
Evaluation of Bio-Polyurethane Foam Synthesized from Liquefied Waste Wood Polyol




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analysis Methods of Wood Meal
2.3. Liquefaction and Evaluation Methods of Liquefied Products
- W: Residue rate (wt.%)
- m1: Residue weight (g)
- m2: The weight of collected sample (g)
- Rw: The ratio of biomass in the liquefaction process (wt.%)
- A: The amount of titrant (mL)
- N: The factor of NaOH ≒ 1
- W: Liquefied product sample weight
2.4. Synthesis and Evaluation Methods of Bio-Polyurethane Foam
- WL: Weight loss (%)
- w1: Initial sample weight
- w2: Sample weight at previous measurement
- w3: Sample weight at newest measurement
- AR: Water adsorption rate (%)
- m1: Initial weight (g)
- m2: The weight after soaking into water (g)
- m3: The weight after drying (g)
- FR: The foaming rate (%)
- H1: The foam height after complete foaming (mm)
- H2: The initial height before foaming (mm)
3. Results and Discussion
3.1. Evaluation of Wood Meal
3.2. Evaluation of Liquefaction and Liquefied Products
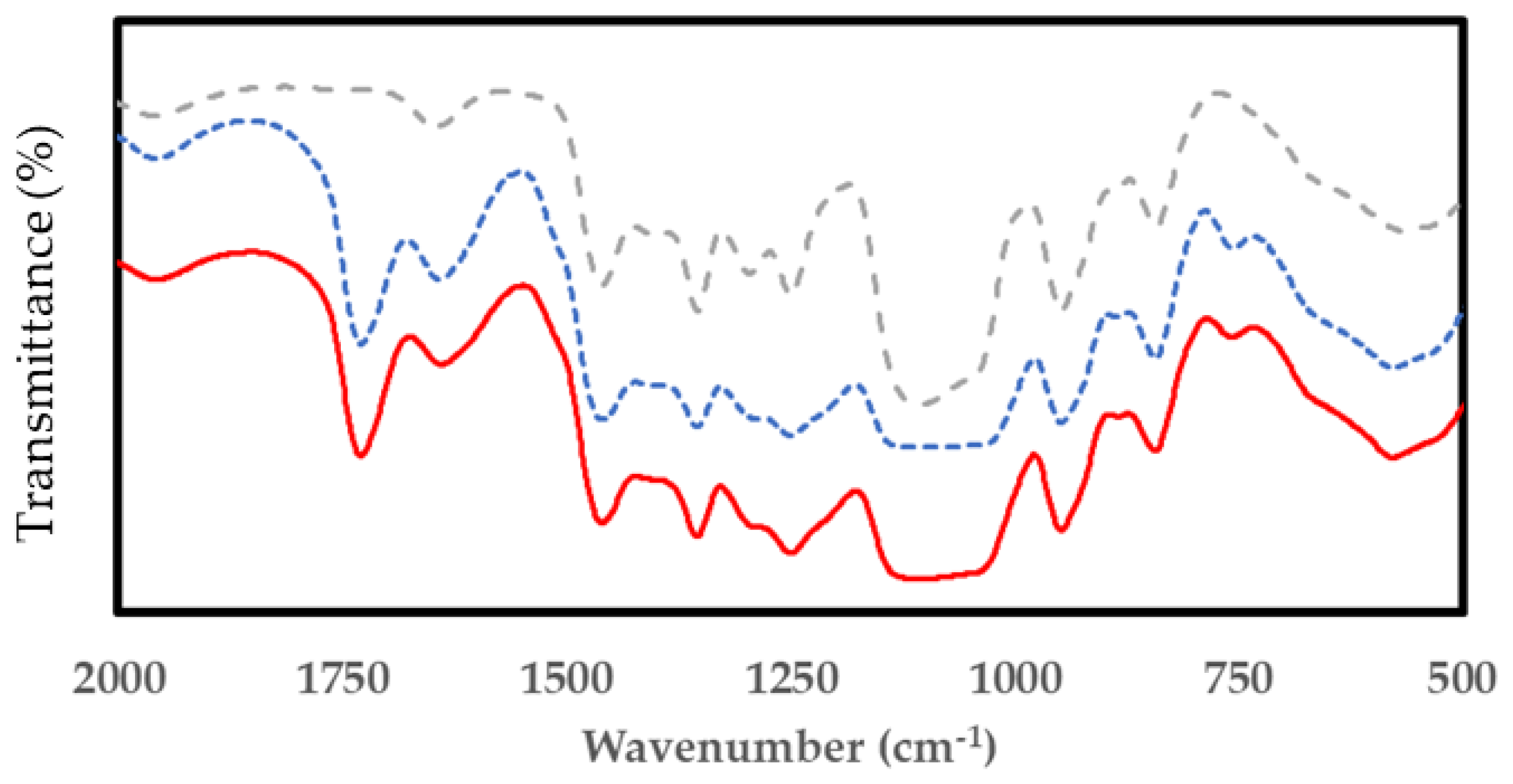
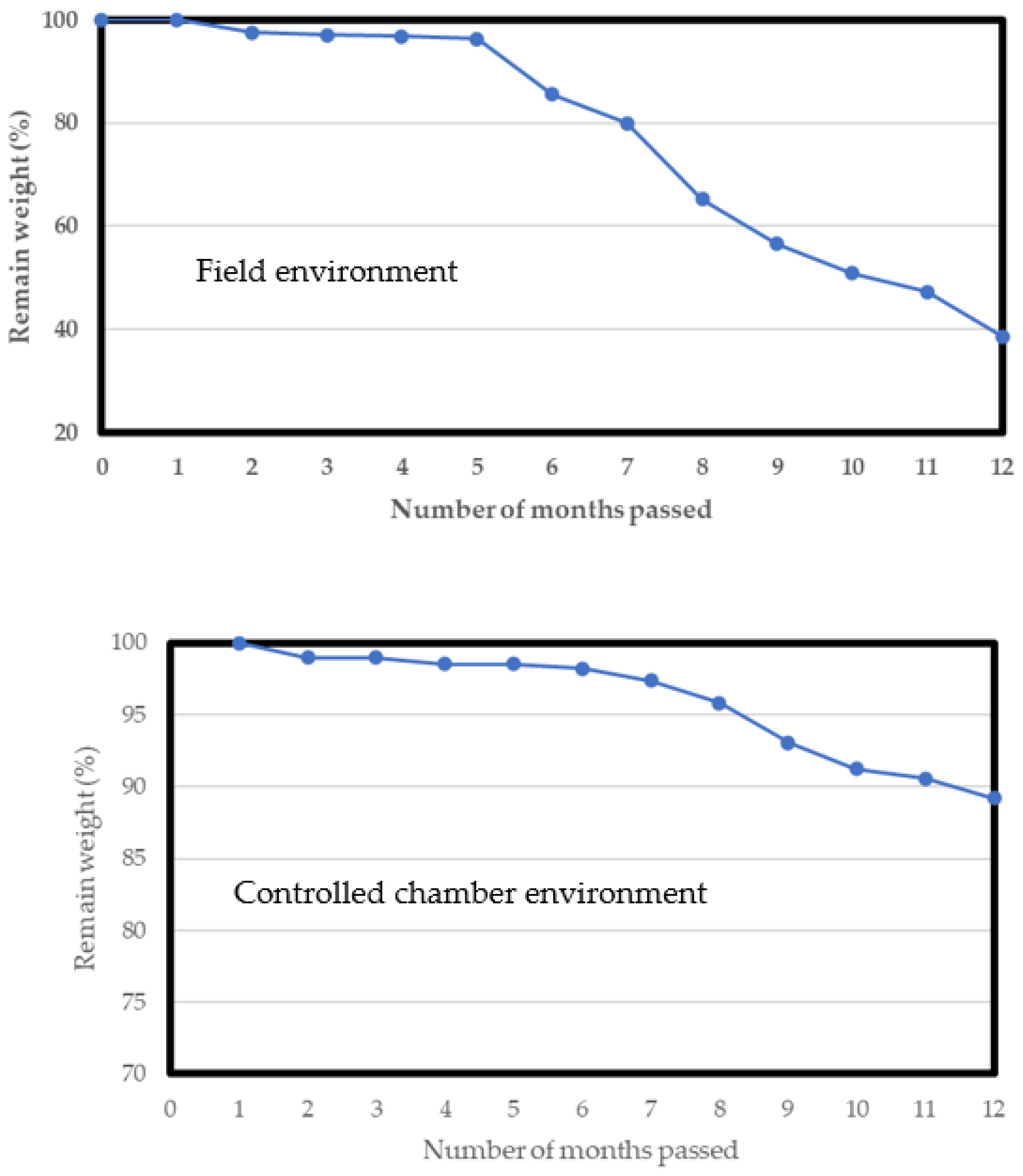
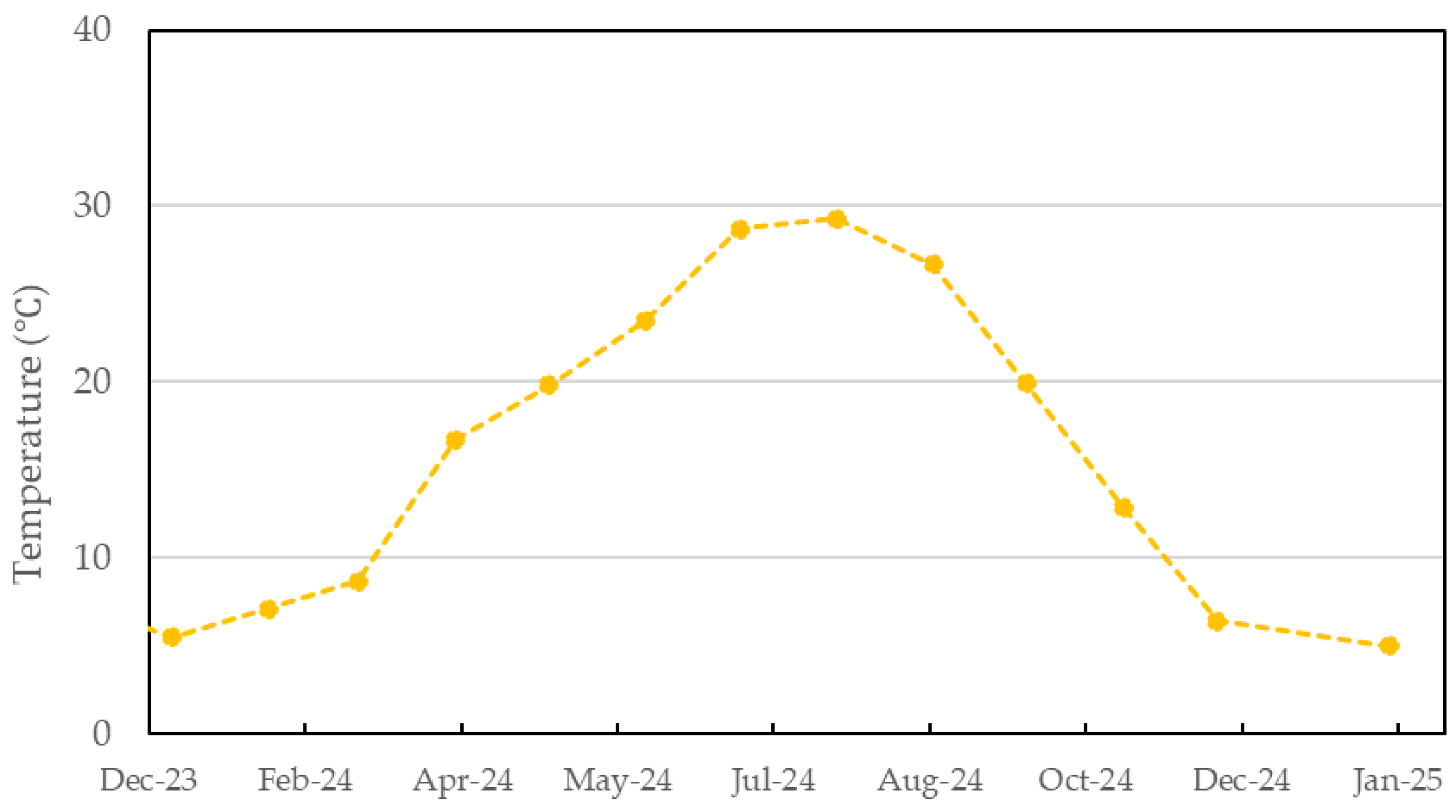
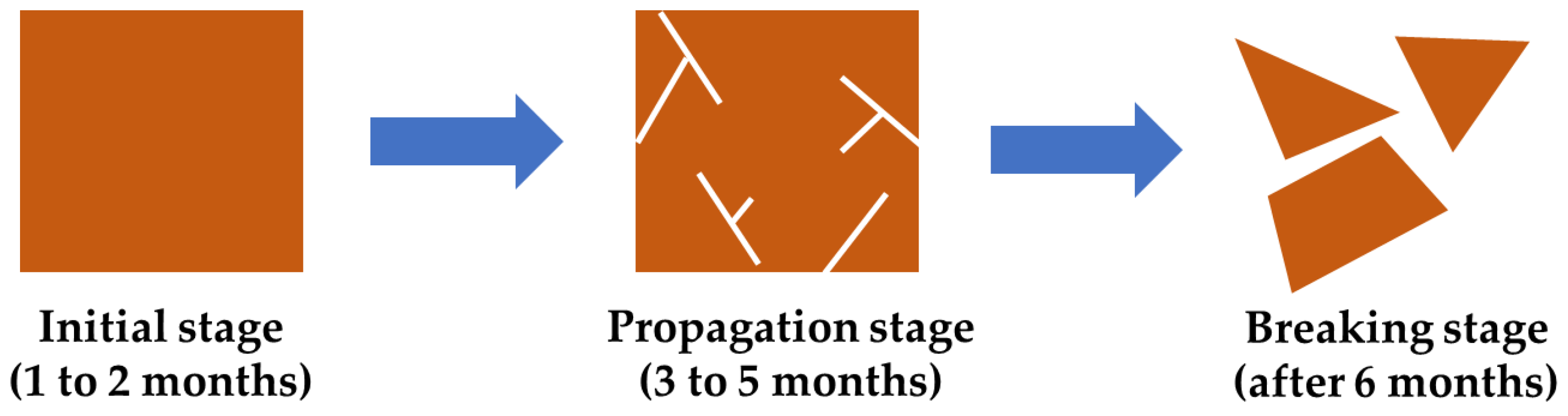
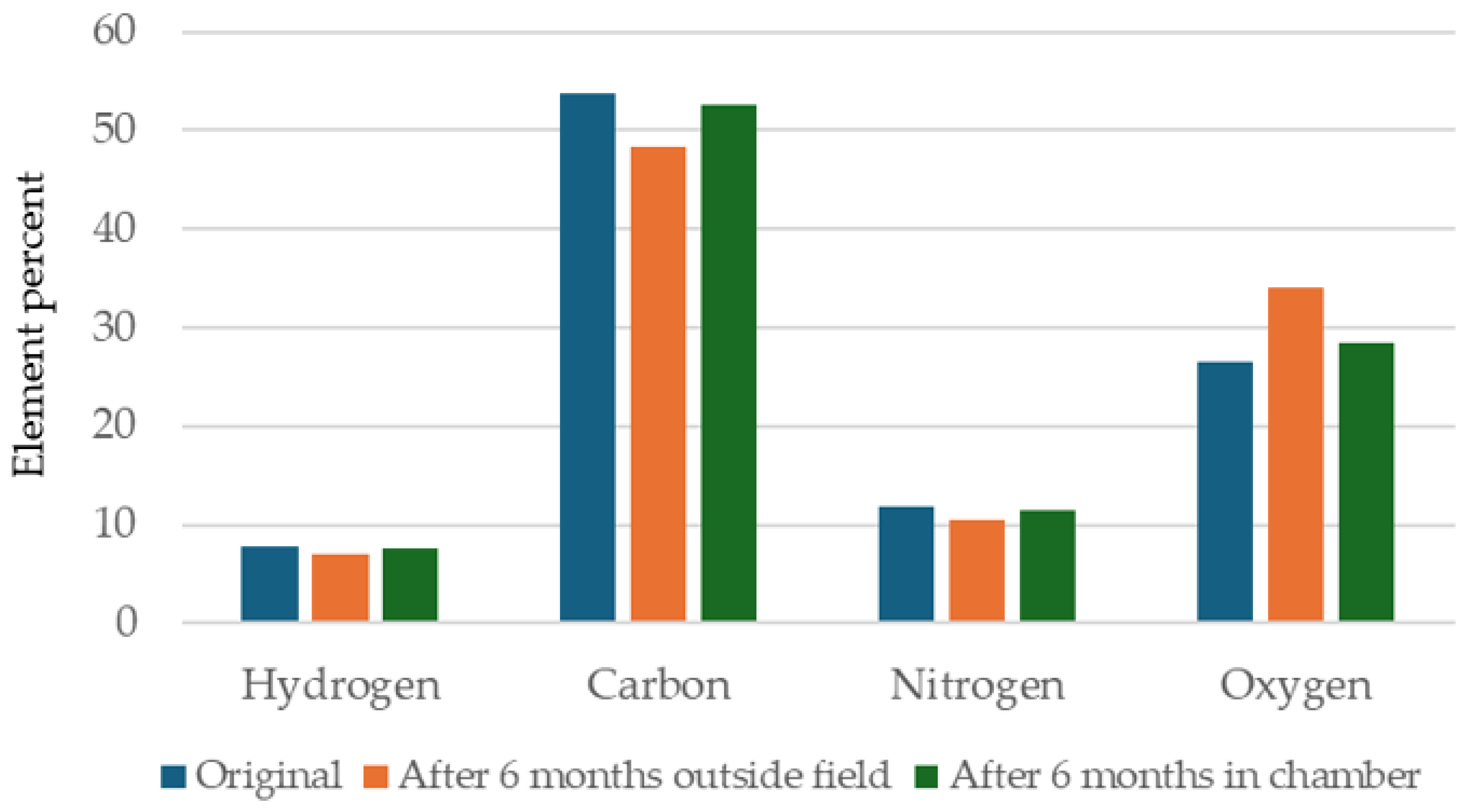
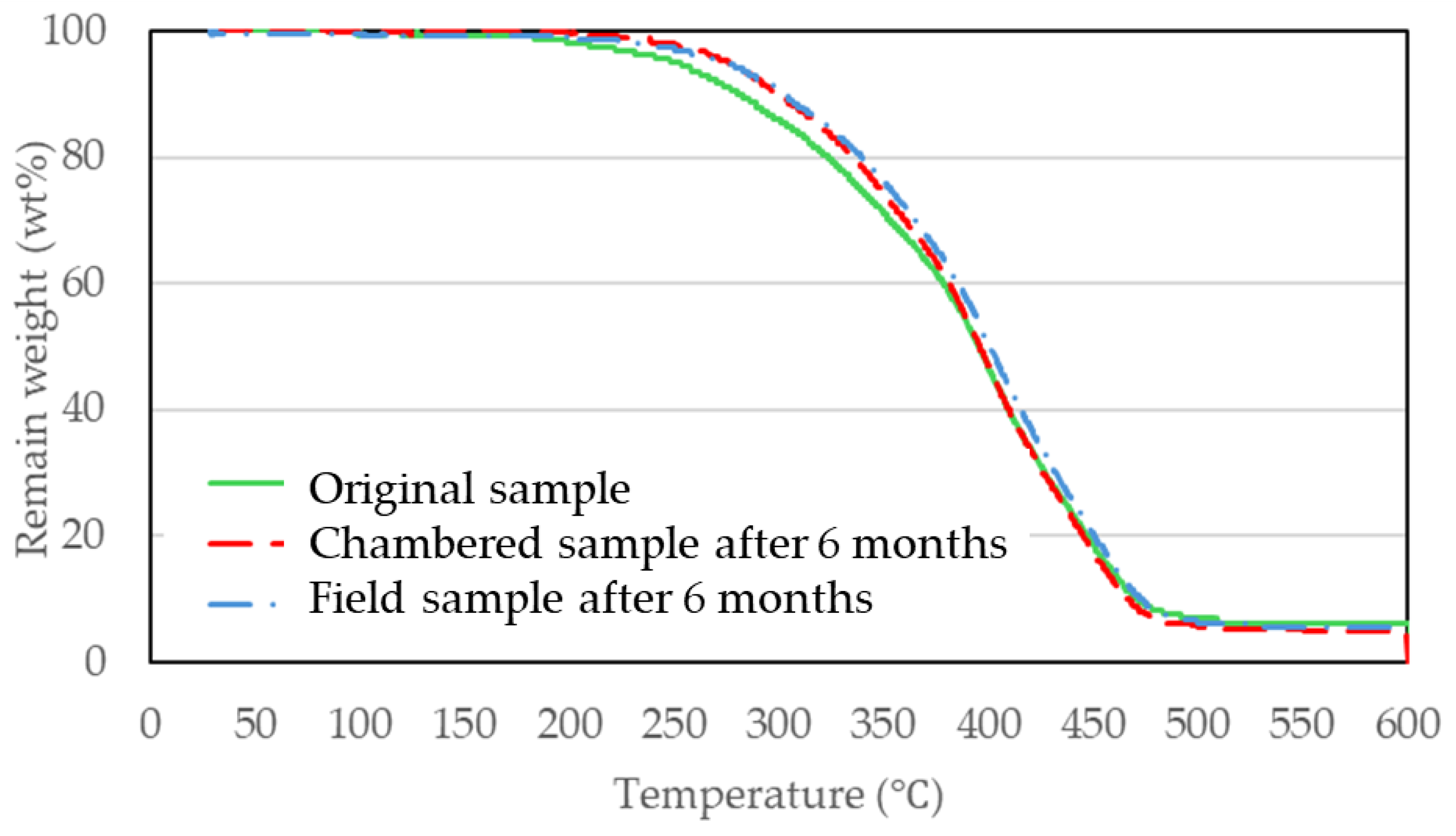
3.3. Evaluation of Bio-Polyurethane Foam
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, S.; Allen, D.; Phoenix, V.R.; Le Roux, G.; Jiménez, P.D.; Simonneau, A.; Binet, S.; Galop, D. Atmospheric transport and deposition of microplastics in a remote mountain catchment. Nat. Geosci. 2019, 12, 339–344. [Google Scholar] [CrossRef]
