Abstract
Lignin-derived carbon nanofibers (LCNFs) formed via the solution blowing of a biopolymer are developed here as a promising replacement for polyacrylonitrile (PAN)-derived carbon nanofibers (PCNFs) formed via electrospinning for such applications as supercapacitor (SC) electrodes. Accordingly, it is demonstrated here that a biopolymer (kraft lignin, which is, essentially, a waste material) can substitute a petroleum-derived polymer (PAN). Moreover, this can be achieved using a much faster and safer fiber-forming method. The present work employs the solution blowing of lignin-derived nonwovens and their carbonization to form electrode materials. These materials are characterized and explored as the electrodes in supercapacitor prototypes. Given the porosity importance of carbon fibers in SC applications, N2 gas adsorption tests were performed for characterization. LCNFs revealed the specific surface area (SSA) and capacitance values as high as 1726 m2/g and 11.95 F/g, which are about one-half of those for PCNFs, 3624 m2/g and 25.5 F/g, respectively. The capacitance values of LCNFs are comparable with those reported in the literature, but the SSA observed here is much higher. Moreover, no further post-carbonization activation steps were performed here in comparison with those materials reported in the literature. It was also found here that fiber pre-oxidation in air prior to carbonization and the addition of zinc chloride affect the SSA and capacitance values of both LCNFs and PCNFs. The electrochemical tests of the SCs prototypes were used to evaluate their capacitance at different charging rates, voltage windows, and the number of cycles. The capacitance of PCNFs decreased by about 47% during fast charging, while the capacitance of LCNFs improved during fast charging, bringing them to the level of only 21% below that of PCNFs. These changes were correlated with the packing density of the electrodes. It should be emphasized that LCNFs revealed a much higher mass yield, which was 4–5 times higher than that of PCNFs. LCNFs also possess a higher packing density, a lower price, and cause a significantly lower environmental impact than PCNFs. The best cell supercapacitor delivered a maximum specific energy of 1.77 Wh/kg and a maximum specific power of 156 kW/kg, surpassing conventional electrochemical supercapacitors. Remarkably, it retained 95.2% of its initial capacitance after 10,000 GCD cycles at a current density of 0.25 A/g, indicating robust stability. Accordingly, kraft lignin, a bio-waste material, holds great promise as a raw material for supercapacitor electrodes.
1. Introduction
The demand for energy increases dramatically with technological progress and population growth. Currently, fossil fuels are the main source of energy accounting for 80% of the global energy consumption. However, fossil fuels are the main source of greenhouse gas emissions [1], and accordingly, renewable sources of energy are being sought. Environmentally friendly energy-harvesting options include solar, wind, wave, hydroelectric, hydrogen-fuel, and others. Because many of these energy sources produce electrical energy intermittently, storage devices are required. For that purpose, supercapacitors are very effective energy storage devices, and they hold great promise in applications requiring fast energy and power supply. Supercapacitors can be used as backup generators, in addition to applications in electronic devices, electric vehicles, and industrial equipment [1,2]. Supercapacitors can provide rapid acceleration to electric vehicles, as well as the storage of energy resulting from car braking losses, thus reducing energy consumption by 15%. Supercapacitors can be used alone or in combination with battery systems to improve performance, given the higher specific power and long lifespan of supercapacitors, as well as high specific energy supplied from batteries [2,3,4].
Supercapacitors can be distinguished by the mechanism of charge storage, such as electric double-layer capacitors (EDLCs), pseudo-capacitors (PCs), and hybrid capacitors (HCs) [2]. The specific capacitance of EDLCs depends on the electrical conductivity, high specific surface area (SSA), and pore-size distribution of the electrodes. Carbon materials, such as activated carbon (AC), graphene (GN), carbon nanotubes (CNTs), and carbon nanofibers (CNFs) are promising candidates for meeting such requirements. Accordingly, they are often employed in EDLC electrodes [3,5]. However, one of the disadvantages of the currently available EDLCs is that they rely on the use of petroleum-derived polymers, such as polyacrylonitrile (PAN), as the precursor of porous carbon nanostructures. Moreover, appropriate carbons are often available in the form of powder, requiring traditional slurry-based methods to form electrodes. These methods involve the use of binders, which inevitably increase the electrical resistance, reduce the ion-transport kinetics, and cause pore blockage, thus reducing the effective SSA due to particle burying [6]. Aiming at an improved performance, the use of self-standing carbon electrodes is gaining significant attention. Now, techniques such as electrospinning, solution blow spinning, and supersonic solution blowing were employed to form nanofibers, which can be further carbonized to become self-standing membranes comprising micro- and mesoporous carbons [7].
Carbon nanofibers (CNFs) are of great importance for energy storage devices, such as supercapacitors [5]. Currently, petroleum-derived polyacrylonitrile (PAN) is the most common precursor for CNFs. PAN-derived CNFs (PCNFs) are efficient and inexpensive but rather environmentally unfriendly, since PAN is a petroleum-derived polymer and its processing releases toxic compounds [8,9]. Therefore, the substitution of PCNFs with a biopolymer such as lignin to form CNFs is desirable. Lignin is the most abundant aromatic biopolymer in nature. It is widely available as a bio-waste from the paper industry, but has been poorly utilized, being mostly consumed as a source of solid fuel [10,11]. Different methods for obtaining lignin-derived carbon fibers (LCNFs) and their characterization have been evaluated to achieve properties like those of PAN-derived CNFs. So far, lignin-derived CNFs have not been able to perform in the same way PAN-derived CNFs, despite the environmental friendliness of the former and their cost being about 40% lower than that of PCNFs. Accordingly, lignin-derived CNFs hold great promise [11].
Lignin is currently in focus in many research works, and multiple strategies have been proposed to explore its application as a CNF precursor for energy storage. The use of CNFs in supercapacitors strongly depends on the specific surface area (SSA), pore size, electrical conductivity, and heating strategies [12,13,14,15]. Electrospinning (ES) is a fiber-forming technique known for producing high-surface area nanotextured nonwoven mats [8]. It is often used to form PAN nanofibers, as well as lignin-derived nanofibers [8,16,17]. Several blends of lignin with other polymers have been used for the electrospinning of lignin-derived nanofibers, e.g., those with polyethylene oxide (PEO), polyacrylonitrile (PAN), polyvinyl alcohol (PVA), and polyvinyl pyrrolidone (PVP) [18,19]. One of the challenges in obtaining nanofibers from lignin is that its solution is insufficiently viscoelastic to sustain continuous stretching during the spinning process, which requires a host polymer for spinnability. Moreover, the content of the guest polymer (lignin) compared to that of the host polymer is very often low. In addition, electrospinning is a relatively slow process, making it less appropriate for large-scale manufacturing. The later drawback can be resolved using solution blowing (SB) [8,20,21]. This method is, in a sense, similar to electrospinning, albeit relying on an aerodynamically driven, rather than electrically driven bending instability to form nanofibers [22]. Accordingly, SB is several times faster than ES, being capable of forming nanofibers at an industrial scale [23].
The review in reference [22] states that PAN-derived carbon nanofibers (PCNFs) reveal SSA as high as 2800 m2/g with capacitance of 107 F/g, with Vanadium being added to increase the electrode conductivity, and KOH used as an electrolyte. Reference [24] demonstrated PAN-derived activated carbon nanofibers (ACNF) with SSA in the 742–1170 m2/g range and Csp in the 141–333 F/g range facilitated by the addition of polypyrrole (PPy) and CNTs. Accordingly, the material and processing choices significantly affect the values of SSA and Csp of carbon electrodes. In addition, carbon-based electrodes are frequently further post-processed for the fine-tuning of their porous structure via physical or chemical activation and heteroatom doping [3]. Variability resulting from the tuning options highlights the need for a study aiming to elucidate the CNF properties and their correlations with specific methodologies.
Although PCNFs are efficient and have attracted significant attention in the literature, non-petroleum-derived green materials are called for in this field to reduce the environmental impact. As an alternative, lignin holds great promise as an eco-friendly, raw material substitute of PAN for electrode manufacturing. Biodegradable lignin is the second most abundant biopolymer. It is widely available, and more importantly, lignin is also available as the bio-waste of several industrial processes. There are different types of lignin, which vary by plant source and extraction process [8]. Many tons of kraft lignin result from the paper industry as a by-product, and yet it is mostly utilized as a cheap fuel source. To increase sustainability and add value to lignin waste, as well as to diminish the use of petroleum-derived polymers, lignin-derived carbon nanofibers (LCNFs) have recently been formed and tested in supercapacitors [9,10,11,12,13].
Motived by these technological challenges, here, nanofibers from PAN, lignin, and lignin with the addition of ZnCl2 as a pore-forming additive were formed, carbonized, and characterized to reveal their morphology, surface chemical composition, graphitization, and the electrical conductivity. Also, the effects of stabilization in air and of the carbonization regimes were elucidated. Specifically, lignin-derived nanofibers (~544 nm in diameter and ~335 nm in diameter if ZnCl2 was added) with a lignin content as high as 90 wt.% in the raw material (with the additional 10 wt.% of nylon 6) were formed via solution blow spinning. The fibers formed from the raw material with 80 wt.% of lignin were the most robust. Accordingly, they were chosen for the production of self-standing electrodes. Moreover, the effect of the carbonization temperature was explored. The fibers were carbonized at different temperatures, 600 °C, 900 °C, and 1200 °C, with or without a pre-oxidation step (PO) in air flow. After carbonization at 1200 °C, LCNFs with diameters as small as 99 nm were formed, while the freestanding fibrous structure was maintained.
