nanoparticles as vaccine adjuvants. Int. J. Nanomed. 2017, 12, 5443–5460. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, L.; Liu, Q.; Jia, J.; Liu, Y.; Zhang, W.; Ma, G.; Su, Z. Polycation-decorated PLA microspheres induce robust immune responses via commonly used parenteral administration routes. Int. Immunopharmacol. 2014, 23, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Benjaminsen, R.V.; Mattebjerg, M.A.; Henriksen, J.R.; Moghimi, S.M.; Andresen, T.L. The possible “proton sponge ” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol. Ther. 2013, 21, 149–157. [Google Scholar] [CrossRef]
- Akinc, A.; Thomas, M.; Klibanov, A.M.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005, 7, 657–663. [Google Scholar] [CrossRef]
- Le Bihan, O.; Chevre, R.; Mornet, S.; Garnier, B.; Pitard, B.; Lambert, O. Probing the in vitro mechanism of action of cationic lipid/DNA lipoplexes at a nanometric scale. Nucleic Acids Res. 2011, 39, 1595–1609. [Google Scholar] [CrossRef]
- Démoulins, T.; Milona, P.; Englezou, P.C.; Ebensen, T.; Schulze, K.; Suter, R.; Pichon, C.; Midoux, P.; Guzmán, C.A.; Ruggli, N.; et al. Polyethylenimine-based polyplex delivery of self-replicating RNA vaccines. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Regnstrom, K.; Ragnarsson, E.G.E.; Koping-Hoggard, M.; Torstensson, E.; Nyblom, H.; Artursson, P. PEI—A potent, but not harmless, mucosal immuno-stimulator of mixed T-helper cell response and FasL-mediated cell death in mice. Gene Ther. 2003, 10, 1575–1583. [Google Scholar] [CrossRef]
- Chen, J.; Li, Z.; Huang, H.; Yang, Y.; Ding, Q.; Mai, J.; Guo, W.; Xu, Y. Improved antigen cross-presentation by polyethyleneimine-based nanoparticles. Int. J. Nanomed. 2011, 6, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Yin, Y.; Huang, L.; Yu, Q.; Yang, Q. H9N2 influenza whole inactivated virus combined with polyethyleneimine strongly enhances mucosal and systemic immunity after intranasal immunization in mice. Clin. Vaccine Immunol. 2015, 22, 421–429. [Google Scholar] [CrossRef]
- Dhanwani, R.; Takahashi, M.; Sharma, S. Cytosolic sensing of immuno-stimulatory DNA, the enemy within. Curr. Opin. Immunol. 2018, 50, 82–87. [Google Scholar] [CrossRef]
- Ma, Y.F.; Yang, Y.W. Delivery of DNA-based cancer vaccine with polyethylenimine. Eur. J. Pharm. Sci. 2010, 40, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Dautzenberg, H.; Kunath, K.; Kissel, T. Poly(diallyldimethylammonium chlorides) and their N-methyl-N-vinylacetamide copolymer-based DNA-polyplexes: Role of molecular weight and charge density in complex formation, stability, and in vitro activity. Int. J. Pharm. 2004, 280, 253–269. [Google Scholar] [CrossRef]
- Alatorre-Meda, M.; Taboada, P.; Krajewska, B.; Willemeit, M.; Deml, A.; Klösel, R.; Rodríguez, J.R. DNA−Poly(diallyldimethylammonium chloride) Complexation and Transfection Efficiency. J. Phys. Chem. B 2010, 114, 9356–9366. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Wu, B.; Zhao, Z.; Yin, F.; Li, S.; Qin, X.; Chen, Q. Dispersion of single-walled carbon nanotubes in poly(diallyldimethylammonium chloride) for preparation of a glucose biosensor. Sens. Actuators B Chem. 2008, 130, 809–815. [Google Scholar] [CrossRef]
- Sanches, L.M.; Petri, D.F.S.; de Melo Carrasco, L.D.; Carmona-Ribeiro, A.M. The antimicrobial activity of free and immobilized poly (diallyldimethylammonium) chloride in nanoparticles of poly (methylmethacrylate). J. Nanobiotechnol. 2015, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Galvão, C.N.; Sanches, L.M.; Mathiazzi, B.I.; Ribeiro, R.T.; Petri, D.F.S.; Carmona-Ribeiro, A.M. Antimicrobial coatings from hybrid nanoparticles of biocompatible and antimicrobial polymers. Int. J. Mol. Sci. 2018, 19, 2965. [Google Scholar] [CrossRef]
- Ribeiro, R.T.; Galvão, C.N.; Betancourt, Y.P.; Mathiazzi, B.I.; Carmona-Ribeiro, A.M. Microbicidal dispersions and coatings from hybrid nanoparticles of poly (methyl methacrylate), poly (diallyl dimethyl ammonium) chloride, lipids, and surfactants. Int. J. Mol. Sci. 2019, 20, 6150. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Biomimetic nanomaterials from the assembly of polymers, lipids, and surfactants. In Surfactants and Detergents; Dutta, A.K., Ed.; IntechOpen: Rijeka, Croatia, 2019; pp. 1–16. ISBN 978-1-78984-661-4. [Google Scholar]