- Dris, R.; Gasperi, J.; Rocher, V.; Saad, M.; Renault, N.; Tassin, B. Microplastic contamination in an urban area: A case study in Greater Paris. Environ. Chem. 2015, 12, 592–599. [Google Scholar] [CrossRef]
- Chen, G.; Feng, Q.; Wang, J. Mini-review of microplastics in the atmosphere and their risks to humans. Sci. Total Environ. 2020, 703, 135504. [Google Scholar] [CrossRef] [PubMed]
- White, W.R.; Durocher, D.T. Recycling of Rigid Polyurethane Articles and Reformulation into a Variety of Polyurethane Applications. J. Cell. Plast. 1997, 33, 477–486. [Google Scholar] [CrossRef]
- Blais, M.; Carpenter, K. Flexible Polyurethane Foams: A Comparative Measurement of Toxic Vapors and Other Toxic Emissions in Controlled Combustion Environments of Foams With and Without Fire Retardants. Fire Technol. 2015, 51, 3–18. [Google Scholar] [CrossRef]
- Tuohedi, N.; Wang, Q. Preparation and Evaluation of Epoxy Resin Prepared from the Liquefied Product of Cotton Stalk. Processes 2021, 9, 1417. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Q.; Qiao, Q.; Sugiyama, K. Process analysis of the waste bamboo by using polyethylene glycol solvent liquefaction. Int. J. Sustain. Dev. Plan. 2014, 9, 647–657. [Google Scholar] [CrossRef]
- Wang, Q.; Tuohedi, N. Polyurethane Foams and Bio-Polyols from Liquefied Cotton Stalk Agricultural Waste. Sustainability 2020, 12, 4214. [Google Scholar] [CrossRef]
- Kurimoto, Y.; Doi, S.; Tamura, Y. Species effects on wood-liquefaction in polyhydric alcohols. Holzforschung 1999, 53, 617–622. [Google Scholar] [CrossRef]
- Kurimoto, Y.; Shirakawa, K.; Yoshioka, M.; Shiraishi, N. Liquefaction of untreated wood with polyhydric alcohols and its application to polyurethane foams. FRI Bull. 1992, 176, 163–172. [Google Scholar]