Here, a sustainable approach starting from a bio-waste biopolymer (kraft lignin) is explored to establish whether lignin could serve as an environmentally friendly inexpensive substitute for petroleum-derived precursors (PAN) in the production of CNFs. This is a novel approach to the utilization of abundant bio-waste for high-end applications such as supercapacitors. The formation of lignin-derived nanofibers via solution blowing is in focus. Lignin was used as a guest polymer blended with nylon 6, a highly spinnable host polymer. Nylon 6 loses about 95% of its weight when carbonized in inert atmosphere at 600 °C [25], also serving as a pore-forming agent [19]. As an additional potential porogen, ZnCl2 was also dissolved in the original solution to be incorporated into the as-blown nanofibers. In this regard, different carbonization protocols were tested, because they can significantly affect CNF properties [26]. In addition, for comparison purposes, electrospun PAN nanofibers were formed, carbonized, and tested in the same manner as the lignin-derived CNFs [26]. The main aim of this work is to form bio-waste-derived materials and establish them as the efficient electrodes for supercapacitor prototypes. Multiple characterization methods employed in this work revealed that that, despite a relatively lower SSA value and specific capacitance than those of carbon nanofibers formed from petroleum-derived polymer polyacrylonitile (PAN), lignin guarantees a much higher mass yield than that of PAN, as well as a higher packing density and a lower price. Lignin-derived carbon nanofibers also reveal an improved performance at fast scan rates, as well as a much lower environmental impact, which is an attractive starting point for supercapacitor electrodes.
2. Materials and Methods
Lignin alkali low sulfonated (Mw = 10 kDa), nylon 6 (CAS No.: 25038-54-4), polyacrylonitrile (PAN) (Mw = 150 kDa), zinc chloride (ZnCl2, >98%), formic acid (≥95%), dimethylformamide (DMF, 99.8%), and sodium perchlorate (NaClO4, ≥98%) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.1. Fiber Precursor Preparation
The lignin-derived nanofibers were prepared via the solution blowing technique. A solution of 30 wt.% of lignin and a solution of 15 wt.% of nylon 6 were prepared in formic acid under magnetic stirring at 50 °C. Both solutions were mixed at different lignin/nylon 6 ratios. In one case, zinc chloride was added to a mixed solution as an additional porosity promoter. Information regarding solution preparation and blowing parameters is listed in Table 1.

Table 1.
Lignin-derived solutions and relevant solutions blowing information. In the right-hand side column, parameters are listed in the following order: pumping rate (mL/h); air pressure (psi); relative humidity (%); and needle gauge (G). Spinning was carried out at room temperature, ~23 °C.
2.2. Fiber Carbonization Protocols
The available literature reports that one of the hurdles in producing lignin-derived CNFs is the need for a very long pre-oxidation time, of up to 26 h; however, it is also reported to be unnecessary due to the presence of the host polymer [12,14,27]. Accordingly, in the present work, nanofibers were carbonized with and without a pre-oxidation step. For PAN nanofibers, the pre-oxidation step is known to be important and could strongly impact the resulting electrical conductivity and mechanical properties [28]. Still, for the sake of comparison, PAN nanofibers were carbonized with and without a pre-oxidation step. The pre-oxidation step for PAN nanofibers was conducted in a muzzle furnace at temperatures of up to 280 °C, with a heating rate of 3 °C/min. Then, PAN nanofibers were kept at 280 °C for 3 h. A similar pre-oxidation step was used for lignin-derived nanofibers, although with a maximum temperature of 240 °C. In the subsequent step, for the carbonization of lignin-derived or PAN nanofibers, three heating profiles were tested to determine the corresponding changes in morphology, composition, the electrical conductivity, weight loss, and the degree of graphitization of the resulting CNFs. These heating profiles are listed in Table 2. Here and hereinafter, the heating profiles are identified by their IDs in Table 2.
Table 2.
Thermal treatment profiles. Legend: Pre-oxidized (PO), used for PAN nanofibers only. Pre-oxidized lignin (POL), used for lignin and lignin/ZnCl2 nanofibers. NO stands for the cases without pre-oxidation step. The notations H1, H2, and H3 stand for different carbonization profiles, with different final temperatures (600 °C, 900 °C, and 1200 °C, respectively) for different specimens.
The nanofiber specimens were identified according to the processes used during their formation. For example, specimen L2_NO_H1 means that an original specimen with 80 wt.% of lignin and 20 wt.% of nylon 6 (L2 specimen of Table 1), without a pre-oxidation step, was carbonized according to the heating profile H1 (Table 2). Another example: P1_PO_H2, means that this specimen originated from a PAN nanofiber mat that was pre-oxidized and then carbonized according to the H2 profile. The flow chart of the methodology used in the present work is depicted in Figure 1 to facilitate general comprehension. The characterization procedures are further described in the Supplementary Materials.
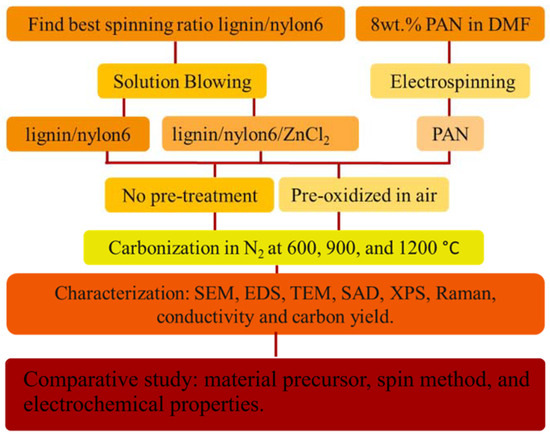
Figure 1.
Flow chart of the present methodology.
2.3. Structural, Chemical, and Morphologic Characterization
The surface area, as well as the pore volume and sizes, were determined via N2 gas adsorption using an Absorption Analyzer Autosorb IQ by Quantachrome Instruments. Before performing measurements, specimens were degassed for 10 h at 110 °C. The surface area was determined according to the Brunauer–Emmett–Teller (BET) theory, with the aid of the micropore assistant and by means of the nonlinear density functional theory (NLDFT), implying the existence of cylindrical and spherical pores [29].
Scanning electron microscope (SEM) and energy dispersive spectroscopy (EDS) were conducted using JEOL (JMS-IT500HR SEM). The images were obtained in secondary electron beam mode at an accelerating voltage of 3–5 kV. The nanofiber mats were sputter-coated with a 4 nm layer of platinum/palladium prior to imaging. Transmission electron microscopy (TEM) and selected area diffraction (SAD) images were obtained using JEOL (JEM-3010). The electron beam was accelerated at a voltage of 300 kV, and samples were ground and placed on a lacey carbon film mesh.
XPS was performed using Kratos (AXIS-165) at a vacuum of 10−9 Pa, with the X-ray charged with a current of 10 mA and accelerated at 12 kV. An initial survey scan was conducted. Then, higher resolution scans were conducted for each specific peak. The results were analyzed by means of the software XPS-PEAK (version 4.1), a Shirley background was traced, and the peaks were fitted via the Gaussian–Lorentz functions.
Raman spectroscopy was performed using Renishaw via reflex Raman, with a red laser at the wavelength of 633 nm, with a laser power of 10%. Three measurements were conducted for each sample at different locations. The data obtained were processed, and Gaussian fitting was performed for the main D and G peaks. The Gaussian fitting results in accurate values for the peak positions, the area, and the corresponding half-width (HW).
2.4. Electrochemistry Assessment
Symmetrical supercapacitors were built using CR2032 coin cells. The carbonized nanofiber specimens were cut in circles of 1.3 cm2 in area with a known thickness and weight. They were placed on both positive and negative electrodes. Here and hereinafter, “S” in tables or figures stands for Supplementary Materials, provided in a separate document. A Celgard 3401 microporous membrane of 25 µm in thickness was used as a separator. A 17 mol/L solution of sodium perchlorate (NaClO4) was used as an electrolyte. The coin cells were closed using a hydraulic press at 0.55 Torr.
Cyclic voltammetry analysis was performed using GAMRY instruments to evaluate the supercapacitor performance via the shape of the cyclic-voltammetry curve, the corresponding capacitance, and its retention. First, the specimens were submitted to a scan of 200 cycles with potential variation in the 0–1.5 V range at a scan rate of 50 mV/s for specimen wetting. Subsequently, tests were conducted while varying the voltage windows from 0 to 1 V, 1.2 V, 1.4 V, 1.6 V, 1.8 V, 2.0 V, and 2.2 V at a scan rate of 20 mV/s to obtain the working voltage window. At the voltage range of 0 to 1.5 V, scan rates of 10, 30, 40, 50, 100, 200, and 500 mV/s were employed. Second, 1000 cycles were performed at a scan rate of 50 mV/s and a voltage of 0 to 1.5 V to verify the capacitance retention. The third cycle was used as the initial value because it is common that the first or second cycle curves present inconsistencies due to the initial wetting. The first few curves of a voltammogram are often discarded because they may not accurately represent the steady-state behavior of a system. During the initial cycles, the electrode surface might undergo changes or the system might not have reached a stable state. Therefore, subsequent cycles, which better represent the stable behavior of the system, are usually considered for analysis. The specific capacitance was calculated via the following equation [15]:
in which is the specific capacitance (F/g), A is the area inside the cyclic voltammetry curve (with the units of Ampere Volt), m (g) is the mass of both electrodes, k is the scan rate (V/s), and is the voltage window of the scan (V). The results were also analyzed by comparing the capacitance in terms of the specimen area and volume. The overall procedure (from synthesis to electrochemical assessment) used in the present work is shown in Figure 2.
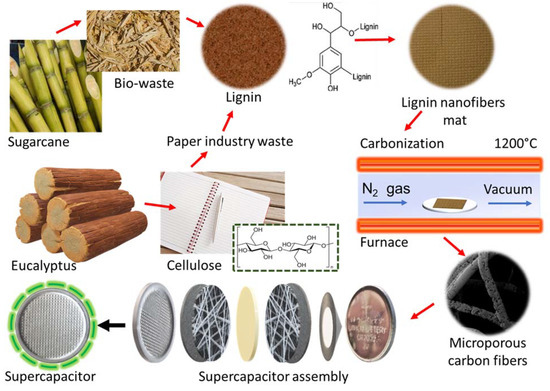
Figure 2.