- Leong, H.S.; Butler, K.S.; Brinker, C.J.; Azzawi, M.; Conlan, S.; Dufés, C.; Owen, A.; Rannard, S.; Scott, C.; Chen, C.; et al. On the issue of transparency and reproducibility in nanomedicine. Nat. Nanotechnol. 2019, 14, 629–635. [Google Scholar] [CrossRef]
- Rehman, Z.U.; Hoekstra, D.; Zuhorn, I.S. Mechanism of polyplex- and lipoplex-mediated delivery of nucleic acids: Real-time visualization of transient membrane destabilization without endosomal lysis. ACS Nano 2013, 7, 3767–3777. [Google Scholar] [CrossRef]
- Rejman, J.; Bragonzi, A.; Conese, M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol. Ther. 2005, 12, 468–474. [Google Scholar] [CrossRef]
- Siewert, C.; Haas, H.; Nawroth, T.; Ziller, A.; Nogueira, S.S.; Schroer, M.A.; Blanchet, C.E.; Svergun, D.I.; Radulescu, A.; Bates, F.; et al. Investigation of charge ratio variation in mRNA—DEAE-dextran polyplex delivery systems. Biomaterials 2019, 192, 612–620. [Google Scholar] [CrossRef]
- Rice, J.M.; Madison, R.M. Subcutaneous injections of vaccine adjuvant DEAE-dextran induce local sarcomas in mice. Nat. New Biol. 1972, 236, 28. [Google Scholar] [CrossRef] [PubMed]
- Houston, W.E.; Crabbs, C.L.; Kremer, R.J.; Springer, J.W. Adjuvant effects of diethylaminoethyl-dextran. Infect. Immun. 1976, 13, 1559–1562. [Google Scholar] [CrossRef] [PubMed]
- Joo, I.; Emod, J. Adjuvant effect of DEAE-dextran on cholera vaccines. Vaccine 1988, 6, 233–237. [Google Scholar] [CrossRef]
- Wittmann, G.; Bauer, K.; Mussgay, M. [Trials of vaccination of pigs with vaccines of inactivated foot-and-mouth disease (FMD) virus. II. Trials with ethyl ethyleneimine (EEI) inactivated virus and diethylaminoethyl dextran as an adjuvant]. Arch. Gesamte Virusforsch. 1970, 29, 139–158. [Google Scholar] [CrossRef]
- Bakrania, A.K.; Variya, B.C.; Patel, S.S. Role of β-interferon inducer (DEAE-dextran) in tumorigenesis by VEGF and NOTCH1 inhibition along with apoptosis induction. Front. Pharmacol. 2017, 8, 930. [Google Scholar] [CrossRef]
- Ji, W.; Panus, D.; Palumbo, R.N.; Tang, R.; Wang, C. Poly(2-aminoethyl methacrylate) with well-defined chain length for DNA vaccine delivery to dendritic cells. Biomacromolecules 2011, 12, 4373–4385. [Google Scholar] [CrossRef]
- Chen, W.; Hong, Y.; Zhang, T.; Kong, D.; Zhang, M.; Zhang, Q.; Wang, C. Star-shaped poly(2-aminoethyl methacrylate)s as non-viral gene carriers: Exploring structure-function relationship. Colloids Surf. B Biointerfaces 2019, 181, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Farris, E.; Brown, D.M.; Ramer-Tait, A.E.; Pannier, A.K. Micro- and nanoparticulates for DNA vaccine delivery. Exp. Biol. Med. 2016, 241, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Mazid, R.; Tan, M.X.; Danquah, M.K. Molecular delivery of plasmids for genetic vaccination. Curr. Pharm. Biotechnol. 2013, 14, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Luhrs, P.; Schmidt, W.; Kutil, R.; Buschle, M.; Wagner, S.N.; Stingl, G.; Schneeberger, A. Induction of specific immune responses by polycation-based vaccines. J. Immunol. 2002, 169, 5217–5226. [Google Scholar] [CrossRef]
- De Koker, S.; De Geest, B.G.; Singh, S.K.; De Rycke, R.; Naessens, T.; Van Kooyk, Y.; Demeester, J.; De Smedt, S.C.; Grooten, J. Polyelectrolyte microcapsules as antigen delivery vehicles to dendritic cells: Uptake, processing, and cross-presentation of encapsulated antigens. Angew. Chem. Int. Ed. 2009, 48, 8485–8489. [Google Scholar] [CrossRef] [PubMed]
- Devriendt, B.; Baert, K.; Dierendonck, M.; Favoreel, H.; De Koker, S.; Remon, J.P.; De Geest, B.G.; Cox, E. One-step spray-dried polyelectrolyte microparticles enhance the antigen cross-presentation capacity of porcine dendritic cells. Eur. J. Pharm. Biopharm. 2013, 84, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Mattner, F.; Fleitmann, J.K.; Lingnau, K.; Schmidt, W.; Egyed, A.; Fritz, J.; Zauner, W.; Wittmann, B.; Gorny, I.; Berger, M.; et al. Vaccination with poly-l-arginine as immunostimulant for peptide vaccines: Induction of potent and long-lasting T-cell responses against cancer antigens. Cancer Res. 2002, 62, 1477–1480. [Google Scholar] [PubMed]
