Variability and Bias in Measurements of Metals Mass Fractions in Automobile Shredder Residue

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Primary Samples
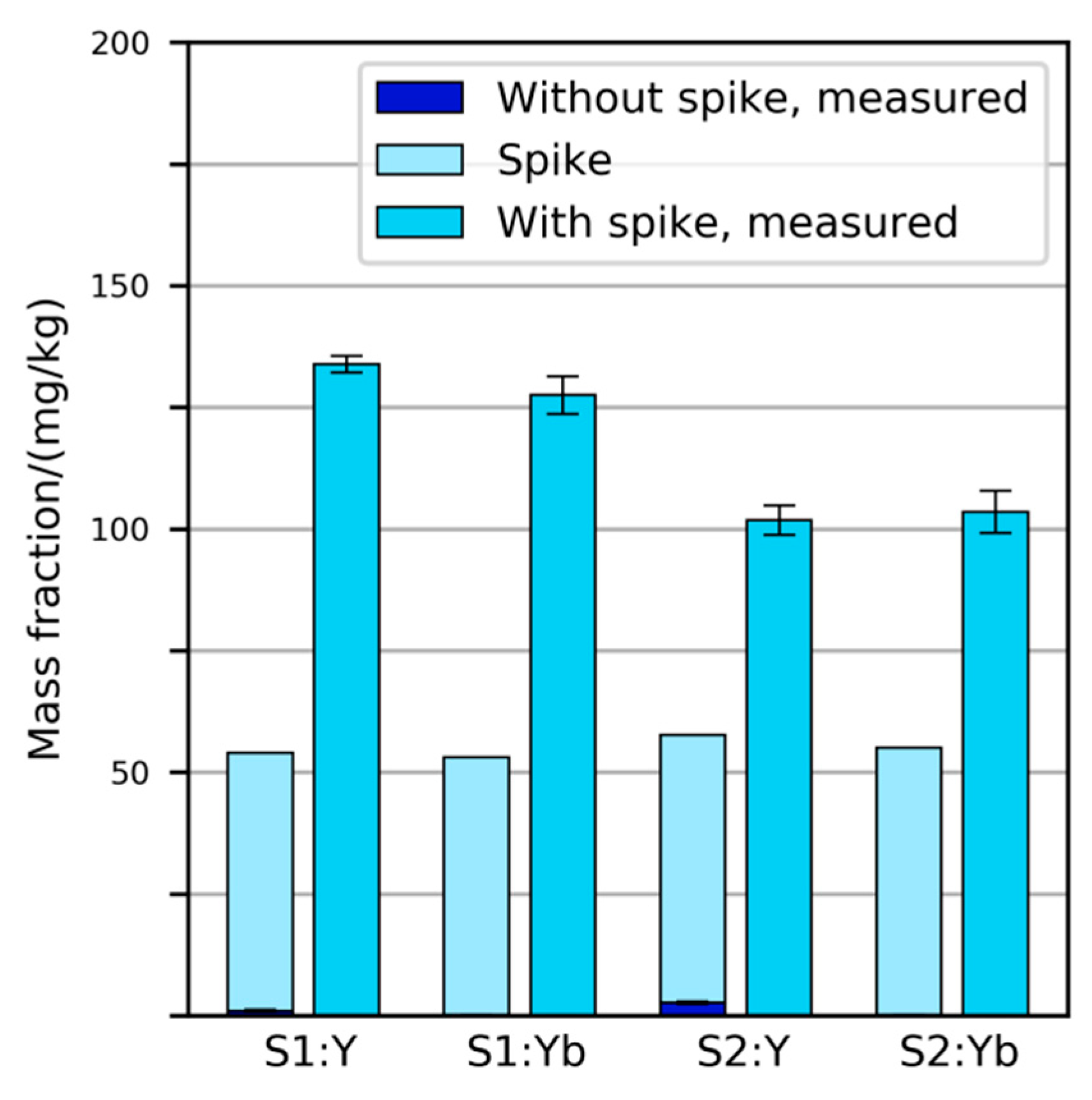
2.2. Spiking of Primary Samples
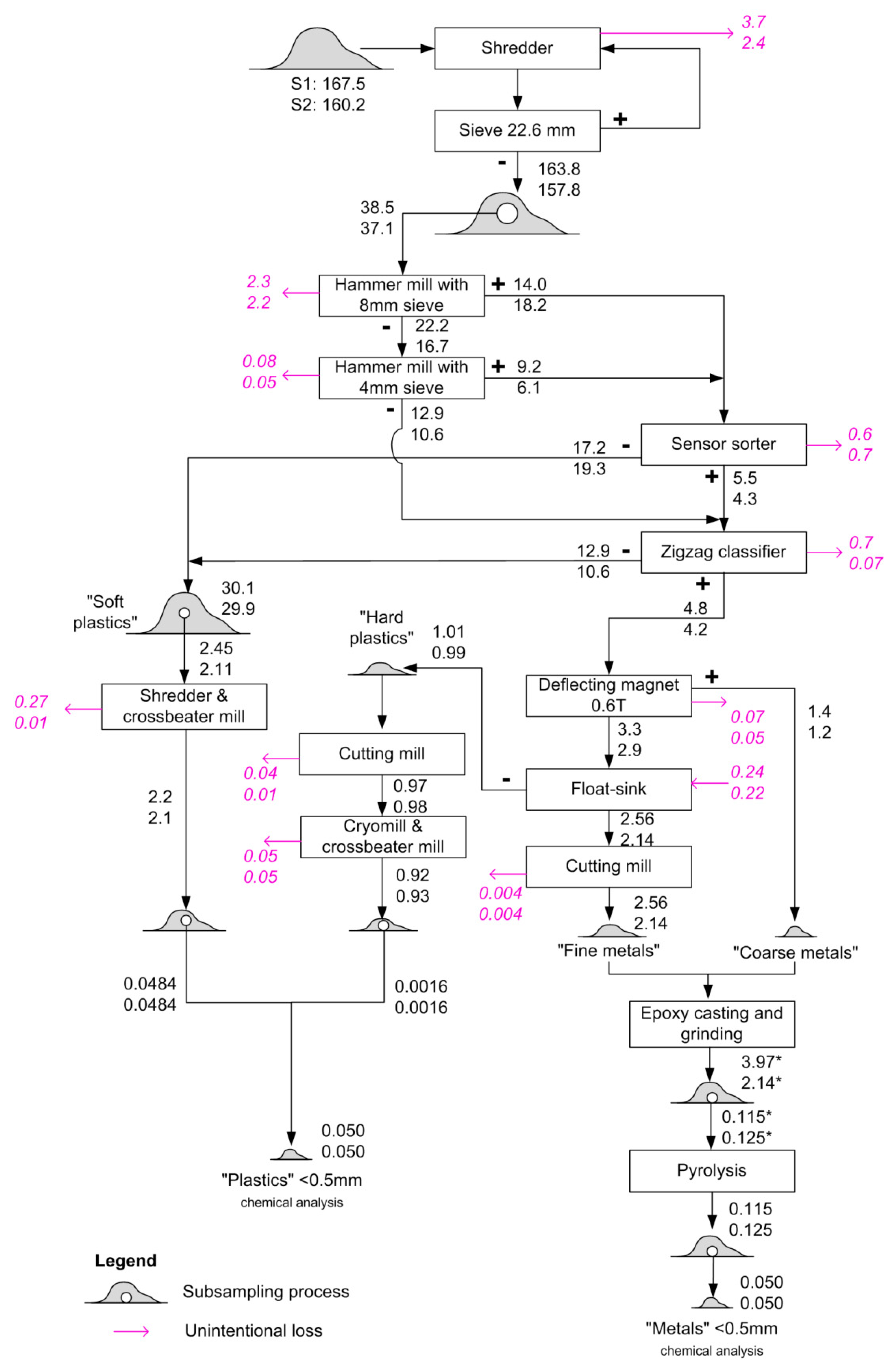
2.3. Physical Sample Preparation
2.4. Chemical Analysis
2.4.1. General Approach
2.4.2. Element Selection
2.4.3. Wavelength Dispersive X-Ray Fluorescence Spectrometry (WD-XRF)
2.4.4. Wet-Chemical Analysis
2.4.5. Data Analysis
3. Results and Discussion
3.1. Physical Sample Preparation
3.2. Comparison of Chemical Analysis Methods
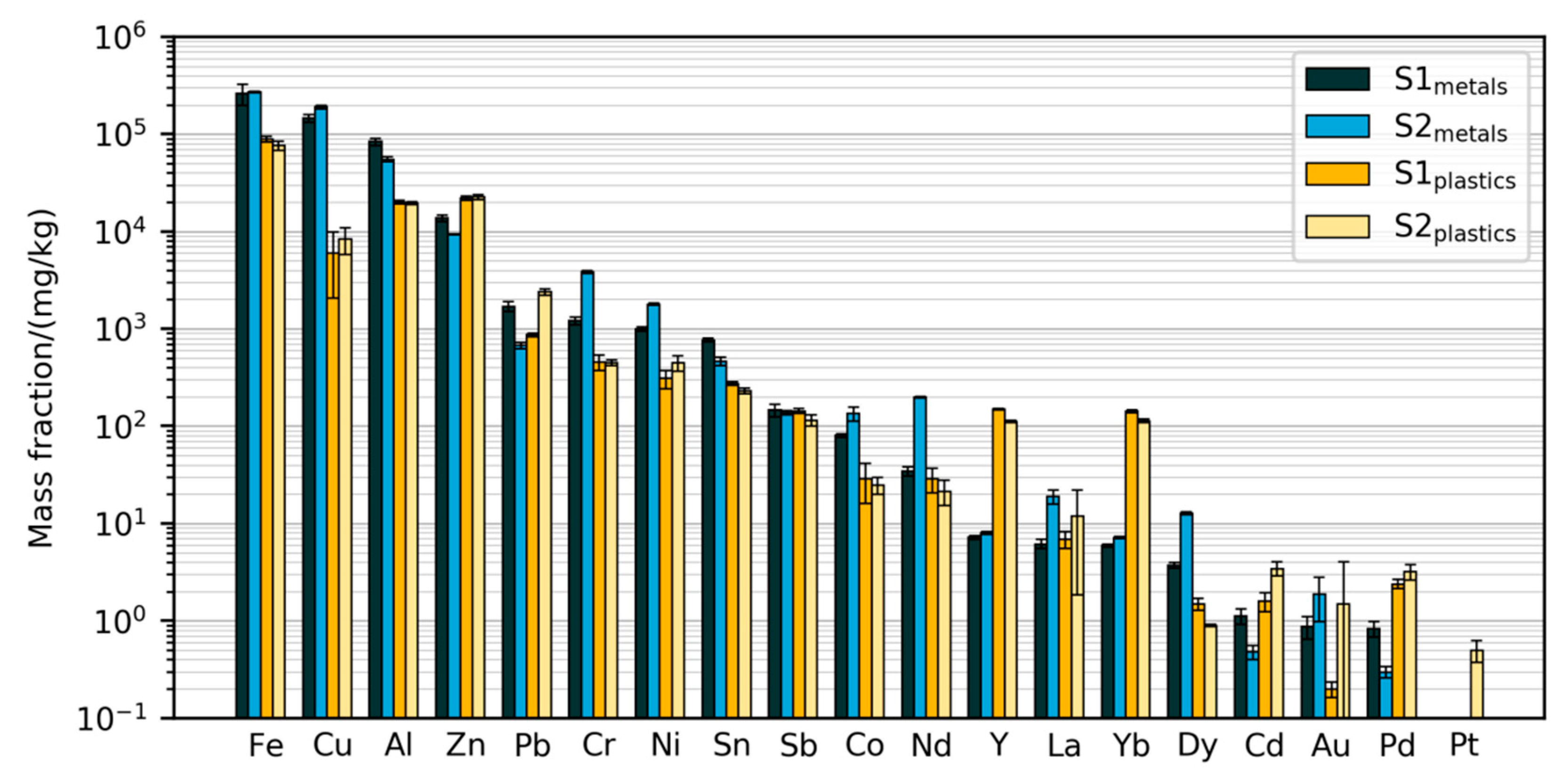
3.2.1. Mass Fractions Measured with Validated Method