Schematic of the overall procedure used in the present work to obtain carbon nanofibers from lignin and PAN for supercapacitors and other applications. In this work, the only lignin used was a raw lignin as purchased. In the figure, the images of sugar cane and eucalyptus are presented solely for an overview of the potential sources of lignin.
3. Results and Discussion
3.1. SEM-EDS
SEM and EDS analyses were performed for all specimens to determine fiber morphology and composition changes. First, the best lignin/nylon 6 nanofibers were chosen. Figure 3a–c depicts the SEM images of lignin-derived nanofibers (specimens L1, L2, and L3). Despite the different initial content of lignin (66 wt.%, 80 wt.%, and 90 wt.%, respectively), all nanofiber specimens possess similar morphologies and cross-sectional fiber diameters. This indicates that the method used to produce nanofibers is quite insensitive to changes in the initial polymer solution. Because lignin is plant-derived and variations in molecular weight are quite common, it is important to use a fiber-forming method unaffected by such a variability. Here, the strategy was to use the low molecular weight lignin as a guest biopolymer while relying on the excellent spinnability of nylon 6 used as a host polymer in the fiber-forming process. In this way, fibrillar structures were even obtained for high loadings of lignin. Despite maintaining their fibrillar morphology, nanofibers lose appropriate mechanical properties at very high lignin loadings. With the aim of creating self-standing electrodes, which eliminates one step in supercapacitor preparation, nanofibers with 80 wt.% of lignin and 20 wt.% of nylon 6, which possessed appropriate mechanical properties, were chosen for further carbonization and analysis. This specimen was chosen due to the high fraction of lignin present in the solution and the convenience of its good handling, which is required for the assembly step of free-standing electrodes in the supercapacitor. Several example of such nanofiber mats (as-blown, after pre-oxidation, and as a freestanding carbon electrode after carbonization) are given in Figure 3d.

Figure 3.
SEM images of as-blown lignin-derived nanofibers. (a) 66 wt.% of lignin (L1); (b) 80 wt.% lignin (L2); and (c) 90 wt.% of lignin (L3). Scale bar is 2 µm. (d) Images of a Z1 specimen: as-blown (left), after pre-oxidation (middle), and as a freestanding carbon electrode after carbonization (right).
As described in the ‘Materials and Methods’ section, PAN, lignin/nylon 6, and lignin/nylon 6/ZnCl2 nanofibers were explored. Because PAN is currently widely used for the production of carbon nanofibers, PAN nanofibers were first characterized for comparisons with the literature. Then, lignin-derived nanofibers were studied, followed by lignin-derived nanofibers incorporating ZnCl2, which were used to explore whether promotes pore formation.
For PAN CNFs, the behavior observed was like that already established in the literature [30]. The stabilization process (pre-oxidation) promoted the maintenance of the fibrous structure, with a significant reduction in the mean nanofiber diameter at an increased temperature (cf. Figure 4a and Figures S1–S3). Morphologically, the non-pre-oxidized nanofibers presented a structure with cohesive fibers (joints). This may lead to a negative effect (reducing surface area), as well as to a positive effect (increasing the electrical conductivity) [31]. The pre-oxidized nanofibers revealed a structure composed of relatively aligned nanofibers, with fiber–fiber merging (joints), which possessed relatively homogeneous average diameters. Thus, carbon nanofibers of 291 ± 53 nm in diameter were obtained after pre-oxidation at 280 °C (specimen P1_PO), and carbon nanofibers of 74 ± 17 nm in diameter were obtained after carbonization at 1200 °C (specimen P1_PO_H3). In contrast, carbon nanofibers without pre-oxidation, which were obtained at 1200 °C (specimen P1_NO_H3), revealed an average diameter of 275 ± 72 nm. These results ascertain the importance of pre-oxidation for PAN-derived carbon nanofibers. The formation of PAN-derived carbon nanofibers is well known [30,32] and involves a stabilization step (pre-oxidation) in an oxidative atmosphere used to induce the cyclization of the nitrile groups, which allows such nanofibers to acquire mechanical strength. Then, during the carbonization step, other non-carbon elements are eliminated in an inert atmosphere (which was confirmed by EDS), thus reducing fiber diameters. The results of the elemental mapping obtained via EDS are presented in Figure 4a, Figures S1 and S2 and in Table S1.
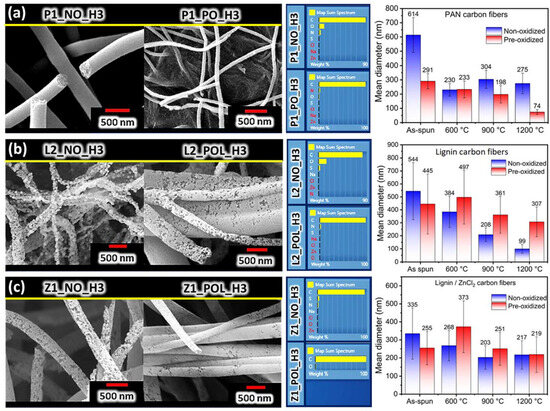
Figure 4.
(a) SEM and EDS of P1_NO_H3 and P1_PO_H3 specimens, and the mean diameter values for all P1 specimens. (b) Specimens L2_NO_H3 and L2_POL_H3: SEM and EDS. The mean diameters of all L2 specimens. (c) Specimens Z1_NO_H3 and Z1_POL_H3: SEM and EDS. The mean diameters of all L2 specimens. The parameters of SEM imagens are: 5 kV, WD 11 mm and 40 kX.
Macro- and mesoporous structures were observed for carbonized PAN nanofibers, for both PO and NO specimens (cf. Figure 4a). These pores were hard to resolve with the currently available SEM resolution at such a magnification. For this reason, TEM was performed to re-evaluate the information regarding pore structure. This information will be discussed below.
The observations of the pre-oxidation effect on the average nanofiber diameter and morphology are discussed next. The lack of linearity in lignin chains is one of the factors that does not allow for the spinnability of pure lignin solutions [33]. Thus, nylon 6 was used in a blend with lignin to facilitate fiber formation [34].
A strong influence of pre-oxidation on the average nanofiber diameter was observed for lignin/nylon 6 nanofibers. Specimens without pre-oxidation (denoted as NO) revealed an accentuated reduction in mean diameter with an increase in carbonization temperature compared to that of the pre-oxidized specimens (cf. Figure 4b and Figures S4–S6). The L2_NO_H3 specimen revealed a diameter reduction of ~82% compared to the L2_NO specimen (the corresponding to blown specimen), i.e., a reduction from 544 ± 220 nm to 99 ± 34 nm. For the L2_POL_H3 specimen, the mean diameter was reduced by ~44% in comparison with the corresponding as-blown precursor nanofibers (L2_POL), i.e., from 544 ± 220 nm to 307 ± 114 nm.
The influence of pre-oxidation (thermal stabilization) treatment on lignin-derived nanofibers is still incompletely understood. Even though it is treated as a key step [9], contradictory data were reported regarding the pre-oxidation effect on carbon nanofibers derived from lignin-containing nanofibers [35,36]. Such differences stem from the complexity of lignin molecules, as well as the preceding purification processes and host polymers used in a blend. Reference [35] reported that morphological changes in electrospun lignin/PEO nanofibers were not observed, except for a decrease in the mean diameter. On the other hand, reference [13] reported that lignin fibers require a long pre-oxidation time prior to carbonization, otherwise revealing the morphological collapse of the fibrous structure. However, in the present work, pre-oxidized or not, the lignin/nylon 6 specimens did not lose their fibrous structure during carbonization. Reference [37] reported the existence of strong hydrogen bonds in lignin and nylon 6 blends, which improved thermal stability, thus indicating that nylon 6 might be responsible for maintaining the fibrous shape. At the same time, nylon 6 might also be responsible for an increase in the nanofiber diameter in the PO specimens. This implies that the long treatment of nanofibers at a temperature higher than the nylon 6 glass transition temperature (Tg) causes macromolecules to acquire mobility and join the adjacent fibers [27]. In a similar way, PO is responsible for the differences observed in the nanofiber morphology, because different heating profiles produce different impacts on the microphase separation of lignin and nylon 6 in the blend [38]. As for the pore formation, it is expected that nylon 6 will have a certain influence because of its very low carbon yield. At the same time, because pure lignin cannot be spun into fibers, it is hard to determine whether porosity originates from nylon 6 or is due to the gasification of lignin heterocompounds. EDS attests that lignin contains components other than just C, O, and N. This is because lignin is a plant-derived material that underwent an extraction process [33]. The EDS for L2 specimens revealed how N, O, S, and Na atoms were eliminated from nanofibers as the temperature increased.
The lignin/nylon 6/ZnCl2 specimen containing the pore-forming agent ZnCl2 without the pre-oxidation step revealed a reduction in the mean diameter, which was approximately proportional to the carbonization temperature. For the PO specimens carbonized at 600 °C, an increase in the average diameter was also observed, as presented by the lignin/nylon 6 specimens (without ZnCl2), which can be explained by the influence of nylon 6 in fiber coalescence, as discussed above. The Z1_POL_H1 specimen revealed some fiber clusterization that might be attributed to the precipitation of ZnCl2 crystallites at a low temperature (600 °C), which may still occur even in an almost inert atmosphere [39]. This would explain the presence of oxygen revealed by the EDS analysis of that specimen. Although the NO specimens carbonized at 1200 °C revealed a slight increase in the average diameter after carbonization, this could be related to the coalescence of some nanofibers, as seen in Figure 4c and Figures S7–S9. However, based on the observed standard deviation, this slight increase in diameter is statistically insignificant. It is more probable that the diameter of Z1 fibers barely changes during carbonization at different temperatures. Both types of specimens (denoted as NO and PO) present macropores on fiber surfaces, which can rule out the effect of pre-oxidation on formation of such pores. Such macroporosity can be caused by the elimination of nylon 6 and/or by the action of ZnCl2 as a porogen.