- Maubant, S.; Banissi, C.; Beck, S.; Chauvat, A.; Carpentier, A.F. Adjuvant properties of cytosine-phosphate-guanosine oligodeoxynucleotide in combination with various polycations in an ovalbumin-vaccine model. Nucleic Acid Ther. 2011, 21, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Lingnau, K.; Egyed, A.; Schellack, C.; Mattner, F.; Buschle, M.; Schmidt, W. Poly-l-arginine synergizes with oligodeoxynucleotides containing CpG-motifs (CpG-ODN) for enhanced and prolonged immune responses and prevents the CpG-ODN-induced systemic release of pro-inflammatory cytokines. Vaccine 2002, 20, 3498–3508. [Google Scholar] [CrossRef]
- Mulens-Arias, V.; Rojas, J.M.; Sanz-Ortega, L.; Portilla, Y.; Perez-Yague, S.; Barber, D.F. Polyethylenimine-coated superparamagnetic iron oxide nanoparticles impair in vitro and in vivo angiogenesis. Nanomedicine 2019, 21, 102063. [Google Scholar] [CrossRef] [PubMed]
- Mulens-Arias, V.; Rojas, J.M.; Pérez-Yagüe, S.; Morales, M.P.; Barber, D.F. Polyethylenimine-coated SPIONs trigger macrophage activation through TLR-4 signaling and ROS production and modulate podosome dynamics. Biomaterials 2015, 52, 494–506. [Google Scholar] [CrossRef]
- Zarei, H.; Kazemi Oskuee, R.; Hanafi-Bojd, M.Y.; Gholami, L.; Ansari, L.; Malaekeh-Nikouei, B. Enhanced gene delivery by polyethyleneimine coated mesoporous silica nanoparticles. Pharm. Dev. Technol. 2019, 24, 127–132. [Google Scholar] [CrossRef]
- Lazarus, G.G.; Singh, M. In vitro cytotoxic activity and transfection efficiency of polyethyleneimine functionalized gold nanoparticles. Colloids Surf. B. Biointerfaces 2016, 145, 906–911. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, H.; Nian, Q.G.; Yang, Y.; Qin, C.F.; Tang, R. Robust vaccine formulation produced by assembling a hybrid coating of polyethyleneimine-silica. Chem. Sci. 2016, 7, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Li, A.W.; Sobral, M.C.; Badrinath, S.; Choi, Y.; Graveline, A.; Stafford, A.G.; Weaver, J.C.; Dellacherie, M.O.; Shih, T.Y.; Ali, O.A.; et al. A facile approach to enhance antigen response for personalized cancer vaccination. Nat. Mater. 2018, 17, 528–534. [Google Scholar] [CrossRef]
- Minigo, G.; Scholzen, A.; Tang, C.K.; Hanley, J.C.; Kalkanidis, M.; Pietersz, G.A.; Apostolopoulos, V.; Plebanski, M. Poly L-lysine-coated nanoparticles: A potent delivery system to enhance DNA vaccine efficacy. Vaccine 2007, 25, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Wusiman, A.; Wang, S.; Zhang, Y.; Liu, Z.; Hu, Y.; Liu, J.; Wang, D. Polyethylenimine-coated PLGA nanoparticles-encapsulated Angelica sinensis polysaccharide as an adjuvant to enhance immune responses. Carbohydr. Polym. 2019, 223, 115128. [Google Scholar] [CrossRef] [PubMed]
- Salvador, A.; Sandgren, K.J.; Liang, F.; Thompson, E.A.; Koup, R.A.; Pedraz, J.L.; Hernandez, R.M.; Lore, K.; Igartua, M. Design and evaluation of surface and adjuvant modified PLGA microspheres for uptake by dendritic cells to improve vaccine responses. Int. J. Pharm. 2015, 496, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Arif, U.; Haider, S.; Haider, A.; Khan, N.; Alghyamah, A.A.; Jamila, N.; Khan, M.I.; Almasry, W.A.; Kang, I.K. Biocompatible polymers and their potential biomedical applications: A review. Curr. Pharm. Des. 2019, 25, 3608–3619. [Google Scholar] [CrossRef] [PubMed]
- Shastri, V.P. Non-degradable biocompatible polymers in medicine: Past, present and future. Curr. Pharm. Biotechnol. 2003, 4, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Calzoni, E.; Cesaretti, A.; Polchi, A.; Di Michele, A.; Tancini, B.; Emiliani, C. Biocompatible polymer nanoparticles for drug delivery applications in cancer and neurodegenerative disorder therapies. J. Funct. Biomater. 2019, 10, 4. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Malikmammadov, E.; Tanir, T.E.; Kiziltay, A.; Hasirci, V.; Hasirci, N. PCL and PCL-based materials in biomedical applications. J. Biomater. Sci. Polym. Ed. 2018, 29, 863–893. [Google Scholar] [CrossRef]
- Lasprilla, A.J.R.; Martinez, G.A.R.; Lunelli, B.H.; Jardini, A.L.; Filho, R.M. Poly-lactic acid synthesis for application in biomedical devices—A review. Biotechnol. Adv. 2012, 30, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Briso, A.L.; Serrano-Aroca, A. Poly(3-hydroxybutyrate-co-3-hydroxyvalerate): Enhancement strategies for advanced applications. Polymers 2018, 10, 732. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Azad, M.A.K.; Lin, Y.; Kim, S.W.; Tian, Y.; Liu, G.; Wang, H. Biological effects and applications of chitosan and chito-oligosaccharides. Front. Physiol. 2019, 10, 516. [Google Scholar] [CrossRef] [PubMed]