- Lin, L.; Yao, Y.; Yoshioka, M.; Shiraishi, N. Liquefaction mechanism of cellulose in the presence of phenol under acid catalysis. Carbohydr. Polym. 2004, 57, 123–129. [Google Scholar] [CrossRef]
- Kobayashi, M.; Asano, T.; Kajiyama, M.; Tomita, B. Analysis on residue formation during wood liquefaction with polyhydric alcohol. J. Wood Sci. 2004, 50, 407–414. [Google Scholar] [CrossRef]
- Zhang, Q.H.; Zhao, G.J.; Yu, L.L.; Jie, S.J. Preparation of liquefied wood-based resins and their application in molding material. For. Stud. China 2007, 9, 51–56. [Google Scholar] [CrossRef]
- Janiszewska-Latterini, D.; Pizzi, A. Application of Liquefied Wood Products for Particleboard Manufacturing: A Meta-analysis Review. Curr. For. Rep. 2023, 9, 291–300. [Google Scholar] [CrossRef]
- Yin, B.; Hakkarainen, M. Green Plasticizers from Liquefied Wood. Waste Biomass Valorization 2014, 5, 651–659. [Google Scholar] [CrossRef]
- Zhang, J.; Hori, N.; Takemura, A. Optimization of agricultural wastes liquefaction process and preparing biobased polyurethane foams by the obtained polyols. Ind. Crops Prod. 2019, 138, 111455. [Google Scholar] [CrossRef]
- Kurimoto, Y.; Takeda, M.; Koizumi, A.; Yamauchi, S.; Doi, S.; Tamura, Y. Mechanical properties of polyurethane films prepared from liquefied wood with polymeric MDI. Bioresour. Technol. 2000, 74, 151–157. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, L.; Li, D.; Yin, J.; Wu, S.; Mao, Z. Mechanical properties of polyurethane foams prepared from liquefied corn stover with PAPI. Bioresour. Technol. 2008, 99, 2265–2268. [Google Scholar] [CrossRef]
- Domingos, I.; Ferreira, J.; Cruz-Lopes, L.; Esteves, B. Polyurethane foams from liquefied orange peel wastes. Food Bioprod. Process. 2019, 115, 223–229. [Google Scholar] [CrossRef]