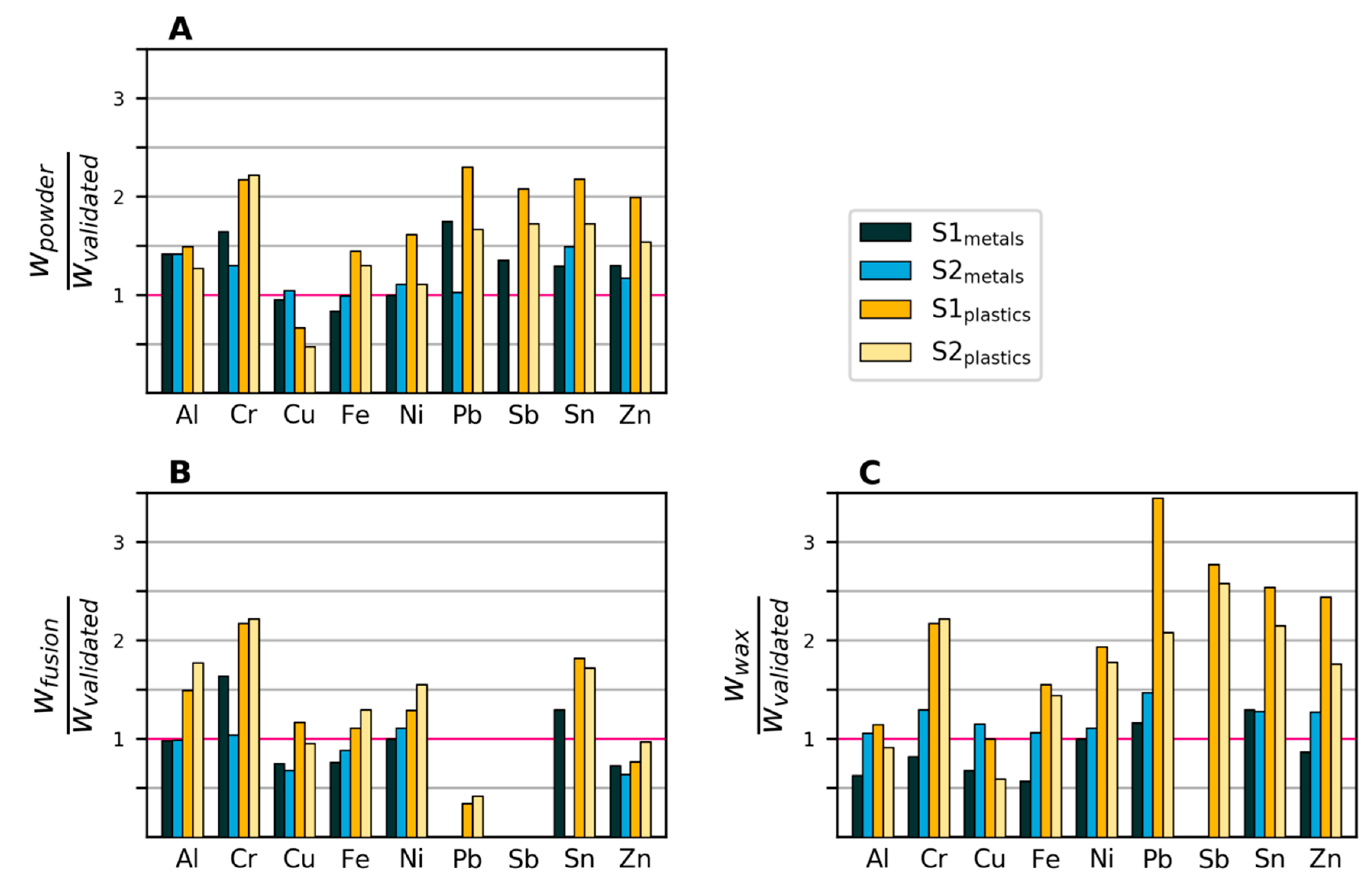
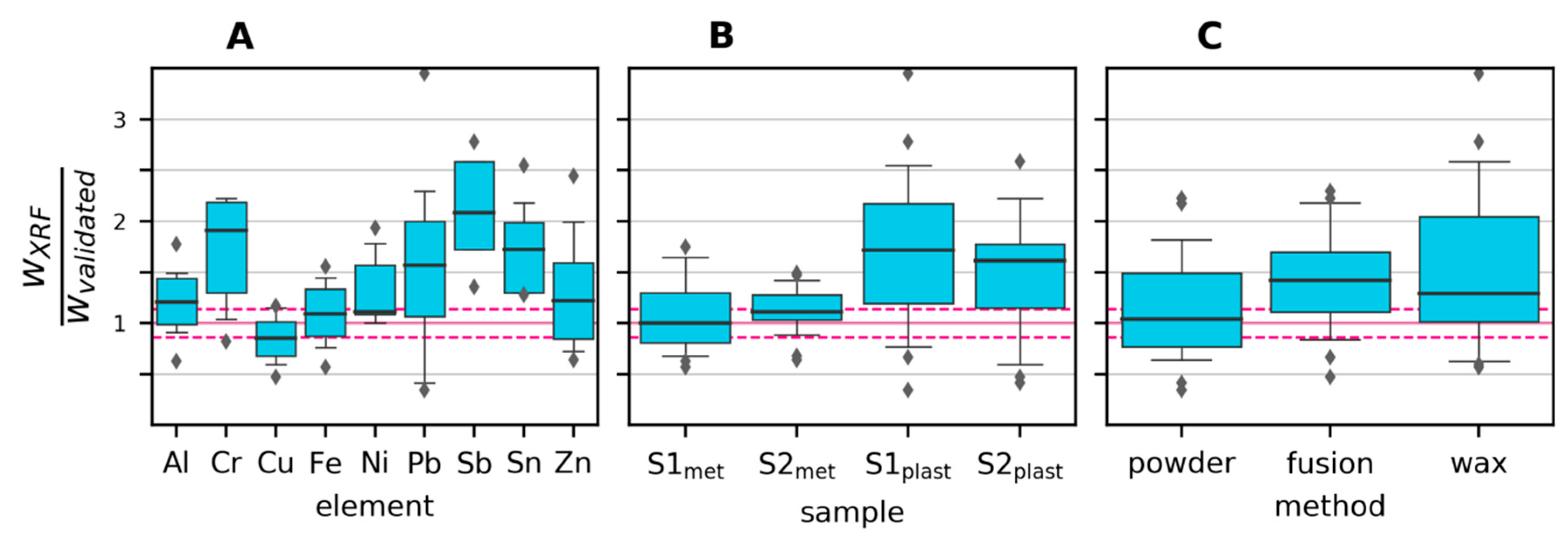
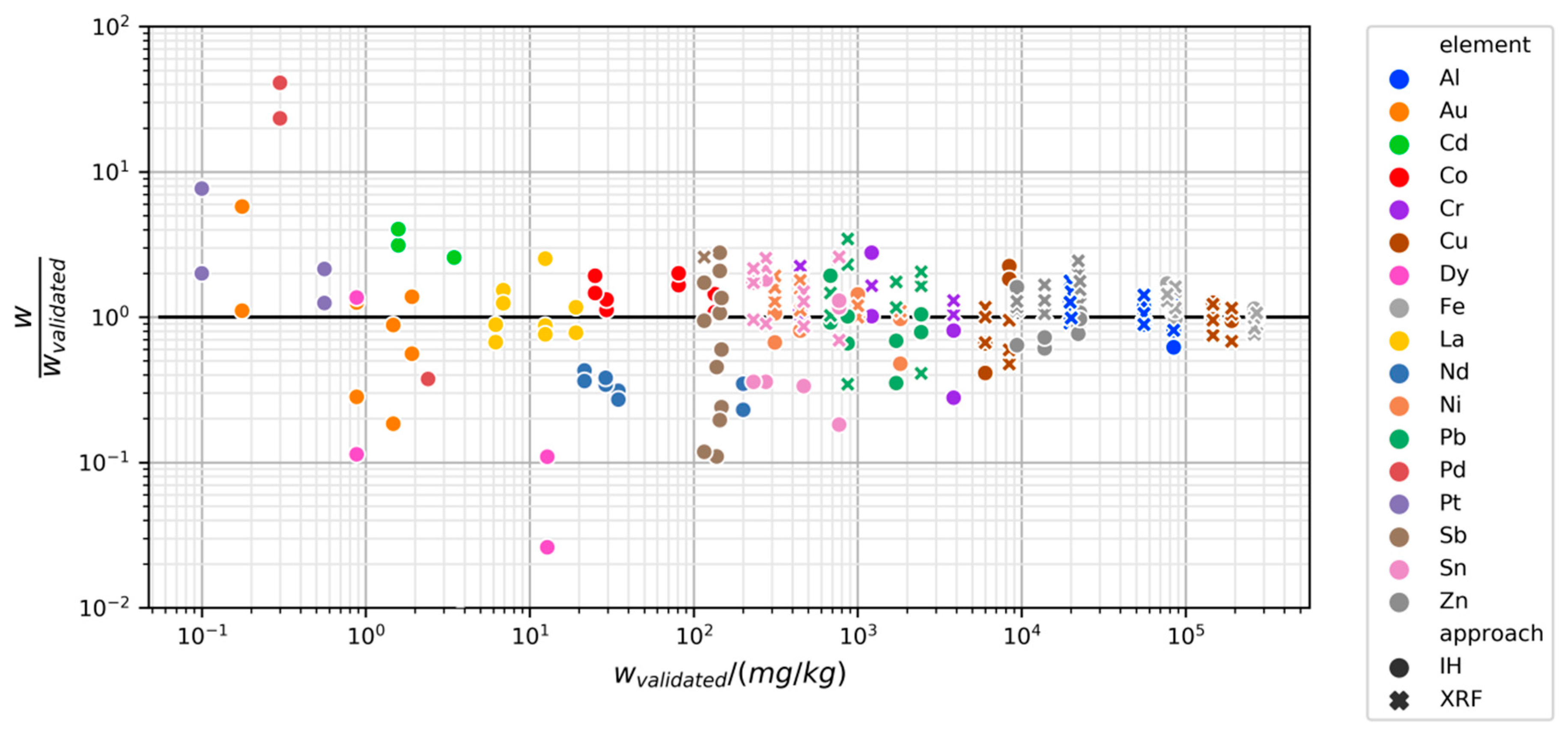
3.2.2. Variability and Bias of XRF Measurements
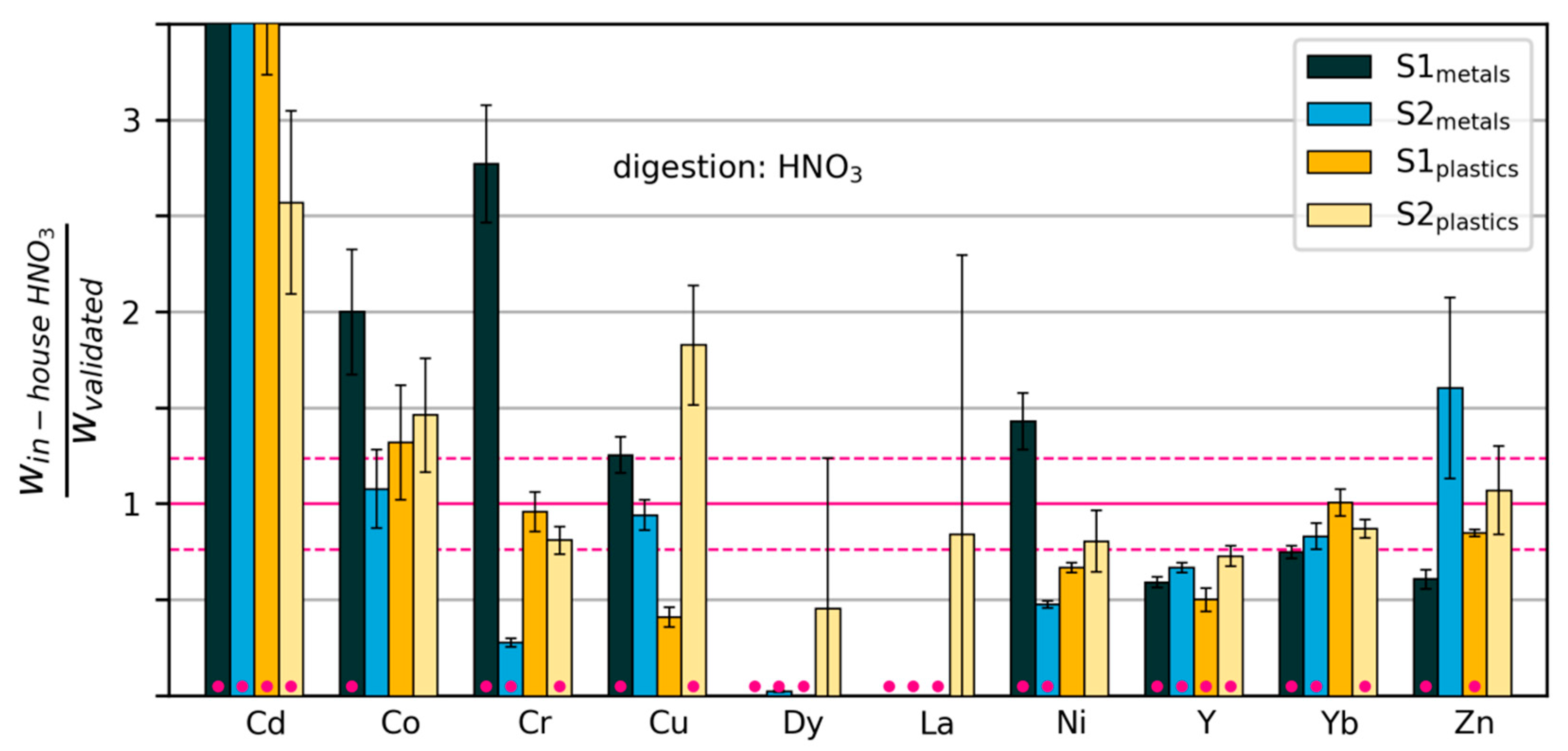
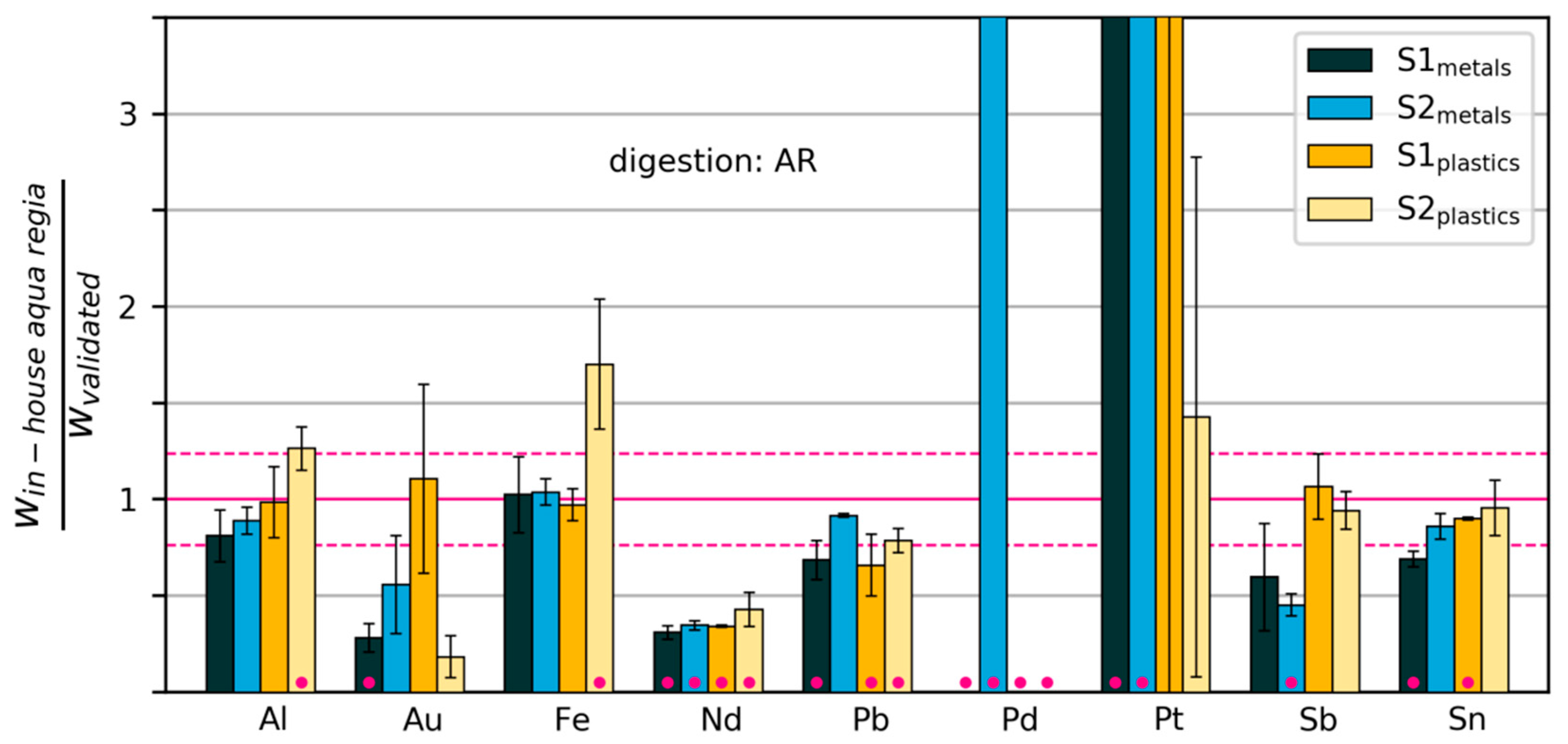
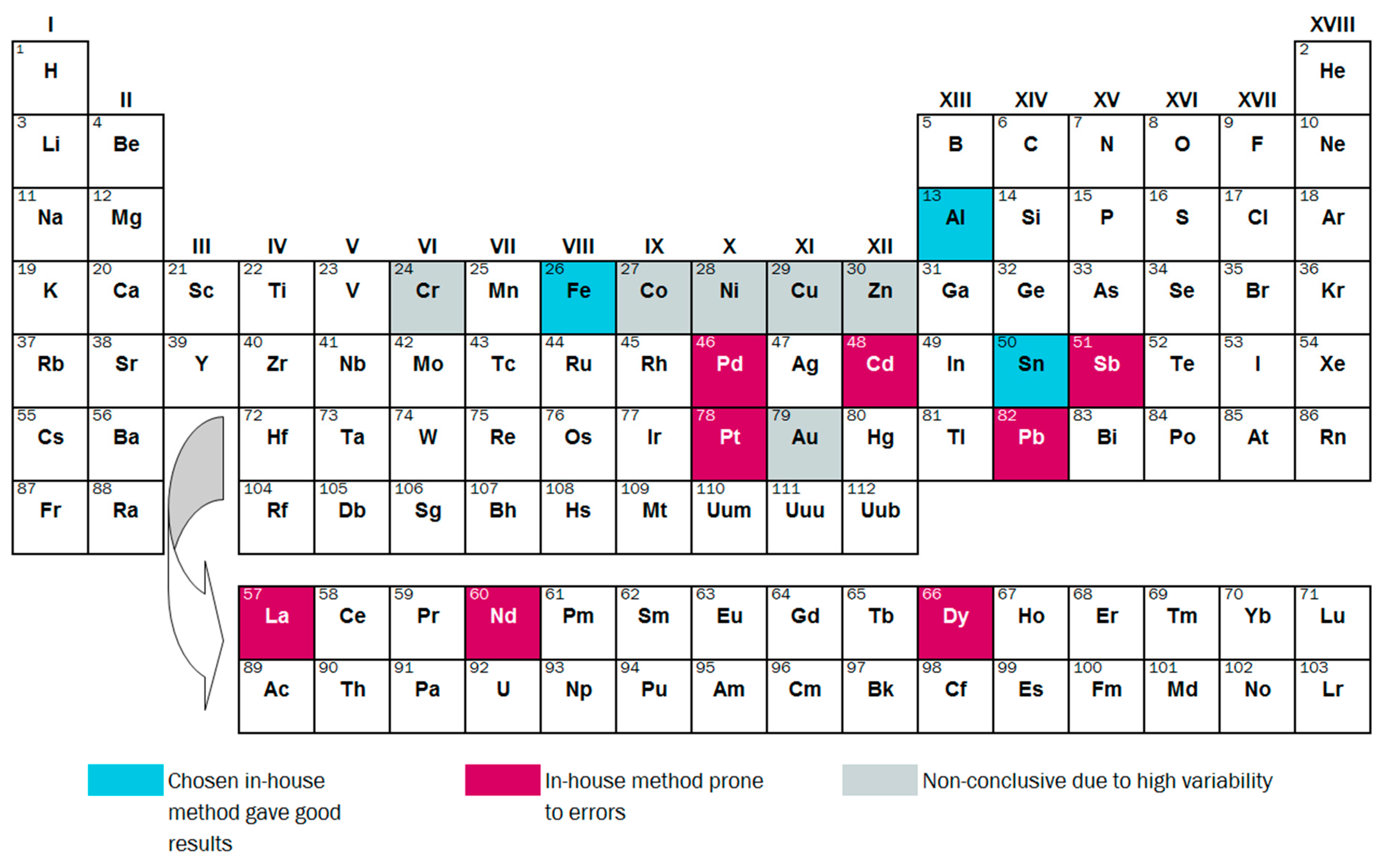
3.2.3. Variability and Bias of In-House Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eurostat End-of-life Vehicle Statistics—Statistics Explained. Available online: http://ec.europa.eu/eurostat/statistics-explained/index.php/End-of-life_vehicle_statistics#Total_number_of_end-of-life_vehicles (accessed on 6 March 2018).