The literature mentions ZnCl2 as a stabilizer and a pore-forming additive and even as an additive that facilitates the formation of thinner fibers [31,40,41]. The present work demonstrates that ZnCl2 added to the lignin-containing solution results in nanofibers with a different morphology: with the diameter is reduced from 544 nm to 335 nm. These nanofibers were very smooth, but with pores formed upon the removal of ZnCl2 at high temperatures. ZnCl2 forms nanocrystals within the as-spun fibers, and their size and distribution affect the pore structure [42]. Figures S7 and S8 show that some regions revealed a more uniform pore distribution, as well pore-size variation, than others. This indicates that the addition of ZnCl2 can influence the formation of pores of meso- and macro-sizes.
Knowing that fiber diameter affects the specific surface area, it is instructive to compare the cross-sectional sizes of fibers obtained via electrospinning and solution blowing. In this work it was possible to obtain similar fibers via the electrospinning of PAN and via the SB of lignin. Comparing the nanofiber diameter observed here with those reported in the literature, one can find that electrospun PAN-derived CFs are widely polydisperse, from tens of nanometers to the micron range [43]. On the other hand, for the lignin-derived fibers, some electrospun and carbonized lignin-derived CFs were reported to have diameters in the 100–800 nm range [26,44,45], while the SB and carbonized fibers were reported to be in the 228–370 nm range [34]. In the present work, PAN-derived CFs were as small as 74 nm when carbonized at 1200 °C. Also, using lignin and nylon 6 blend, the carbonized fiber diameters were 99 nm and 220 nm, respectively, for electrospinning and SB in the present work.
3.2. TEM Analysis
Because the fibers that experienced a pre-oxidation step had a higher degree of graphitization, as well as a higher carbon content, TEM analysis was performed to obtain a better resolution of pores and more detailed information about the graphitic structure. In fact, mesopores can be revealed via the TEM images. PAN and pure lignin fibers possess mesopores that are smaller than 20 nm. On the other hand, the lignin/ZnCl2 nanofibers had a higher macroporosity, with pores of about 50 nm, related to ZnCl2 nanocrystals, as described before.
Another interesting observation was that the porosity distributions of the P1 and Z1 specimens were quite uniform, while the L2 specimen revealed a higher porosity at the center. For supercapacitor performance, the distribution of pore sizes is important because it is correlated to the specific surface area (SSA). On one hand, micropores of <2 nm are desirable in order to obtain larger surface areas. At the same time, micro- and mesopores also play an important role, as such larger pores facilitate easier fluid access and, therefore, lower hydraulic resistance and the better accessibility of ions to the electrode surface. The SEM and TEM images reveal diversity in the pore distributions, yet these data are only qualitative.
As seen in the TEM images (Figure 5), it is possible to resolve graphitic carbon layers on the nanofiber surface. Even though all the specimens revealed a similar level of graphitization via Raman analysis (discussed previously), only the lignin-derived specimen (L2_POL_H3) possessed a long-range graphitic structure, which could be detected via the SAD mode. The pattern observable in Figure 5d was most likely caused by a small casting portion of the solution, which does not have a fiber-like shape. The three diffraction rings that can be observed in the inset in Figure 5e correspond to the sizes of 0.335, 0.197, and 0.117 nm, which are reminiscent of (002), (101), and (112) in Miller’s indices of graphite [46]. The diffraction spots indicate a polycrystalline graphite [14]. In Figure S10a,b, the diffraction pattern, corresponding to that of amorphous carbon, is revealed at the fiber surface. Using Raman spectroscopy data, the BSU (basic structure unit) was estimated to be of the order of ~2 nm. In comparison, in Figure 5c,f,i, the graphitic planes can be observed, and they are of the order of 2 nm. Small graphitic domains were also detected for amorphous carbon using SAD [47].
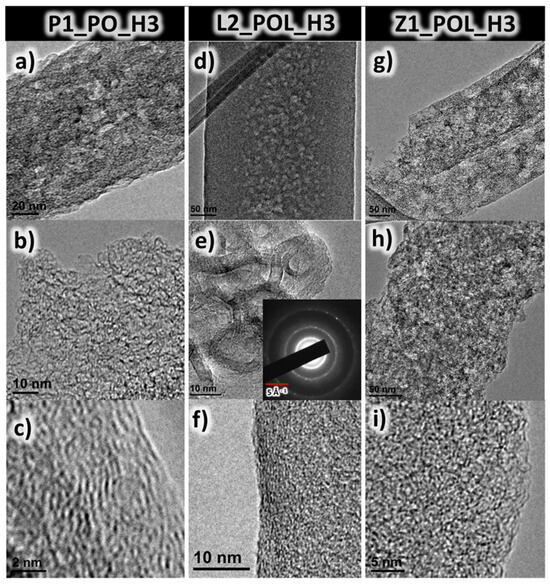
Figure 5.
TEM images for specimens P1_PO_H3 (a–c), L2_POL_H3 (d–f), and Z1_POL_H3 (g–i). In panel (e) the SAD diffraction pattern for the respective location is included in the inset.
3.3. Raman Spectroscopy
A mixture of graphitic and amorphous carbons is called turbostratic. In the turbostratic carbon materials, the small and ordered domains is called the “basic structure unit” (BSU). These units constitute 3–4 turbostratically stacked carbon polyaromatic layers, with an average area of ~9 nm2 and an interplanar distance of ~0.35 nm [48]. Turbostratic carbon plays an important role in energy storage systems, with a BSU size corresponding to a higher degree of graphitization, which contributes to an enhanced electrical conductivity. Raman spectroscopy is very effective for the characterization of carbon materials [49]. The Raman spectrum of a turbostratic material can be split into two or more peaks. The main ones are the D peak (~1350 cm−1), which corresponds to the vibration of BSU edges, and therefore translates into the defect band, whereas the G peak (~1575 cm−1) corresponds to the sp2 vibration of graphite BSU, i.e., is related to the degree of graphitization [47]. The intensity, position, and half-width of the peaks yield information about the molecular structure of carbon material and can be used to estimate the BSU size [50]. The results of the Raman spectra of the H3 specimens are presented in Figure 6a. The plots of the full Raman spectra, as well as the corresponding peaks, are depicted in Figures S11–S14, where Figure S11 demonstrates that all specimens revealed a broad peak at ~2800 cm−1, which corresponds to the 2D band and indicates the stacking of graphite planes [51].
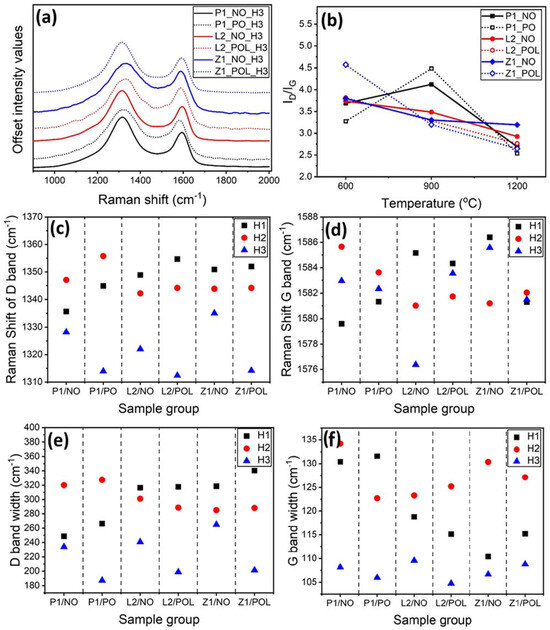
Figure 6.
Raman spectroscopy analysis. (a) Ratio of the D and G peak areas vary with the carbonization temperature. (b) Raman spectroscopy results for specimens carbonized in the heat-treatment regime H3. The spectra and the corresponding peaks for all the specimens are presented in the Supplementary Materials (cf. Figures S11–S14). (c) The Raman shift for the D-peak around 1340 cm−1. (d) The Raman shift for the G-peak around 1582 cm−1. The peak half-widths for all specimens carbonized at different temperatures: (e) the D-peak half-width and (f) the G-peak half-width.
3.3.1. Effect of Carbonization Temperature on the I(D)/I(G) Ratio
The I(D)/I(G) ratio is used to estimate the degree of graphitization of carbonized materials. The larger this value is, the larger the defects and/or amorphous domains are. On the other hand, lower values of the I(D)/I(G) ratio indicate a higher degree of carbon graphitization. In general, the I(D)/I(G) ratio decreases as the carbonization temperature increases. This is expected because of dehydrogenation and denitrogenation taking place during heating and forming a ladder structure and, consequently, the BSU of carbon fibers [18]. In the present work, such a trend was observed for the L2_NO, L2_POL, Z1_NO, and Z1_POL specimens (cf. Figure 6b). On the other hand, this trend was not observed for PAN nanofibers: P1 specimens actually revealed a higher value of the I(D)/I(G) ratio for H2 (900 °C) for both NO and PO specimens. This was unexpected, and several potential reasons for this result are discussed below.