- Bontaş, M.G.; Diacon, A.; Călinescu, I.; Rusen, E. Lignocellulose Biomass Liquefaction: Process and Applications Development as Polyurethane Foams. Polymers 2023, 15, 563. [Google Scholar] [CrossRef]
- Shiraishi, N. Liquefaction of Wood and Its Several Applications. J. Oleo Sci. 1997, 46, 1127–1236. [Google Scholar]
- Yang, W.; Han, Y.; Zhang, W.; Zhang, D. High-Strength and Low-Cost Biobased Polyurethane Foam Composites Enhanced by Poplar Wood Powder Liquefaction. Polymers 2021, 13, 2999. [Google Scholar] [CrossRef] [PubMed]
- Phosri, S.; Kunjiek, T.; Mukkhakang, C.; Suebthep, S.; Sinsup, W.; Phornsirigarn, S.; Charoeythornkhajhornchai, P. Biodegradability of bioplastic blown film in a marine environment. Front. Mar. Sci. 2022, 9, 917397. [Google Scholar] [CrossRef]
- Ruggero, F.; Onderwater, R.C.A.; Carretti, E.; Roosa, S.; Benali, S.; Raquez, J.-M.; Gori, R.; Lubello, C.; Wattiez, R. Degradation of Film and Rigid Bioplastics During the Thermophilic Phase and the Maturation Phase of Simulated Composting. J. Polym. Environ. 2021, 29, 3015–3028. [Google Scholar] [CrossRef]
- Payanthoth, N.S.; Mut, N.N.N.; Samanta, P.; Li, G.; Jung, J. A review of biodegradation and formation of biodegradable microplastics in soil and freshwater environments. Appl. Biol. Chem. 2024, 67, 110. [Google Scholar] [CrossRef]
- Tong, H.; Zhong, X.; Duan, Z.; Yi, X.; Cheng, F.; Xu, W.; Yang, X. Micro- and nanoplastics released from biodegradable and conventional plastics during degradation: Formation, aging factors, and toxicity. Sci. Total Environ. 2022, 833, 15527. [Google Scholar] [CrossRef]