- Eurostat Treatment of Materials from Shredding within the Member State, by Waste Category, Treatment Type, Country and Year, in Tonnes. Available online: http://ec.europa.eu/eurostat/web/waste/key-waste-streams/elvs (accessed on 9 March 2018).
- González-Fernández, O.; Hidalgo, M.; Marguí, E.; Carvalho, M.L.; Queralt, I. Heavy metals’ content of automotive shredder residues (ASR): Evaluation of environmental risk. Environ. Pollut. 2008, 153, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Tanigaki, N.; Takahashi, S.; Sakai, S. Brominated flame retardants and heavy metals in automobile shredder residue (ASR) and their behavior in the melting process. J. Mater. Cycles Waste Manag. 2008, 10, 93–101. [Google Scholar] [CrossRef]
- Mancini, G.; Viotti, P.; Luciano, A.; Fino, D. On the ASR and ASR thermal residues characterization of full scale treatment plant. Waste Manag. 2014, 34, 448–457. [Google Scholar] [CrossRef] [PubMed]
- The Commission of the European Communities. Commission Decision of 3 May 2000 Replacing Decision 94/3/EC Establishing a List of Wastes Pursuant to Article 1(a) of Council Directive 75/442/EEC on Waste and Council Decision 94/904/EC Establishing a List of Hazardous Waste Pursuant to Article 1(4) of Council Directive 91/689/EEC on Hazardous Waste; The Publications Office of the European Union: Luxembourg, 2000. [Google Scholar]