The I(D)/I(G) ratio is calculated using the peak areas. Therefore, considering the D peak width of the P1 specimens, it is possible to observe that the larger width was for the P1 specimens at the temperature corresponding to the heating regime H2. This indicates that there was no error in the curve fitting. In fact, all the Raman measurements were performed in triplicates, at different locations of the nanofibers and demonstrated perfect repeatability. On the other hand, one can also observe that the EDS of the P1_NO_H2 specimens revealed that these nanofibers contained a larger amount of N than those heat-treated in the H1 regime and a larger amount of Na for P1_PO_H2, which could be responsible for a lower degree of graphitization. The XPS survey for P1_PO_H2 was performed as an example (cf. Figure S15 and Table S2, discussed in the Supplementary Materials). This allows one to recognize the intense peaks corresponding to N and Na. Nitrogen is characteristic of PAN, although Na is unexpected. The surge of Na can be explained by the fact that the PAN- and lignin-derived specimens were carbonized simultaneously in the same tube furnace, and perhaps some of the Na-containing vapor expelled from lignin was condensed on the PAN-derived fibers. This would explain the variation in the data for PAN-derived fibers carbonized at H2, and therefore, this is not discussed further. The comparison of the I(D)/I(G) ratios for the NO and PO specimens with those for the L2 and Z1 specimens reveals that the ratio increased for PO specimens in the heat-treatment regime H1 and decreased for PO specimens in the H2 and H3 heat-treatment regimes.
In general, higher heating temperatures and the PO step improved the degree of graphitization and, consequently, the electrical conductivity (Figure S18). The specimens revealed similar values of the I(D)/I(G) ratios in the heat-treatment regime H3. The values varied from 2.68 to 3.19 for the NO specimens and from 2.53 to 2.76 for the PO specimens when calculated by area, as well as from 1.18 to 1.28 and from 1.44 to 1.54, respectively, when calculated solely using the peak intensity (cf. Table S3). This result indicates that the carbonized lignin-derived, as well as lignin/ZnCl2-derived nanofibers are very similar to the PAN-derived CNFs in terms of the degree of graphitization. The ratios obtained are comparable to those reported in the literature [10,11,13,52].
Reference [53] reported how the Raman I(D)/I(G) ratio can be used to estimate the BSU size, specifically, , with K = 4.4 nm for a laser wavelength of 514.5 nm. On the other hand, for a laser with a wavelength of 633 nm, K = 5.8 nm [11]. Using the smaller and larger values of K and the present values of the I(D)/I(G) ratios, the BSU unit size length is estimated to be in the 1.82 to 2.29 nm range. Even though the above-mentioned estimate of is straightforward, it is known to be inaccurate for BSU < 2 nm [54], which makes the present result slightly ambiguous.
3.3.2. Temperature Effect on the Peak Shift
In Raman spectroscopy, the peak shift can be related to the level of organization, i.e., to the BSU size, as well as to the strain embedded during carbonization [49,50]. In the present work, the D-band position was higher for the lower temperatures (in the heat-treatment regime H1) and lower for the higher temperatures (in the heat-treatment regime H3) for both types of specimens derived from either lignin or lignin/ZnCl2. The D-peak shift toward the lower values is associated with the higher degree of graphitization, and for most of the specimens, the observed order in the peak shift agreed with the I(D)/I(G) ratio. Reference [50] reported how the higher values corresponding to the D-peak position translate into the BSU value as small as 1 nm. Figure 6c illustrates how the position of the D-peaks corresponding to the heat-treatment regime H3 reaches the values even lower than 1320 cm−1, which, according to reference [50], corresponds to the BSU of ~2 nm to 4 nm. The latter range is in good agreement with the range established in the preceding section using the I(D)/I(G) ratio. Reference [50] also reported how the shift in the G-peak is not fully correlated to the BSU size. Indeed, the shift in the G-peak observed in the present work was uncorrelated with temperature (cf. Figure 6d).
The PO specimens revealed a clear distinction in the D-peak positions. Specifically, the PO specimens revealed the peak shifts to the lower values for the heat-treatment regime H3. However, the shift in the D-peak to higher the values was observed for the H1 and H2 heat-treatment regimes, being larger for H1 than for H2. It seems that pre-oxidation (PO) is beneficial for graphitization in the heat-treatment regime H3, although this conclusion is unsubstantiated by the lower temperature data.
3.3.3. Temperature Effect on the Half-Peak Width
According to Raman spectroscopy, the narrower the peak width, the higher the degree of graphitization and the larger the BSU size. In general, the higher temperature resulted in a narrower D-band width. The D-peak width was also reduced because of the pre-oxidation (PO) step for all specimens in the heat-treatment regime H3. The pre-oxidation (PO) altered the D-peak width in a similar fashion to that of the G-peak (manifesting the defects) (cf. Figure 6e,f).
In sum, the higher temperatures (the heat-treatment regime H3) resulted in the highest degree of graphitization, and accordingly, in the largest BSU. The degree of graphitization increased with the temperature for the L2 and Z1 specimen groups. The fact that the same trend was not observed for the PAN specimens is associated with sodium contamination during carbonization. It should be emphasized that sodium is a common impurity present in kraft lignin specimens, such as sodium carbonate and sodium sulfate forms. So, most probably, this sodium originated from lignin. The pre-oxidation (PO) process seemingly has a positive impact on the degree of graphitization of the specimens heat-treated at 1200 °C.
3.4. Nitrogen Adsorption
The surface area and pore sizes were evaluated using the N2 adsorption isotherm measurements. The BET and NLDFT methods reveal the isotherms differently, thus their results can differ. Both methods measure SSA, but NLDFT takes into consideration the pore shape and could result in more realistic data [29]. Therefore, here the values measured by both methods are reported for the sake of completeness.
The N2 adsorption measurements can be sensitive to material morphology. It is known that for materials with a soft structure, gas adsorption might reveal unreliable results. The vacuumization at the degasification stage, as well as the gas pressure during the adsorption experiments can cause the collapse of a microporous structure, interfering with the volume of gas adsorbed [55]. Biochars, such as those derived from lignin, are an example of relatively soft materials. In fact, at low carbonization temperatures, e.g., at 600 °C, the results for the SSA values for the lignin-derived carbon nanofibers were quite low (cf. Figure 7, which is discussed in detail in Section 3.1). These low values can be explained by the fact that the lignin-derived fibers might not yet be fully carbonized at 600 °C and/or they might not have acquired enough structural strength to sustain the conditions of the gas adsorption analysis. Consequently, the adsorption isotherm obtained for the nanofibers carbonized at 600 °C would be hardly reliable. Therefore, the corresponding BET and NLDFT data should be compared cautiously.
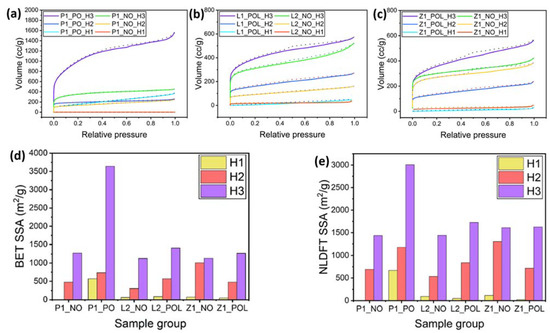
Figure 7.
Adsorption isotherm measured using N2 adsorption. (a) PAN-derived CNFs. (b) Lignin-derived CNFs. (c) Lignin/ZnCl2-derived CNFs. The dotted lines depict the desorption curves. (d) BET results for SSA. (e) NLDFT results for SSA.
3.4.1. Specific Surface Area (SSA)
The BET analysis for the P1_NO_H1 specimens resulted in a relatively low value of the specific surface area (SSA), only 0.017 m2/g, while the one for P1_PO_H1 revealed the SSA of 568 m2/g. The pre-oxidation (PO) step in the carbonization of PAN nanofibers is known to have a strong impact on the stability of the nanofiber structure, enhancing its mechanical stiffness [21]. This fact supports the possibility of micropore collapse in the P1_NO nanofibers carbonized at low temperature. For the L2 specimens, the SSA value measured by BET also increased for pre-oxidized (PO) specimens. The corresponding SEM images of the L2_NO_H1 and L2_POL_H1 specimens (Figures S4 and S5), reveal that the pre-oxidized specimens possess a rougher surface than the non-pre-oxidized ones, hence the higher SSA values. This stems from the fact that the process of the intermolecular cyclization of the nitrile groups leads to a greater shrinkage of the fibers, which, in turn, causes the shrinkage of the surface and an increase in its roughness [19]. The comparison of the specimens Z1_NO_H1 and Z1_POL_H1 demonstrated that the pre-treated specimens (PO) revealed a lower surface area than those without pre-oxidation. The average thickness of the carbonized fibers was 373 nm for Z1_POL_H1 specimens and 268 nm for Z1_NO_H1 specimens. This shows that for the lignin-derived fibers, carbonized at low temperatures, the SSA is most likely to be dependent on the fiber diameter and surface roughness. Accordingly, one can conclude that the collapse of micropores could be one of the reasons for the relatively low SSA values corresponding to the carbonization regime H1. Another reason might be that there were no or only a few micropores formed under the carbonization H1 regime. The corresponding adsorption isotherms and the values of the SSA are presented in Figure 7.
For the specimens carbonized in the regimes H2 and H3, porosity increases due to release of volatiles resulting from non-carbonizable components, such as H2O, NH3, CO, HCN, CO2, and N2. This well-developed porosity can be better detected using a gas adsorption technique such as NLDFT, which is used to estimate pore sizes and volume. Irrespective of what gas measurement technique was used (BET or NLDFT), the values of SSA increased with carbonization temperature. The SSA values also increased due to the pre-oxidation (PO) step. Note also that according to the EDS results (discussed before), SSA is related to the carbon content. For the specimens carbonized at the highest temperature (regime H3), a complete volatile release would be expected. Among these, the pre-oxidized (PO) specimens revealed a higher carbon content than the non-pre-oxidized (NO) ones, thus indicating that PO has a strong impact on removal of heteroatoms, which is also directly correlated with a higher SSA value. On the other hand, the specimens Z1_NO_H3 and Z1_POL_H3 revealed the smallest differences in the carbon content of ~4% and their SSA values were similar, irrespective of the NO or PO pre-treatment steps. The specimens derived from lignin carbonized using regime H3 revealed a larger difference in the carbon content, which can also be associated with the pre-treatment regimes, NO or PO. A significant increase in the SSA values of P1_NO_H3 to P1_PO_H3 specimens reveals that the pre-oxidation step plays a crucial role in pore formation during the carbonization of PAN nanofibers (cf. Figure 7a,b and Table S4).