- Younes, I.; Rinaudo, M. Chitin and chitosan preparation from marine sources. Structure, properties and applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed]
- HPS, A.K.; Saurabh, C.K.; Adnan, A.S.; Nurul Fazita, M.R.; Syakir, M.I.; Davoudpour, Y.; Rafatullah, M.; Abdullah, C.K.; Haafiz, M.K.M.; Dungani, R. A review on chitosan-cellulose blends and nanocellulose reinforced chitosan biocomposites: Properties and their applications. Carbohydr. Polym. 2016, 150, 216–226. [Google Scholar]
- Ali, U.; Karim, K.J.B.A.; Buang, N.A. A review of the properties and applications of poly (methyl methacrylate) (PMMA). Polym. Rev. 2015, 55, 678–705. [Google Scholar] [CrossRef]
- Zaman, M.; Skwarczynski, M.; Malcolm, J.M.; Urbani, C.N.; Jia, Z.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Self-adjuvanting polyacrylic nanoparticulate delivery system for group A streptococcus (GAS) vaccine. Nanomedicine 2011, 7, 168–173. [Google Scholar] [CrossRef]
- O’Hagan, D.T.; Singh, M. Microparticles as vaccine adjuvants and delivery systems. Expert Rev. Vaccines 2003, 2, 269–283. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Midmore, B.R. Synthetic bilayer adsorption onto polystyrene microspheres. Langmuir 1992, 8, 801–806. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; de Moraes Lessa, M. Interactions between bilayer membranes and latex. Colloids Surf. A Physicochem. Eng. Asp. 1999, 153, 355–361. [Google Scholar] [CrossRef]
- Xavier, G.R.S.; Carmona-Ribeiro, A.M. Cationic biomimetic particles of polystyrene/cationic bilayer/gramicidin for optimal bactericidal activity. Nanomaterials 2017, 7, 422. [Google Scholar] [CrossRef] [PubMed]
- Moura, S.P.; Carmona-Ribeiro, A.M. Cationic bilayer fragments on silica at low Ionic strength: Competitive adsorption and colloid stability. Langmuir 2003, 19, 6664–6667. [Google Scholar] [CrossRef]
- Ribeiro, R.T.; Braga, V.H.A.; Carmona-Ribeiro, A.M. Biomimetic cationic nanoparticles based on silica: Optimizing bilayer deposition from lipid films. Biomimetics 2017, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.M.A.; Kosaka, P.M.; Rosa, H.; Vieira, D.B.; Kawano, Y.; Petri, D.F.S.; Carmona-Ribeiro, A.M. Hybrid materials from intermolecular associations between cationic lipid and polymers. J. Phys. Chem. B 2008, 112, 9301–9310. [Google Scholar] [CrossRef]
- Melo, L.D.; Palombo, R.R.; Petri, D.F.S.; Bruns, M.; Pereira, E.M.A.; Carmona-Ribeiro, A.M. Structure-activity relationship for quaternary ammonium compounds hybridized with poly(methyl methacrylate). ACS Appl. Mater. Interfaces 2011, 3, 1933–1939. [Google Scholar] [CrossRef]
- Naves, A.F.; Palombo, R.R.; Carrasco, L.D.M.; Carmona-Ribeiro, A.M. Antimicrobial particles from emulsion polymerization of methyl methacrylate in the presence of quaternary ammonium surfactants. Langmuir 2013, 29, 9677–9684. [Google Scholar] [CrossRef]
- Mathiazzi, B.I.; Carmona-Ribeiro, A.M. Hybrid nanoparticles of poly (methyl methacrylate) and antimicrobial quaternary ammonium surfactants. Pharmaceutics 2020, 12, 340. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Self-assembled antimicrobial nanomaterials. Int. J. Environ. Res. Public Health 2018, 15, 1408. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Biomimetic lipid polymer nanoparticles for drug delivery. In Nanoparticles in Biology and Medicine. Methods in Molecular Biology; Ferrari, E., Soloviev, M., Eds.; Springer Nature: New York, NY, USA, 2020; Volume 2118, pp. 45–60. ISBN 978-1-0716-0318-5. [Google Scholar]
- Han, J.; Zhao, D.; Li, D.; Wang, X.; Jin, Z.; Zhao, K. Polymer-Based Nanomaterials and Applications for Vaccines and Drugs. Polymers 2018, 10, 31. [Google Scholar] [CrossRef]
- Bose, R.J.; Kim, M.; Chang, J.H.; Paulmurugan, R.; Moon, J.J.; Koh, W.G.; Lee, S.H.; Park, H. Biodegradable polymers for modern vaccine development. J. Ind. Eng. Chem. 2019, 77, 12–24. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Skwarczynski, M.; Toth, I. Polymers for subunit vaccine delivery. Eur. Polym. J. 2019, 114, 397–410. [Google Scholar] [CrossRef]
- Pavot, V.; Berthet, M.; Rességuier, J.; Legaz, S.; Handké, N.; Gilbert, S.C.; Paul, S.; Verrier, B. Poly(lactic acid) and poly(lactic-co-glycolic acid) particles as versatile carrier platforms for vaccine delivery. Nanomedicine 2014, 9, 2703–2718. [Google Scholar] [CrossRef]