- Gironi, F.; Piemonte, V. Bioplastics and Petroleum-based Plastics:Strengths and Weaknesses. Energy Sources Part A 2011, 33, 1949–1959. [Google Scholar] [CrossRef]
- Boey, J.Y.; Lee, C.K.; Tay, G.S. Factors Affecting Mechanical Properties of Reinforced Bioplastics: A Review. Polymers 2022, 14, 3737. [Google Scholar] [CrossRef]
- Omar, A.A.; Hanafi, M.H.M.; Razak, N.H.; Ibrahim, A.; Afwanisa’Ab Razak, N. A Best-Evidence Review of Bio-based Plasticizer and the Effects on the Mechanical Properties of PLA. Chem. Eng. Trans. 2021, 89, 241–246. [Google Scholar]
- Shashoua, Y.; Peydaei, A.; Mortensen, M.N.; Kanstrup, A.B.; Gregory, D.J. Real time degradation studies on polyurethane household sponges in Danish weather and marine environments. Mar. Pollut. Bull. 2022, 184, 114128. [Google Scholar] [CrossRef]
- Szpiłyk, M.; Lubczak, R.; Lubczak, J. The biodegradable cellulose-derived polyol and polyurethane foam. Polym. Test. 2021, 100, 107250. [Google Scholar] [CrossRef]
- Gunawan, N.R.; Tessman, M.; Schreiman, A.C.; Simkovsky, R.; Samoylov, A.A.; Neelakantan, N.K.; Bemis, T.A.; Burkart, M.D.; Pomeroy, R.S.; Mayfield, S.P. Rapid biodegradation of renewable polyurethane foams with identification of associated microorganisms and decomposition products. Bioresour. Technol. Rep. 2020, 11, 100513. [Google Scholar] [CrossRef]
- Pires, J.R.A.; Souza, V.G.L.; Fuciños, P.; Pastrana, L.; Fernando, A.L. Methodologies to Assess the Biodegradability of Bio-Based Polymers—Current Knowledge and Existing Gaps. Polymers 2022, 14, 1359. [Google Scholar] [CrossRef]
- Folino, A.; Karageorgiou, A.; Calabrò, P.S.; Komilis, D. Biodegradation of Wasted Bioplastics in Natural and Industrial Environments: A Review. Sustainability 2020, 12, 6030. [Google Scholar] [CrossRef]