At the 900 °C carbonization temperature (the heat-treatment regime H2), specimens P1 and L2 revealed an increase in the SSA values because of the pre-oxidation (PO) step, which was subsequently accompanied by an increase in the carbon content. On the contrary, the Z1 specimens revealed a decrease in the SSA values resulting from pre-oxidation (PO), as well as an ultimate decrease in the carbon content. These results imply a close correlation between the SSA value and the carbon content, which applies to all specimens, except for P1_PO_H3. Note that the latter revealed major weight loss during carbonization of ~96% in comparison to ~81% for lignin (Figure 8a,b and Table S4).
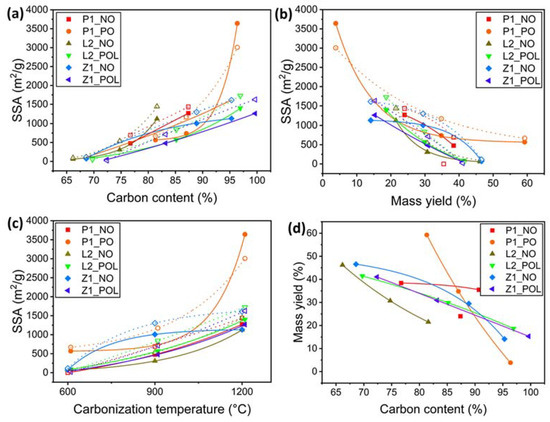
Figure 8.
Dependences of SSA and mass yield on process parameters. (a) SSA vs. carbon content. (b) SSA vs. mass yield. (c) SSA vs. carbonization temperature. (d) Mass yield vs. carbon content. The duplicated curves of the same color correspond to the same specimen groups. Full symbols and solid lines represent the values obtained by BET; empty symbols and dotted lines represent the values obtained by NLDFT. The curves represent the fittings by descending or ascending exponential functions (fitting with R2 = 0.99).
The overall results indicate that there is a correlation between the SSA value, weight loss, temperature, and the carbon content (C%). The acquired data for specimens from different groups are presented and compared in Figure 8a–c, which displays the SSA values versus the corresponding carbon content, mass yield, and carbonization temperature, as well as in Figure 8d, which presents the mass yield versus carbon content. Accordingly, a higher SSA value is associated with a high carbon content and increases with PO and the carbonization temperature. SSA is also correlated with a low mass yield (cf. Figure 8b) due to the elimination of non-carbonizable components and a higher carbon content (cf. Figure 8d).
3.4.2. Pore-Size Distribution
Figure S19a–c illustrate the relationship between the SSA values per unit length (measured in m2/nm/g) and the pore size for the H1, H2, and H3 carbonization regimes in different specimen groups. Figure S19d illustrates the results for all specimens carbonized in the H2 and H3 regimes in more detail. These results reveal that the PO step increased the specific SSA. NLDFT detected pores as large as 50 nm, although they were infrequent, as the distributions in Figure S19 attest. Comparing the data for different groups of specimens, one can observe that P1_PO_H3 specimens reveal distinct pore-size distributions compared to the other specimens. Specifically, the pore-size distributions for P1_PO_H3 specimens, which possess high values of the SSA per unit length, reveal pores of the sizes ~1.6 nm, 2.9 nm, 6.2 nm, and 8.8 nm, associated with wide peaks in the pore-size distributions. On the other hand, the lignin-derived specimens, which possess similar values of the SSA per unit length, reveal porosity associated with pores of sizes ~1 nm, 1.5 nm, and 2 nm, and much narrower peaks in the pore-size distributions, hence resulting in a lower total SSA (cf. Figure S19).
Note also that all the PO specimens, carbonized at 1200 °C, revealed an increase in the frequency of mesopores in the 2–5 nm range: specifically by a factor of 2.09 for PAN, 1.20 for L2, and 1.01 for Z1 (cf. Figure S20).
3.4.3. ZnCl2 Effect on Porosity
To evaluate how the addition of ZnCl2 contributed to pore formation, the pore-size distributions in the L2 and Z1 specimen groups (the latter with ZnCl2) carbonized at 1200 °C were explored. By comparing the non-pre-oxidized (NO) with the pre-oxidized (PO) specimens, it was found that Z1_NO_H3 possessed 25% more micropores than L2_NO_H3. For the PO specimens, it was found that the micropore content was very similar (~58%), the same for the pores in the 2 to 5 nm range, whereas for pores larger than 5 nm, an increase in 6.8–8.3% was observed.
The data for the SSA in Figure 8 reveal that Z1_NO was the only specimen group that displayed distinct behavior (for the data corresponding to carbonization at 900 °C). The Z1_H2 specimen group was the only group in which the SSA value was higher for Z1_NO_H2 than for the pre-oxidized version Z1_POL_H2. This indicates that the pre-oxidation process interferes with the ability of ZnCl2 to form pores at the carbonization temperature of 900 °C. As discussed above, the PO step can cause variations in the size and distribution of Zn within the fibers, which could be responsible for a higher porosity found at a lower carbonization temperature for Z1_NO_H2.
The EDS data for Z1_NO_H2 and Z1_POL_H2 specimens reveal that Z1_POL_H2 had a higher oxygen content. This is probably because the PO step caused a delay in the oxygen elimination, and therefore, a lower SSA value was observed. It should be emphasized that during the high-temperature carbonization regime H3, the NO and PO specimens reached a very similar carbon content, 95.3% and 99.7%, for Z1_NO and Z1_PO, respectively, as well as similar SSA values. This indicates that ZnCl2 increased the SSA value when nanofibers were carbonized in the intermediate-temperature heat-treatment regime H2, whereas ZnCl2 did not affect SSA values when specimens were carbonized in the high-temperature heat-treatment regime H3 (cf. Figure 8d). Nevertheless, on the pore level, one can observe that NO specimen is composed predominantly of micropores, while Z1_PO specimen revealed micro- and mesoporous (cf. Figures S19 and S20). This implies that the pre-oxidation (PO) step affects the distribution of Zn and, thus, the pore-size distribution. Indeed, the Z1_NO_H3 specimen possessed 89% micropores, while Z1_PO_H3 specimen possessed only 58% (cf. Figure S20e,f).
3.5. Electrochemical Assessment
In supercapacitors, such parameters as the SSA and pore-size distribution, the electrical conductivity, the degree of graphitization, and the elemental content, as well as the electrode thickness and the type of electrolyte can affect the capacitive performance [27,33]. Accounting for characterization (morphological, structural, and chemical), carbon nanofibers derived from pre-oxidized PAN, lignin, and lignin/ZnCl2 specimens revealed similar characteristics in terms of the degree of graphitization, the elemental content, and the electrical conductivity. However, the PCNFs possessed two to three times higher SSA values than those of the LCNFs, which is expected to have an impact on the corresponding capacitance. On the other hand, the PCNFs revealed a carbon yield of only ~4%. Therefore, although the LCNFs did not reveal such high values of SSA, they have the advantage of a much higher carbon mass yield, i.e., ~15–18.5% (Table S5).
In this section, the specimens carbonized at the high-temperature regime H3 were used to build symmetrical supercapacitors. Cyclic voltammetry was used to evaluate the capacitance and how it is affected by the potential range, the scan rate, and the number of cycles. Then, the results are compared with the differences observed in the pore-size distribution, packing density, and due to pre-oxidation. In the literature, it is common to find statements that a few cycles are required to ensure the complete wetting of specimens before initiating the electrochemical measurements. Here, 200 cycles were performed, but no significant changes in the cyclic-voltammetry curve were observed.
A water-in-salt electrolyte (WiSE) using sodium perchlorate was chosen in the present experiments. Such electrolytes possess a high salt concentration; hence, they behave completely differently from other dilute salt-in-water electrolytes. The use of WiSE enables the employment of high cell potential ranges of up 2.3 V [28]. Yet, recent studies recommend potential values up to 1.8 V to avoid electrolyte deterioration [30]. Figure 9 demonstrates how the cyclic-voltammetry curves begin to deviate from a squared ideal shape at higher potential values. This is the major difference which appears between the yellow and green curves, from 1.4 to 1.6 V. For this reason, to ensure the maintenance of the electrolyte properties and low ohmic resistance, the potential of 1.5 V was chosen in the subsequent tests. Figure 9 demonstrates that the lignin-derived specimens (Figure 9b,c,e,f) reveal a bump in the curves with potential 1.8 V or higher. This is indicative of the fact that a reduction reaction is taking place at higher potentials, which occurs due to the presence of heteroatoms, such as O, N, or S previously observed in small quantities [31].
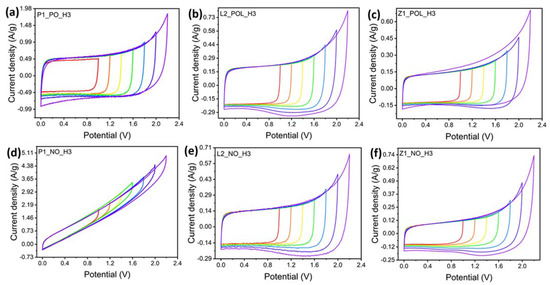
Figure 9.
Cyclic voltammetry tests at potentials varying in the 0–1 V range and up to the 0–2.2 V range at a scan rate of 20 mV/s. (a) P1_PO_H3, (b) L2_POL_H3, (c) Z1_POL_H3, (d) P1_NO_H3, (e) L2_NO_H3, (f) Z1_NO_H3. Legend: red 0–1 V; orange 0–1.2 V; yellow 0–1.4 V; green 0–1.6 V; blue 0–1.8 V; dark blue 0–2 V; violet 0–2.2 V.