- Moran, H.B.T.; Turley, J.L.; Andersson, M.; Lavelle, E.C. Immunomodulatory properties of chitosan polymers. Biomaterials 2018, 184, 1–9. [Google Scholar] [CrossRef]
- Ebrahimian, M.; Hashemi, M.; Maleki, M.; Abnous, K.; Hashemitabar, G.; Ramezani, M.; Haghparast, A. Induction of a balanced Th1/Th2 immune responses by co-delivery of PLGA/ovalbumin nanospheres and CpG ODNs/PEI-SWCNT nanoparticles as TLR9 agonist in BALB/c mice. Int. J. Pharm. 2016, 515, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimian, M.; Hashemi, M.; Maleki, M.; Hashemitabar, G.; Abnous, K.; Ramezani, M.; Haghparast, A. Co-delivery of dual toll-like receptor agonists and antigen in poly(lactic-co-glycolic) acid/polyethylenimine cationic hybrid nanoparticles promote efficient in vivo immune responses. Front. Immunol. 2017, 8, 1077. [Google Scholar] [CrossRef]
- Kasturi, S.P.; Sachaphibulkij, K.; Roy, K. Covalent conjugation of polyethyleneimine on biodegradable microparticles for delivery of plasmid DNA vaccines. Biomaterials 2005, 26, 6375–6385. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, B.; Yu, X.; Zha, X.; Zhang, X.; Wang, X.; Jin, Y.; Wu, Y.; Chen, Y.; Shan, Y.; et al. Controlled release of PEI/DNA complexes from PLGA microspheres as a potent delivery system to enhance immune response to HIV vaccine DNA prime/MVA boost regime. Eur. J. Pharm. Biopharm. 2008, 68, 589–595. [Google Scholar] [CrossRef]
- Duran, V.; Yasar, H.; Becker, J.; Thiyagarajan, D.; Loretz, B.; Kalinke, U.; Lehr, C.M. Preferential uptake of chitosan-coated PLGA nanoparticles by primary human antigen presenting cells. Nanomedicine 2019, 21, 102073. [Google Scholar] [CrossRef]
- Pawar, D.; Mangal, S.; Goswami, R.; Jaganathan, K.S. Development and characterization of surface modified PLGA nanoparticles for nasal vaccine delivery: Effect of mucoadhesive coating on antigen uptake and immune adjuvant activity. Eur. J. Pharm. Biopharm. 2013, 85, 550–559. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.Y.; Wang, L.Y.; Liu, Y.; Weifeng, Z.; Fan, B.; Ma, X.; Yuan, Q.; Ma, G.; Su, Z. Enhanced humoral and cell-mediated immune responses generated by cationic polymer-coated PLA microspheres with adsorbed HBsAg. Mol. Pharm. 2014, 11, 1772–1784. [Google Scholar] [CrossRef]
- Chaturvedi, M.; Kumar, M.; Pathak, K. A review on mucoadhesive polymer used in nasal drug delivery system. J. Adv. Pharm. Technol. Res. 2011, 2, 215–222. [Google Scholar] [CrossRef]
- Slütter, B.; Bal, S.; Keijzer, C.; Mallants, R.; Hagenaars, N.; Que, I.; Kaijzel, E.; van Eden, W.; Augustijns, P.; Löwik, C.; et al. Nasal vaccination with N-trimethyl chitosan and PLGA based nanoparticles: Nanoparticle characteristics determine quality and strength of the antibody response in mice against the encapsulated antigen. Vaccine 2010, 28, 6282–6291. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Shin, B.A.; Oh, I.J. Poly(l-lactic acid)/polyethylenimine nanoparticles as plasmid DNA carriers. Arch. Pharm. Res. 2008, 31, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.S.; Lee, S.K.; Park, Y.M.; Lee, Y.B.; Shin, S.C.; Lee, K.C.; Oh, I.J. Physicochemical characterization of poly(l-lactic acid) and poly(d,l-lactide-co-glycolide) nanoparticles with polyethylenimine as gene delivery carrier. Int. J. Pharm. 2005, 298, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wen, T.; Cao, P.; Shan, L.; Li, L. Alginate-chitosan coated layered double hydroxide nanocomposites for enhanced oral vaccine delivery. J. Colloid Interface Sci. 2019, 556, 258–265. [Google Scholar] [CrossRef]
- Jaafar, M.H.M.; Hamid, K.A. Chitosan-Coated Alginate Nanoparticles Enhanced Absorption Profile of Insulin Via Oral Administration. Curr. Drug Deliv. 2019, 16, 672–686. [Google Scholar] [CrossRef]
- Biswas, S.; Chattopadhyay, M.; Sen, K.K.; Saha, M.K. Development and characterization of alginate coated low molecular weight chitosan nanoparticles as new carriers for oral vaccine delivery in mice. Carbohydr. Polym. 2015, 121, 403–410. [Google Scholar] [CrossRef]
- Borges, O.; Silva, M.; de Sousa, A.; Borchard, G.; Junginger, H.E.; Cordeiro-da-Silva, A. Alginate coated chitosan nanoparticles are an effective subcutaneous adjuvant for hepatitis B surface antigen. Int. Immunopharmacol. 2008, 8, 1773–1780. [Google Scholar] [CrossRef]