- Gunawan, N.R.; Tessman, M.; Zhen, D.; Johnson, L.; Evans, P.; Clements, S.M.; Pomeroy, R.S.; Burkart, M.D.; Simkovsky, R.; Mayfield, S.P. Biodegradation of renewable polyurethane foams in marine environments occurs through depolymerization by marine microorganisms. Sci. Total Environ. 2022, 850, 158761. [Google Scholar] [CrossRef] [PubMed]
- Masuda, G.; Nagao, A.; Wang, W.; Wang, Q. Evaluation of Polyurethane Foam Derived from the Liquefied Driftwood Approaching for Untapped Biomass. Processes 2023, 11, 2929. [Google Scholar] [CrossRef]
- Masuda, G.; Enyoh, C.E.; Wang, W.; Suzuki, M.; Honda, Y.; Wang, Q. Synthesis and Characterization of Bio-Based Polyurea Derived from Liquefied Wood of Wooden Musical Instrument Offcuts. Macromol 2024, 4, 739–752. [Google Scholar] [CrossRef]
- Wang, Q.; Kawamura, S. Decayed Woody Material from Mushroom Cultivation: Characterization of Liquefaction. In Sustainable Development and Planning X; WIT Press: Hampshire, UK, 2018; pp. 481–492. [Google Scholar]
- Morohoshi, N.; Hong, C.-C.; Morimoto, M.; Oohashi, T.; Matumoto, H. Polymers and Environment I. Biodegradability of Flexible Polyurethane Foams. Kobunshi Ronbunshu 1993, 50, 731–738. [Google Scholar] [CrossRef]
- Taourit, S.; Le Gac, P.Y.; Fayolle, B. Relationship between network structure and ultimate properties in polyurethane during a chain scission process. Polym. Degrad. Stab. 2022, 201, 109971. [Google Scholar] [CrossRef]
- Di Bisceglie, F.; Quartinello, F.; Vielnascher, R.; Guebitz, G.M.; Pellis, A. Guebitz and Alessandro Pellis Cutinase-Catalyzed Polyester-Polyurethane Degradation Elucidation of the Hydrolysis Mechanism. Polymers 2022, 14, 411. [Google Scholar] [CrossRef]