- Ministry of Environment Japan. Report on 2010 Survey to Identify the Characteristics of Automotive Shredder Residue; Ministry of Environment: Tokyo, Japan, 2010. Available online: https://www.env.go.jp/recycle/car/pdfs/h22_report01_mat.pdf (accessed on 30 January 2018).
- Widmer, R.; Du, X.; Haag, O.; Restrepo, E.; Wäger, P.A. Scarce Metals in Conventional Passenger Vehicles and End-of-Life Vehicle Shredder Output. Environ. Sci. Technol. 2015, 49, 4591–4599. [Google Scholar] [CrossRef] [PubMed]
- Mallampati, S.R.; Lee, C.H.; Truc, N.T.T.; Lee, B.-K. Quantitative analysis of precious metals in automotive shredder residue/combustion residue via EDX fluorescence spectrometry. Int. J. Environ. Anal. Chem. 2015, 95, 1081–1089. [Google Scholar] [CrossRef]
- Mallampati, S.R.; Lee, B.H.; Mitoma, Y.; Simion, C. Sustainable recovery of precious metals from end-of-life vehicles shredder residue by a novel hybrid ball-milling and nanoparticles enabled froth flotation process. J. Clean. Prod. 2018, 171, 66–75. [Google Scholar] [CrossRef]
- Eurachem, EUROLAB, CITAC, Nordtest and the RSC Analytical Methods Committee. Measurement Uncertainty Arising from Sampling: A Guide to Methods and Approaches, 1st ed.; Ramsey, M.H., Ellison, S.L.R., Eds.; 2007; ISBN 978-0-948926-26-6. Available online: https://eurachem.org/images/stories/Guides/pdf/UfS_2007.pdf (accessed on 31 August 2016).