- Yuan, J.; Guo, L.; Wang, S.; Liu, D.; Qin, X.; Zheng, L.; Tian, C.; Han, X.; Chen, R.; Yin, R. Preparation of self-assembled nanoparticles of epsilon-polylysine-sodium alginate: A sustained-release carrier for antigen delivery. Colloids Surf. B Biointerfaces 2018, 171, 406–412. [Google Scholar] [CrossRef]
- Setty, C.M.; Sahoo, S.S.; Sa, B. Alginate-coated alginate-polyethyleneimine beads for prolonged release of furosemide in simulated intestinal fluid. Drug Dev. Ind. Pharm. 2005, 31, 435–446. [Google Scholar] [CrossRef]
- Wang, G.D.; Tan, Y.Z.; Wang, H.J.; Zhou, P. Autophagy promotes degradation of polyethyleneimine-alginate nanoparticles in endothelial progenitor cells. Int. J. Nanomed. 2017, 12, 6661–6675. [Google Scholar] [CrossRef]
- Li, P.; Luo, Z.; Liu, P.; Gao, N.; Zhang, Y.; Pan, H.; Liu, L.; Wang, C.; Cai, L.; Ma, Y. Bioreducible alginate-poly(ethylenimine) nanogels as an antigen-delivery system robustly enhance vaccine-elicited humoral and cellular immune responses. J. Control. Release 2013, 168, 271–279. [Google Scholar] [CrossRef]
- Tencomnao, T.; Klangthong, K.; Pimpha, N.; Chaleawlert-Umpon, S.; Saesoo, S.; Woramongkolchai, N.; Saengkrit, N. Acceleration of gene transfection efficiency in neuroblastoma cells through polyethyleneimine/poly(methyl methacrylate) core-shell magnetic nanoparticles. Int. J. Nanomed. 2012, 7, 2783–2792. [Google Scholar]
- Zhu, J.; Tang, A.; Law, L.P.; Feng, M.; Ho, K.M.; Lee, D.K.L.; Harris, F.W.; Li, P. Amphiphilic core-shell nanoparticles with poly(ethylenimine) shells as potential gene delivery carriers. Bioconjug. Chem. 2005, 16, 139–146. [Google Scholar] [CrossRef]
- Castaldello, A.; Brocca-Cofano, E.; Voltan, R.; Triulzi, C.; Altavilla, G.; Laus, M.; Sparnacci, K.; Ballestri, M.; Tondelli, L.; Fortini, C.; et al. DNA prime and protein boost immunization with innovative polymeric cationic core-shell nanoparticles elicits broad immune responses and strongly enhance cellular responses of HIV-1 tat DNA vaccination. Vaccine 2006, 24, 5655–5669. [Google Scholar] [CrossRef] [PubMed]
- Santo, D.; Mendonça, P.V.; Lima, M.S.; Cordeiro, R.A.; Cabanas, L.; Serra, A.; Coelho, J.F.J.; Faneca, H. Poly(ethylene glycol)- block-poly(2-aminoethyl methacrylate hydrochloride)-Based Polyplexes as Serum-Tolerant Nanosystems for Enhanced Gene Delivery. Mol. Pharm. 2019, 16, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Dombu, C.; Carpentier, R.; Betbeder, D. Influence of surface charge and inner composition of nanoparticles on intracellular delivery of proteins in airway epithelial cells. Biomaterials 2012, 33, 9117–9126. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Becker, K.W.; Knight, F.C.; Baljon, J.J.; Sevimli, S.; Shae, D.; Gilchuk, P.; Joyce, S.; Wilson, J.T. Poly(propylacrylic acid)-peptide nanoplexes as a platform for enhancing the immunogenicity of neoantigen cancer vaccines. Biomaterials 2018, 182, 82–91. [Google Scholar] [CrossRef]
- Lynn, G.M.; Sedlik, C.; Baharom, F.; Zhu, Y.; Ramirez-Valdez, R.A.; Coble, V.L.; Tobin, K.; Nichols, S.R.; Itzkowitz, Y.; Zaidi, N.; et al. Peptide–TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat. Biotechnol. 2020, 38, 320–332. [Google Scholar] [CrossRef]
- Woodrow, K.A.; Bennett, K.M.; Lo, D.D. Mucosal vaccine design and delivery. Annu. Rev. Biomed. Eng. 2012, 14, 17–46. [Google Scholar] [CrossRef]
- Borchard, G.; Lueβen, H.L.; de Boer, A.G.; Verhoef, J.C.; Lehr, C.M.; Junginger, H.E. The potential of mucoadhesive polymers in enhancing intestinal peptide drug absorption. III: Effects of chitosan-glutamate and carbomer on epithelial tight junctions in vitro. J. Control. Release 1996, 39, 131–138. [Google Scholar] [CrossRef]
- Men, Y.; Tamber, H.; Audran, R.; Gander, B.; Corradin, G. Induction of a cytotoxic T lymphocyte response by immunization with a malaria specific CTL peptide entrapped in biodegradable polymer microspheres. Vaccine 1997, 15, 1405–1412. [Google Scholar] [CrossRef]
- Storni, T.; Kundig, T.M.; Senti, G.; Johansen, P. Immunity in response to particulate antigen-delivery systems. Adv. Drug Deliv. Rev. 2005, 57, 333–355. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Maharjan, S.; Jiang, T.; Kang, S.K.; Choi, Y.J.; Cho, C.S. Combinatorial approach of antigen delivery using M cell-homing peptide and mucoadhesive vehicle to enhance the efficacy of oral vaccine. Mol. Pharm. 2015, 12, 3816–3828. [Google Scholar] [CrossRef]