Figure 9 also reveals that all the specimens, except P1_NO_H3, result in an almost ideal rectangular shape of the cyclic-voltammetry curve characteristic of carbon-based EDLC supercapacitors. The P1_NO_H3 specimen was strongly affected by the lack of the pre-oxidation (PO) step. The corresponding curve is greatly deformed, indicating a high electrical resistance, thus re-enforcing the need for the pre-oxidation (PO) step for PAN-derived carbon nanofibers to increase their electrical conductivity.
For the lignin-derived carbon nanofibers, the cyclic-voltammetry curves do not reveal any major distortions (for either PO or NO specimens), although a decrease in the current density was observed for the latter. As previously discussed, in Raman spectroscopy, it was detected that both L2_NO_H3 and Z1_NO_H3 specimen groups possessed lower degrees of graphitization than their pre-oxidized (PO) counterparts. Accordingly, the low degree of graphitization could be responsible for the observed lower current density.
The cyclic-voltammetry scanning (Figure 10) of supercapacitors at different scan rates affects deviations from the squared shape as the ion diffusion rate increases. Accordingly, a higher electrical resistance can be observed. Figure 10 and Figure 11a reveal that the non-pre-oxidized (NO) specimens were less affected by fast charging, and the lignin-derived specimens were, roughly speaking, also less affected by fast charging in comparison to the PAN-derived ones. Figure 11a reveals that the P1_PO_H3 specimen reached its highest capacitance (26 F/g) at 30 mV/s, descending to 13.8 F/g at 500 mV/s, being the most affected specimen, with its capacitance reducing by 46% lower than its maximum. Similarly, Z1_NO_H3 was the specimen that was least affected by fast charging, with its highest capacitance (11.1 F/g) achieved at 200 mV/s, being reduced by only 2% when charged at 500 mV/s (10.9 F/g).
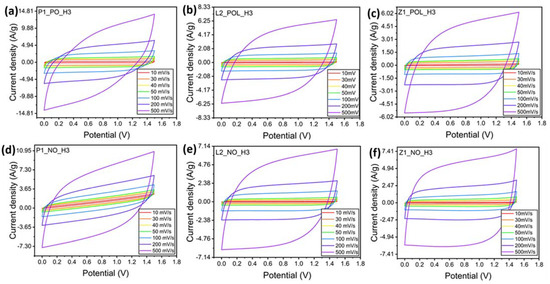
Figure 10.
Cyclic voltammetry tests in the potential range of 0–1.5 V, with the scan rates varying in the 10–500 mV/s range. The scan rates are listed as a legend in each panel. Specimens: (a) P1_PO_H3, (b) L2_POL_H3, (c) Z1_POL_H3, (d) P1_NO_H3, (e) L2_NO_H3, (f) Z1_NO_H3.
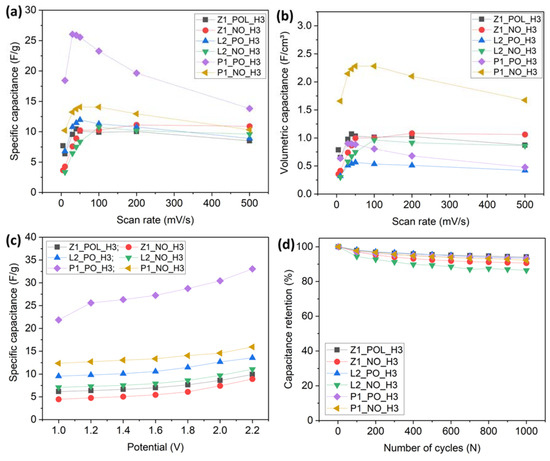
Figure 11.
Capacitance measured under different scan rates: (a) specific capacitance; (b) volumetric capacitance. Capacitance measured under different conditions: (c) varying the potential; (d) varying the number of cycles.
For fast charging, Table 3 reveals that the non-pre-oxidized (NO) specimens performed better than their pre-oxidized (PO) counterparts. The review in reference [27] discussed how specimens with a higher packing density improved the volumetric capacitance, including that in fast-charging/discharging cycles. Accordingly, a high electrode packing density would create an interconnected porous architecture, ensuring the rapid and efficient transport of electrolyte ions, which lowers the interfacial resistance, improving ion diffusion and, therefore, possessing higher capacitance at fast-scan rates. This phenomenon was confirmed by the present results. Indeed, the NO specimens possessed a higher packing density (Table S6), and, accordingly, Figure 11a and Table 3 demonstrate the higher capacitance of these specimens at high scanning rates. Figure 11b and Table S6 illustrate the volumetric capacitance and packing density; the specimens with a higher packing density revealed a higher volumetric capacitance. It should be emphasized that the volumetric capacitance of a supercapacitor is a critical parameter as it provides a measure of the device’s ability to store charge per unit volume. This is particularly important for applications where space is a constraint and high energy density is required. Reporting the volumetric capacitance allows for a more accurate comparison of performance across different supercapacitors. It also aids in understanding the fundamental limits on volumetric capacitance and the factors that influence it, such as the nature of the ions.
Table 3.
Specific capacitance values. (a) Maximum specific capacitance values recorded within the respective specimen group; the corresponding scan rate is shown in column (b). (c) Capacitance values at the high scan rate of 500 mV/s. (d) Percentage reduction between the maximum capacitance values and the corresponding values recorded at 500 mV/s.
The non-pre-oxidized (NO) specimens also revealed a higher percentage of ultra-micropores and micropores than their PO counterparts. At the same time, the performance of the NO carbon nanofibers could be strongly improved by a high SSA generated by micropores. It is unclear whether these pores are a part of an effective SSA, and whether they are accessible to electrolytes. The efficiency of the EDLC charge storage is also related to the dimensions of solvated ions and pore size. It is known that the EDLC charge storage is efficient when the ion and pore sizes are similar. This is the case in the present work, because the solvated ions and are 0.098 and 1.48 nm, respectively [25]. The Z1_PO_H3 specimen possesses a similar packing density as that of the Z1_NO_H3 specimen (cf. Figure 11c), but its performance at a high rate of charging was not as high as that of the Z1_NO_H3 specimen. Therefore, in the case of micropores accessible to the electrolyte ions, a higher packing density and the pore-size distribution might be a synergistical factor facilitating the improvement of the fast-charging capacitance.
In addition, Figure 11c illustrates how the capacitance increased with the scan potential. In spite of an improved performance of NO specimens at fast charging rate in Figure 11a,b,d, it is seen that the capacitance retention was lower for NO specimens than for the counterpart PO specimen groups. The PAN- and lignin-derived PO specimens revealed very similar retention abilities, with Z1_POL_H3 being the specimen that revealed the best retention at 1000 cycles, i.e., 94.2%, followed by L2_POL_H3 at 93.7% and P1_PO_H3 at 93.4%. However, lower values were obtained for NO specimens, P1_NO_H3, L2_NO_H3, and Z1_NO_H3, which retained 92.2%, 86.5%, and 90.65%, respectively. It is known that in spite of their better retention, specimens with high percentage of micropores can cause faster electrolyte damage due to an elevated exposition. This could be one of the reasons for the lower capacitance retention rate observed for such specimens. Another possible explanation is that the repetitive cyclization could cause actual physical damage to the porous structure [26].
3.6. Evaluation after the Cycling Test
Carbon nanofibers typically possess a relatively high strength. In particular, the mechanical properties of lignin-based nanofibers have been reported [56]. In the present case, the coin cells were closed under high pressure. For this reason, it is of interest to explore how the freestanding electrodes could withstand that high pressure mechanically. Note that the repetitive cyclization, which could cause actual physical damage to the porous structure (on the scale < 2 nm), cannot be responsible for the complete breakage of fibers on the scale of 100 µm. Figure 12 displays SEM images of the withdrawn carbon nanofibers after their use in supercapacitors. The fibrillar structure was maintained for all specimens. It is known that the lignin-derived carbon nanofibers do not reach as high mechanical strength as the PAN-derived ones [34]. Figure 12 demonstrates that the lignin-derived carbon nanofibers reveal some broken fiber ends. This indicates that even though the macrostructure and fibrillar dimensions were maintained, some breakage happened. Solution blowing techniques forms fluffier fibers, with lower density and larger inter-fiber pores than those formed by electrospinning, which implies that the present original solution-blown nanofiber mats are weaker mechanically than their counterparts formed by electrospinning [35]. Accordingly, the images in Figure 12 also reveal that due to the larger electrode thickness and a larger free volume (a more open porosity), the lignin-derived carbon nanofibers are more susceptible to compaction and, thus, crushing.

Figure 12.
SEM images of the freestanding carbon electrodes removed from the supercapacitor coins after 1000 cycles of cyclic voltammetry at the rate of 50 mV/s.
4. Discussion
The comparison of the present results on carbon nanofiber performance as electrodes in supercapacitors with those in the literature is desirable but should be carried out cautiously. Any material performance is very susceptible to a series of minor details in its preparation. Performance can be affected by the batch of the precursor used, as well as by different processing techniques and the equipment used for the analysis. Keeping that in mind, to achieve a fair comparison, PAN-derived and lignin-derived carbon nanofibers were produced here using similar procedures to those used for porous electrodes in supercapacitor prototypes. Comparisons were first performed between the present data and the available literature (discussed in Section 4.1). Then, the comparisons were carried out within the present data for the PAN- and lignin-derived carbon nanofibers obtained in the present and the previous work [57] of the present group (Section 4.2). All the data are discussed in the following sections.