- Pellizzi, E.; Lattuati-Derieux, A.; Lavédrine, B.; Cheradame, H. Degradation of polyurethane ester foam artifacts: Chemical properties, mechanical properties and comparison between accelerated and natural degradation. Polym. Degrad. Stab. 2014, 107, 255–261. [Google Scholar] [CrossRef]
- Skleničková, K.; Suchopárová, E.; Abbrent, S.; Pokorný, V.; Kočková, O.; Nevoralová, M.; Cajthaml, T.; Strejček, M.; Uhlík, O.; Halecký, M.; et al. Biodegradation of aliphatic polyurethane foams in soil: Influence of amide linkages and supramolecular structure. Sci. Total Environ. 2024, 912, 169062. [Google Scholar] [CrossRef] [PubMed]
- Kurańska, M.; Leszczyńska, M.; Kubacka, J.; Prociak, A.; Ryszkowska, J. Effects of Modified Used Cooking Oil on Structure and Properties of Closed-Cell Polyurethane foams. J. Polym. Environ. 2020, 28, 2780–2788. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Vo, D.K.; Nguyen, K.T.D.; Tran, D.Q.; Nguyen, D.M.; Nguyen, N.T.; Vu, T.T.; Nguyen, V.T.; Hoang, D. Lightweight Biobased Polyurethane Composites Derived fromLiquefied Polyol Reinforced by Biomass Sources with HighMechanical Property and Enhanced Fire-Resistance Performance. ACS Omega 2024, 9, 19182–19192. [Google Scholar] [CrossRef]
- Lubczak, R.; Kus-Liśkiewicz, M.; Lubczak, J.; Szpiłyk, M.; Broda, D.; Bobko, E. Biodegradable Polyurethane Foams Based on Polyols Obtained from Cellulose and Its Hydroxypropyl Derivative. Materials 2024, 17, 5490. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Setting Parameter | Setting Point |
|---|---|
| Wood meal weight | 10 g |
| PEG400/Glycerol (7/3) | 50 g |
| Solvent weight ratio (vs. wood meal) | 6 times or 7 times |
| 95% sulfuric acid | 1.5 g |
| Stirring speed | 700 rpm |