- Gupta, N.K.; Tomar, P.; Sharma, V.; Dixit, V.K. Development and characterization of chitosan coated poly-(varepsilon-caprolactone) nanoparticulate system for effective immunization against influenza. Vaccine 2011, 29, 9026–9037. [Google Scholar] [CrossRef]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid-polymer hybrid nanoparticles as a next-generation drug delivery platform: State of the art, emerging technologies, and perspectives. Int. J. Nanomed. 2019, 14, 1937–1952. [Google Scholar] [CrossRef]
- Dave, V.; Tak, K.; Sohgaura, A.; Gupta, A.; Sadhu, V.; Reddy, K.R. Lipid-polymer hybrid nanoparticles: Synthesis strategies and biomedical applications. J. Microbiol. Methods 2019, 160, 130–142. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Herrington, T.M. Phospholipid adsorption onto polystyrene microspheres. J. Colloid Interface Sci. 1993, 156, 19–23. [Google Scholar] [CrossRef]
- Rapuano, R.; Carmona-Ribeiro, A.M. Physical adsorption of bilayer membranes on Silica. J. Colloid Interface Sci. 1997, 193, 104–111. [Google Scholar] [CrossRef]
- Rapuano, R.; Carmona-Ribeiro, A.M. Supported bilayers on silica. J. Colloid Interface Sci. 2000, 226, 299–307. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Bilayer vesicles and liposomes as interface agents. Chem. Soc. Rev. 2001, 30, 241–247. [Google Scholar] [CrossRef]
- Pacheco, L.F.; Carmona-Ribeiro, A.M. Effects of synthetic lipids on solubilization and colloid stability of hydrophobic drugs. J. Colloid Interface Sci. 2003, 258, 146–154. [Google Scholar] [CrossRef]
- Moura, S.P.; Carmona-Ribeiro, A.M. Biomimetic particles for isolation and reconstitution of receptor function. Cell Biochem. Biophys. 2006, 44, 446–452. [Google Scholar] [CrossRef]
- Pereira, E.M.A.; Vieira, D.B.; Carmona-Ribeiro, A.M. Cationic bilayers on polymeric particles: Effect of low NaCl concentration on surface coverage. J. Phys. Chem. B 2004, 108, 11490–11495. [Google Scholar] [CrossRef]
- Moura, S.P.; Carmona-Ribeiro, A.M. Biomimetic particles: Optimization of phospholipid bilayer coverage on silica and colloid stabilization. Langmuir 2005, 21, 10160–10164. [Google Scholar] [CrossRef]
- Lincopan, N.; Carmona-Ribeiro, A.M. Lipid-covered drug particles: Combined action of dioctadecyldimethylammonium bromide and amphotericin B or miconazole. J. Antimicrob. Chemother. 2006, 58, 66–75. [Google Scholar] [CrossRef]
- Barbassa, L.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Supramolecular assemblies of rifampicin and cationic bilayers: Preparation, characterization and micobactericidal activity. BMC Biotechnol. 2011, 11, 40. [Google Scholar] [CrossRef]
- Carvalho, C.A.; Olivares-Ortega, C.; Soto-Arriaza, M.A.; Carmona-Ribeiro, A.M. Interaction of gramicidin with DPPC/DODAB bilayer fragments. Biochim. Biophys. Acta 2012, 1818, 3064–3071. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Carrasco, L.D.M. Fungicidal assemblies and their mode of action. OA Biotechnol. 2013, 2, 25. [Google Scholar] [CrossRef]
- Ragioto, D.A.M.T.; Carrasco, L.D.M.; Carmona-Ribeiro, A.M. Novel gramicidin formulations in cationic lipid as broad-spectrum microbicidal agents. Int. J. Nanomed. 2014, 9, 3183–3192. [Google Scholar]
- Carmona-Ribeiro, A.M.; de Melo Carrasco, L.D. Novel formulations for antimicrobial peptides. Int. J. Mol. Sci. 2014, 15, 18040–18083. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L.D.; Bertolucci, R.J.; Ribeiro, R.T.; Sampaio, J.L.M.; Carmona-Ribeiro, A.M. Cationic nanostructures against foodborne pathogens. Front. Microbiol. 2016, 7, 1804. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L.D.; Santos, H.C.; Sampaio, J.L.; Carmona-Ribeiro, A.M. Self-assembled antibiotic nanoparticles against intracellular bacteria. Drug Deliv. Lett. 2017, 7, 39–47. [Google Scholar]
- Mamizuka, E.M.; Carmona-Ribeiro, A.M. Cationic liposomes as antimicrobial agents. In Communicating Current Research and Educational Topics and Trends in Applied Microbiology; Méndez Vilas, A., Ed.; Formatex: Badajoz, Spain, 2007; Volume 2, pp. 636–647. ISBN 9788461194223. [Google Scholar]
- Moura, S.P.; Carmona-Ribeiro, A.M. Adsorption behavior of DODAB/DPPC vesicles on silica. J. Colloid Interface Sci. 2007, 313, 519–526. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Barbassa, L.; Melo, L. Antimicrobial Biomimetics. In Biomimetic Based Applications; George, A., Ed.; IntechOpen: Rijeka, Croatia, 2011; pp. 227–284. ISBN 978-953-307-195-4. [Google Scholar]