- Gy, P.M. Sampling for Analytical Purposes; John Wiley & Sons Ltd.: Chichester, UK, 1998; ISBN 978-0-471-97956-2. [Google Scholar]
- Pineau, J.L.; Kanari, N.; Menad, N. Representativeness of an automobile shredder residue sample for a verification analysis. Waste Manag. 2005, 25, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Anzano, M.; Collina, E.; Piccinelli, E.; Lasagni, M. Lab-scale pyrolysis of the Automotive Shredder Residue light fraction and characterization of tar and solid products. Waste Manag. 2017, 64, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Khodier, A.; Williams, K.; Dallison, N. Challenges around automotive shredder residue production and disposal. Waste Manag. 2017, 73, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Haydary, J.; Susa, D.; Gelinger, V.; Čacho, F. Pyrolysis of automobile shredder residue in a laboratory scale screw type reactor. J. Environ. Chem. Eng. 2016, 4, 965–972. [Google Scholar] [CrossRef]
- Totland, M.; Jarvis, I.; Jarvis, K.E. An assessment of dissolution techniques for the analysis of geological samples by plasma spectrometry. Chem. Geol. 1992, 95, 35–62. [Google Scholar] [CrossRef]
- Joint Committee for Guides in Metrology GUM: Guide to the Expression of Uncertainty in Measurement. Available online: http://www.bipm.org/en/publications/guides/gum.html (accessed on 17 February 2016).
- Gy, P.M. Sampling of discrete materials—A new introduction to the theory of sampling: I. Qualitative approach. Chemom. Intell. Lab. Syst. 2004, 74, 7–24. [Google Scholar] [CrossRef]
- Agenzia Nazionale per la Protezione dell’Ambiente (ANPA). La Caratterizzazione del Fluff di Frantumazione Dei Veicoli; Agenzia Nazionale per la Protezione dell’Ambiente (ANPA): Rome, Italy, 2002; ISBN 88-448-0057-8. [Google Scholar]
- Agenzia per la protezione dell’ambiente e per i servizi tecnici (APAT). Studio APAT/ARPA Sul Fluff di Frantumazione Degli Autoveicoli; Agenzia per la Protezione Dell’ambiente e per i Servizi Tecnici (APAT): Rome, Italy, 2006; ISBN 88-448-0181-7. [Google Scholar]
- Ahmed, N.; Wenzel, H.; Hansen, J.B. Characterization of Shredder Residues generated and deposited in Denmark. Waste Manag. 2014, 34, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Börjeson, L.; Löfvenius, G.; Hjelt, M.; Johansson, S.; Marklund, S. Characterization of automotive shredder residues from two shredding facilities with different refining processes in Sweden. Waste Manag. Res. 2000, 18, 358–366. [Google Scholar] [CrossRef]
- Day, M.; Shen, Z.; Cooney, J.D. Pyrolysis of auto shredder residue: Experiments with a laboratory screw kiln reactor. J. Anal. Appl. Pyrolysis 1999, 51, 181–200. [Google Scholar] [CrossRef]
- Galvagno, S.; Fortuna, F.; Cornacchia, G.; Casu, S.; Coppola, T.; Sharma, V.K. Pyrolysis process for treatment of automobile shredder residue: Preliminary experimental results. Energy Convers. Manag. 2001, 42, 573–586. [Google Scholar] [CrossRef]
- Mancini, G.; Tamma, R.; Viotti, P. Thermal process of fluff: Preliminary tests on a full-scale treatment plant. Waste Manag. 2010, 30, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Mayyas, M.; Pahlevani, F.; Handoko, W.; Sahajwalla, V. Preliminary investigation on the thermal conversion of automotive shredder residue into value-added products: Graphitic carbon and nano-ceramics. Waste Manag. 2016, 50, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Mirabile, D.; Pistelli, M.I.; Marchesini, M.; Falciani, R.; Chiappelli, L. Thermal valorisation of automobile shredder residue: Injection in blast furnace. Waste Manag. 2002, 22, 841–851. [Google Scholar] [CrossRef]
- Morselli, L.; Santini, A.; Passarini, F.; Vassura, I. Automotive shredder residue (ASR) characterization for a valuable management. Waste Manag. 2010, 30, 2228–2234. [Google Scholar] [CrossRef]
- Notarnicola, M.; Cornacchia, G.; De Gisi, S.; Di Canio, F.; Freda, C.; Garzone, P.; Martino, M.; Valerio, V.; Villone, A. Pyrolysis of automotive shredder residue in a bench scale rotary kiln. Waste Manag. 2017, 65, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.; Chaala, A. Vacuum pyrolysis of automobile shredder residues. Resour. Conserv. Recycl. 2001, 32, 27. [Google Scholar] [CrossRef]
- Santini, A.; Morselli, L.; Passarini, F.; Vassura, I.; Di Carlo, S.; Bonino, F. End-of-Life Vehicles management: Italian material and energy recovery efficiency. Waste Manag. 2011, 31, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Zolezzi, M.; Nicolella, C.; Ferrara, S.; Iacobucci, C.; Rovatti, M. Conventional and fast pyrolysis of automobile shredder residues (ASR). Waste Manag. 2004, 24, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Lyn, J.A.; Ramsey, M.H.; Fussell, R.J.; Wood, R. Measurement uncertainty from physical sample preparation: Estimation including systematic error. Analyst 2003, 128, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Korf, N.; Løvik, A.N.; Figi, R.; Schreiner, C.; Kuntz, C.; Mählitz, P.M.; Rösslein, M.; Wäger, P.; Rotter, V.S. Multi-element chemical analysis of printed circuit boards—Challenges and pitfalls. Waste Manag. 2019, 92, 124–136. [Google Scholar] [CrossRef] [PubMed]