| Sampling points | 15, 30, 60, 90, 120, 180 min |
| Setting oil bath temperature | 150 °C |
| Element | Hydrogen | Carbon | Nitrogen | Oxygen | Ash |
|---|---|---|---|---|---|
| Content (%) | 5.8 | 44.5 | 0.3 | 46.1 | 3.3 |
| Component | Organic Solvent-Soluble Content | Holocellulose | Lignin |
|---|---|---|---|
| Content (%) | 11.9 | 64.5 | 28.4 |
| Residue Content (%) | 5 | |
|---|---|---|
| Liquefaction time (min) | 60 | 180 |
| Acid number (mg KOH/g) | 14 | 16 |
| Hydroxyl number (mg KOH/g) | 324 | 265 |
| Residue Content (%) | 5 | |
|---|---|---|
| Liquefaction time (min) | 60 | 180 |
| Mw | 1010 | 1080 |
| Mn/Mw | 1.80 | 1.90 |
| Polyol | PEG400 | Liquefied Product |
|---|---|---|
| Foaming rate (times) | 9.9 | 14.0 |
| Water adsorption (%) | 10.2 | 16.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masuda, G.; Enyoh, C.E.; Ishidoya, K.; Wang, W.; Wang, Q. Evaluation of Bio-Polyurethane Foam Synthesized from Liquefied Waste Wood Polyol. Recycling 2025, 10, 126. https://doi.org/10.3390/recycling10040126
Masuda G, Enyoh CE, Ishidoya K, Wang W, Wang Q. Evaluation of Bio-Polyurethane Foam Synthesized from Liquefied Waste Wood Polyol. Recycling. 2025; 10(4):126. https://doi.org/10.3390/recycling10040126
Chicago/Turabian StyleMasuda, Go, Christian Ebere Enyoh, Keiju Ishidoya, Weiqian Wang, and Qingyue Wang. 2025. "Evaluation of Bio-Polyurethane Foam Synthesized from Liquefied Waste Wood Polyol" Recycling 10, no. 4: 126. https://doi.org/10.3390/recycling10040126
APA StyleMasuda, G., Enyoh, C. E., Ishidoya, K., Wang, W., & Wang, Q. (2025). Evaluation of Bio-Polyurethane Foam Synthesized from Liquefied Waste Wood Polyol. Recycling, 10(4), 126. https://doi.org/10.3390/recycling10040126