- Tapias, G.N.; Sicchierolli, S.M.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Interactions between cationic vesicles and Escherichia coli. Langmuir 1994, 10, 3461–3465. [Google Scholar] [CrossRef]
- Martins, L.M.S.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Cationic Vesicles as Bactericides. Langmuir 1997, 13, 5583–5587. [Google Scholar] [CrossRef]
- Lima, E.G.; Gomes, L.R.; Carmona-Ribeiro, A.M. Stable indomethacin dispersions in water from drug, ethanol, cationic lipid and carboxymethyl-cellulose. Pharm. Nanotechnol. 2016, 4, 126–135. [Google Scholar] [CrossRef]
- Grabowski, E.; Morrison, I. Particle size distribution from analysis of quasi-elastic light scattering data. In Measurement of Suspended Particles by Quasi-Elastic Light Scattering; John Wiley & Sons: New York, NY, USA, 1983; pp. 199–236. [Google Scholar]
- Andersson, M.; Hammarstroem, L.; Edwards, K. Effect of Bilayer Phase Transitions on Vesicle Structure, and its Influence on the Kinetics of Viologen Reduction. J. Phys. Chem. 1995, 99, 14531–14538. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Castuma, C.E.; Sesso, A.; Schreier, S. Bilayer structure and stability in dihexadecyl phosphate dispersions. J. Phys. Chem. 1991, 95, 5361–5366. [Google Scholar] [CrossRef]
- Gall, D. The adjuvant activity of aliphatic nitrogenous bases. Immunology 1966, 11, 369–386. [Google Scholar]
- Dailey, M.O.; Hunter, R.L. The role of lipid in the induction of hapten-specific delayed hypersensitivity and contact sensitivity. J. Immunol. 1974, 112, 1526–1534. [Google Scholar] [PubMed]
- Klinguer-Hamour, C.; Libon, C.; Plotnicky-Gilquin, H.; Bussat, M.C.; Revy, L.; Nguyen, T.; Bonnefoy, J.Y.; Corvaïa, N.; Beck, A. DDA adjuvant induces a mixed Th1/Th2 immune response when associated with BBG2Na, a respiratory syncytial virus potential vaccine. Vaccine 2002, 20, 2743–2751. [Google Scholar] [CrossRef]
- Jain, V.; Gupta, A.; Pawar, V.K.; Asthana, S.; Jaiswal, A.K.; Dube, A.; Chourasia, M.K. Chitosan-assisted immunotherapy for intervention of experimental leishmaniasis via amphotericin B-loaded solid lipid nanoparticles. Appl. Biochem. Biotechnol. 2014, 174, 1309–1330. [Google Scholar] [CrossRef] [PubMed]
- Bernocchi, B.; Carpentier, R.; Lantier, I.; Ducournau, C.; Dimier-Poisson, I.; Betbeder, D. Mechanisms allowing protein delivery in nasal mucosa using NPL nanoparticles. J. Control. Release 2016, 232, 42–50. [Google Scholar] [CrossRef]
- Khademi, F.; Derakhshan, M.; Yousefi-Avarvand, A.; Najafi, A.; Tafaghodi, M. A novel antigen of Mycobacterium tuberculosis and MPLA adjuvant co-entrapped into PLGA:DDA hybrid nanoparticles stimulates mucosal and systemic immunity. Microb. Pathog. 2018, 125, 507–513. [Google Scholar] [CrossRef]
- Rose, F.; Wern, J.E.; Ingvarsson, P.T.; van de Weert, M.; Andersen, P.; Follmann, F.; Foged, C. Engineering of a novel adjuvant based on lipid-polymer hybrid nanoparticles: A quality-by-design approach. J. Control. Release 2015, 210, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Sahdev, P.; Ochyl, L.J.; Akerberg, J.; Moon, J.J. Cationic liposome-hyaluronic acid hybrid nanoparticles for intranasal vaccination with subunit antigens. J. Control. Release 2015, 208, 121–129. [Google Scholar] [CrossRef]
- Liu, L.; Ma, P.; Wang, H.; Zhang, C.; Sun, H.; Wang, C.; Song, C.; Leng, X.; Kong, D.; Ma, G. Immune responses to vaccines delivered by encapsulation into and/or adsorption onto cationic lipid-PLGA hybrid nanoparticles. J. Control. Release 2016, 225, 230–239. [Google Scholar] [CrossRef] [PubMed]




















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona-Ribeiro, A.M.; Pérez-Betancourt, Y. Cationic Nanostructures for Vaccines Design. Biomimetics 2020, 5, 32. https://doi.org/10.3390/biomimetics5030032
Carmona-Ribeiro AM, Pérez-Betancourt Y. Cationic Nanostructures for Vaccines Design. Biomimetics. 2020; 5(3):32. https://doi.org/10.3390/biomimetics5030032
Chicago/Turabian StyleCarmona-Ribeiro, Ana Maria, and Yunys Pérez-Betancourt. 2020. "Cationic Nanostructures for Vaccines Design" Biomimetics 5, no. 3: 32. https://doi.org/10.3390/biomimetics5030032
APA StyleCarmona-Ribeiro, A. M., & Pérez-Betancourt, Y. (2020). Cationic Nanostructures for Vaccines Design. Biomimetics, 5(3), 32. https://doi.org/10.3390/biomimetics5030032