- International Organization for Standardization (ISO). ISO/IEC 17025:2005: General Requirements for the Competence of Testing and Calibration Laboratories; International Organization for Standardization: Geneva, Switzerland, 2005. [Google Scholar]
- International Organization for Standardization (ISO). ISO 11466: Soil Quality—Extraction of Trace Elements Soluble in Aqua Regia; International Organization for Standardization: Geneva, Switzerland, 1995. [Google Scholar]
- European Commission. Study on the Review of the List of Critical Raw Materials; European Commission: Brussels, Belgium, 2017. [Google Scholar]
- Bunge, R. Mechanische Aufbereitung: Primär—und Sekundärrohstoffe; 1. Auflage.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; ISBN 978-3-527-33209-0. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Vehicle Cohort Years | Mass/kg |
|---|---|---|
| S1 | 1988–2000 | 167.5 |
| S2 | 2001–2015 | 160.2 |
| Element Group | Elements Measured |
|---|---|
| Base metals | Al, Cu, Fe, Zn |
| Precious metals | Ag *, Au, Pd, Pt |
| Rare earth elements | Ce *, Dy, La, Nd, Sm *, Y, Yb |
| Other elements | Be *, Cd, Co, Cr, Ga *, Ge *, In *, Mg *, Ni, Pb, Sb, Sn |
| In-House Method | Validated Method | |||||||
|---|---|---|---|---|---|---|---|---|
| Microwave digestion | ||||||||
| Device name | CEM MarsXpress | MLS START | ||||||
| Acid 1): mixture | 6 mL HNO3 69% (Roth suprapur) + 1 mL H2O2 35% (Merck pro analysi) | 6 mL HNO3 67–69% (VWR Chemicals Normatom)+ 1 mL H2O2 30% (Merck suprapur) | ||||||
| Acid 1): elements measured | Al, Ag, Au, Be, Cd, Ce, Co, Cr, Cu, Dy, Fe, Ga, Ge, In, La, Mg, Nd, Ni, Pb, Pd, Pt, Sb, Sm, Sn, Y, Yb, Zn (all) | Cd, Co, Cr, Cu, Dy, La, Ni, Y, Yb, Zn | ||||||
| Acid 2): mixture | 5.25 mL HCl 35% (Roth suprapur) + 1.75 mL HNO3 (Roth suprapur) | 5.25 mL HCl 30% (Merck suprapur) +1.75 mL HNO3 67–69% (VWR Chemicals Normatom) | ||||||
| Acid 2): elements measured | Al, Ag, Au, Be, Cd, Ce, Co, Cr, Cu, Dy, Fe, Ga, Ge, In, La, Mg, Nd, Ni, Pb, Pd, Pt, Sb, Sm, Sn, Y, Yb, Zn (all) | Al, Au, Fe, Nd, Pb, Pd, Pt, Sb, Sn | ||||||
| Digestion programme | Step | Hold time/min | Power/W | Temp/°C | Step | Hold time/min | Power/W | Temp/°C |
| 1 | 6 | 1600 | 150 | 1 | 6 | 700 | 150 | |
| 2 | 5 | 1600 | 200 | 2 | 5 | 800 | 200 | |
| 3 | 18 | 1600 | 215 | 3 | 18 | 800 | 215 | |
| 4 | 25 | 1600 | 0 | 4 | 5 | 800 | 240 | |
| Sample mass digested | approximately 0.1 g | 0.2 g | ||||||
| Number of digestions | n = 3 | n = 5 | ||||||
| Final volume and dilution | 50 mL, filled up with 0.5 mol/L HNO3 for acid mixtures 1) and 2) | 50 mL, filled up with H2O. for 1) and with 2 mol/L HCl for 2) | ||||||
| Validation | element spikes added before digestion | element spikes added before digestion; certified standard reference material | ||||||
| Analytical determination | ||||||||
| ICP-OES device name | Thermo Scientific iCap 6000 Series | Varian Vista Pro Radial | ||||||
| ICP-OES elements | Al, Ag, Au, Be, Cd, Ce, Co, Cr, Cu, Dy, Fe, Ga, Ge, In, La, Mg, Nd, Ni, Pb, Pd, Pt, Sb, Sm, Sn, Y, Yb, Zn (all elements) | Al, Cr, Cu, Fe, Ni, Pb and Zn | ||||||
| ICP-MS device name | Thermo Scientific iCap Q | Agilent 8800 QQQ | ||||||
| ICP-MS collision mode | He | He | ||||||
| ICP-MS elements | Au, Ce, In, La, Y | Au, Cd, Co, Dy, Nd, La, Pd, Pt, Sb, Sn, Y, Yb | ||||||
| |δ| > 20% | |δ| < 20% | |||
|---|---|---|---|---|
| Significant | Insignificant | Significant | Insignificant | |
| HNO3 | 24 (55%) | 5 (11%) | 5 (11%) | 6 (14%) |
| aqua regia | 16 (44%) | 5 (14%) | 3 (8%) | 12 (33%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Løvik, A.N.; Figi, R.; Schreiner, C.; Rösslein, M.; Widmer, R.; Bunge, R.; Pohl, T.; Korf, N.; Kuntz, C.; Mählitz, P.M.; et al. Variability and Bias in Measurements of Metals Mass Fractions in Automobile Shredder Residue. Recycling 2019, 4, 34. https://doi.org/10.3390/recycling4030034
Løvik AN, Figi R, Schreiner C, Rösslein M, Widmer R, Bunge R, Pohl T, Korf N, Kuntz C, Mählitz PM, et al. Variability and Bias in Measurements of Metals Mass Fractions in Automobile Shredder Residue. Recycling. 2019; 4(3):34. https://doi.org/10.3390/recycling4030034
Chicago/Turabian StyleLøvik, Amund N., Renato Figi, Claudia Schreiner, Matthias Rösslein, Rolf Widmer, Rainer Bunge, Thomas Pohl, Nathalie Korf, Claudia Kuntz, Paul Martin Mählitz, and et al. 2019. "Variability and Bias in Measurements of Metals Mass Fractions in Automobile Shredder Residue" Recycling 4, no. 3: 34. https://doi.org/10.3390/recycling4030034
APA StyleLøvik, A. N., Figi, R., Schreiner, C., Rösslein, M., Widmer, R., Bunge, R., Pohl, T., Korf, N., Kuntz, C., Mählitz, P. M., Rotter, V. S., & Wäger, P. (2019). Variability and Bias in Measurements of Metals Mass Fractions in Automobile Shredder Residue. Recycling, 4(3), 34. https://doi.org/10.3390/recycling4030034