4.1. Comparison with the Existing Literature
The SSA values and specific capacitance are very often used for comparison between different works. In the present data, for the PAN-derived carbon nanofibers, a SSA value as high as 3641 m2/g and a capacitance of 26 F/g were reached. The present SSA value was higher than those of 2800 m2/g, achieved in reference [22], where the capacitance was higher, i.e., 107 F/g. However, note that in reference [22], additional methods were employed to increase the electrical conductivity and porosity, whereas in the present case, the approach was straightforward. On the other hand, despite an ultra-high-SSA in the present work, it yielded lower specific capacitance values than the ones reported in reference [24]. The carbon fibers in that study revealed SSA values in the 742–1170 m2/g range, lower than that obtained here, but with a higher capacitance, in the 141 F/g–333 F/g range, achieved due to the addition of polypyrrole (PPy) and CNTs, which increased the electrical conductivity. In summary, the present straightforward approach was successful in forming PAN-derived carbon nanofibers with ultra-high-surface area, and the capacitance values were comparable to those reported in the literature. To achieve higher capacitance values, methods to increase the electrical conductivity of the electrodes should be employed.
The second highest value for the SSA measured in the present work, 1726 m2/g, was for lignin-derived carbon nanofiber specimen L2_POL_H3. This value was about one-half of the SSA value measured here for the PAN-derived carbon nanofibers. Table 4 reveals that the maximum values for the lignin-derived specimens that were not activated (i.e., their porosity was not increased after carbonization in certain post-processing methods) was 583 m2/g, while for the activated ones it was as high as 2170 m2/g, albeit with total production times of 147 and 19 h, respectively. The comparison of those results with the present ones reveals that the present straightforward approach to form lignin-derived carbon nanofibers, with no prior activation steps, could yield SSA values as high as those for activated specimens reported in the literature. Also, the specimen preparation time is much shorter in the present case and the required procedures are simpler.
Table 4.
Properties of LCNFs reported in the literature and measured in the present work. * CB = carbon black.
The capacitance value measured at 50 ms/V for the present L2_POL_H3 specimen is 11.95 F/g. The values in the literature, listed in Table 4, vary within the 64–429 F/g range. The lowest limit reported in reference [26] was 64 F/g, even though a different electrolyte and electrochemical assembly method than those employed here, and also, both specimens in our study were carbonized at 1200 °C and did not undergo activation treatment. It should be emphasized that the capacitance values measured here for L2_POL_H3 specimen and in reference [26] are for different materials: 80 wt.% lignin with 20 wt.% nylon 6 and 70 wt.% lignin with 30 wt.% PVA, respectively. The capacitance values were different as well the SSA values (1726 m2/g and 583 m2/g, respectively). Note that even though in reference [26] the values of the electrical conductivity are unavailable, it was reported that the fibers underwent a long stabilization (pre-oxidation) process for 26 h, whereas in the present work the pre-oxidation time for L2_POL_H3 was only ~4 h. That explains why even though the present samples revealed SSA values about three times higher, they could not reach as high a specific capacitance as those in reference [26]. Summarizing, one can conclude that besides the high SSA values of the present lignin-derived carbon nanofibers, the resulting capacitance is still lower because of the short pre-oxidation step, which probably significantly affected the electrical conductivity.
The electrical conductivity reported in reference [44] was two orders of magnitude higher than that in the present work: respectively, 27 and ~0.2 S/cm. On the other hand, the SSA value in reference [26] was by two orders of magnitude lower (12.8 m2/g) than the one in the present work (1726 m2/g). Despite the low SSA, the material in reference [44] revealed a specific capacitance of 102 F/g, while the present L2_POL_H3 specimen showed a lower value.
The available data imply that the electrical conductivity plays an important role for the specific capacitance. Note that in reference [44], fibers were stabilized in air for 126 h. The general understanding is that long stabilization periods might be necessary to improve the electrical conductivity of lignin-derived fibers. Additional measures could be useful, such as the incorporation of carbon black. Low electrical conductivity could also affect PAN-derived carbon nanofibers. The PAN-derived carbon nanofibers, for which high values of capacitance were reported, relied on the incorporation of carbon black or the use of nickel foam to improve the conductivity of the system. In the present work, using a straightforward method, PCNFs and LCNFs revealed ultra-high SSA values, but their potential was not fully exploited due to relatively low electrical conductivities.
As the P1_PO_H3 sample presented the best physicochemical properties and promising initial capacitance results, a more detailed electrochemical study was undertaken using other techniques applied to the symmetric coin cell. Figure 13a reveals the results for the galvanostatic charge–discharge (GCD) curves obtained through different current densities in the 0.1–5 A/g range. The symmetrical triangular profile can be observed, which indicates the super-capacitive behavior for carbon-based materials and suggests a double-layer energy storage process and outstanding reversibility during charging and discharging [59].
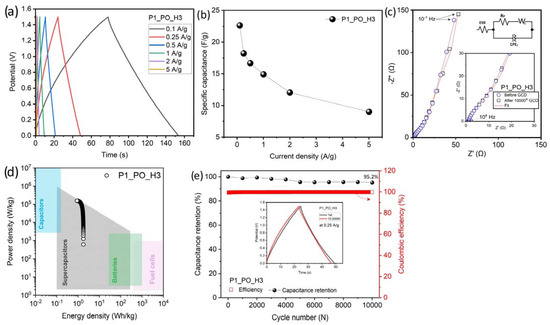
Figure 13.
Electrochemical properties for the specimen P1_PO_H3: (a) GCD curves at different current density; (b) specific capacitance calculated via GCD curves; (c) impedance spectra collected in the 104–0.1 Hz frequency range (the equivalent circuit used for fitting the spectra is inserted); (d) Ragone plot; (e) capacitance retention and coulombic efficiency after 10,000 GCD curves at a current density of 0.25 A/g.
The supercapacitor cell capacitance values were calculated from the GCD curves using the following equation [17]:
where I (A) is the discharge current, m (g) is the total mass of the active materials in both electrodes, and dV/dt (V/s) is the slope of the discharge curve after IR drop. The calculated values obtained from each GCD curve were 22.6, 18.2, 16.6, 14.9, 12, and 9 F/g at 0.1, 0.25, 0.5, 1, 2, and 5 A/g, respectively, as shown in Figure 13b. A reduction in capacitance with an increased current density is related to the porous structure of carbon nanofibers. In other words, at higher current densities, the ions cannot move as quickly to the micropores, thus these regions end up contributing more to the capacitance at lower current densities [3,60].
The spectroscopy impedance electrochemical (EIS) was collected for the P1_PO_H3 supercapacitor cell in the 0.1–104 Hz frequency range and the voltage amplitude of 10 mV. To fit the spectra, an equivalent circuit model of the Randles type was used, consisting of the solution impedance (ESR), polarization resistance (RP), Warburg (WS) impedance, and a constant phase element (CPE) to simulate an ideal capacitor. Figure 13c presents the results obtained before and after the GCD cyclability test, revealing very similar spectra, which suggests that the electrical characteristics of the supercapacitor remained completely preserved after cycling. From the fit of the Nyquist plot (Figure 13c), the ESR and RP values were 1.1 and 32 Ω, respectively, for both spectra (before and after GCD cyclability).
The Ragone plot is a crucial tool for assessing the performance of energy storage and conversion in a system for practical uses, as it illustrates the energy and power a cell delivers upon demand. Therefore, the maximum specific energy (Emax, measured in Wh/kg), and the maximum specific power (Pmax, measured in W/kg) were calculated using the equations provided in reference [61]:
Here, is the specific capacitance obtained via GCD, is the voltage window, and ESR is the equivalent series resistance previously obtained via EIS. Then, the Ragone plot for the EDLC supercapacitors was calculated using a numerical solution proposed in reference [62] to find values for “PL” and “EL” between 0 and Pmax using the following equation:
Figure 13d presents the Ragone plot for the P1_PO_H3 specimen. It reveals that the cell delivered a maximum specific energy of 1.77 Wh/kg, placing it within the supercapacitors range. Furthermore, it achieved an impressive maximum specific power of 156 kW/kg, surpassing the values for conventional electrochemical supercapacitors based on electrospun carbon nanofibers-based cells [63]. Finally, the long-term cycle stability of the P1_PO_H3 cell supercapacitor is commendable (Figure 13e). The data indicate that the cell maintains 95.2% of its initial capacitance even after 10,000 GCD cycles at a current density of 0.25 A/g with coulombic efficiency ~100%, demonstrating its robust stability.
4.2. Comparison of PAN- and Lignin-Derived Carbon Nanofibers Formed in the Present Work
In this subsection the results for LCNFs and PCNFs (the lignin-derived and PAN-derived carbon nanofibers, respectively) formed and tested under similar conditions are compared and discussed. The carbon electrodes revealed the electric conductivities of the same order of magnitude and the very close retention of capacitance, 93% for PCNFs and 94% for LCNFs. Significant differences between PCNFs and LCNFs are found in their SSA and capacitance values, 3641 m2/g and 25.55 F/g for P1_PO_H3 (PCNF), in comparison to 1726 m2/g and 11.95 F/g for L2_POL_H3 (LCNF). These values were measured at a moderate scan rate of 50 mV/s. Both the SSA and capacitance values of the LCNFs were about one-half of those of PCNFs. Bearing in mind a desirable high mass yield, the latter comparison presents a dilemma. Indeed, Figure 10b reveals that the higher the SSA value, the lower the mass yield. According to our data [57], one can observe that even though L2_POL_H3 had a ~50% lower SSA, it revealed a mass yield of 18.6%, while P1_POL_H3 had only 3.8%. Accordingly, for the same amount of precursor, for example 1 g, the PAN-derived P1_PO_H3 would reveal a surface area of only 138.4 m2, whereas the lignin-derived L2_POL_H3 would reveal 321.0 m2 for L2_POL_H3. Therefore, from this point of view, given the lower environmental impact, as well as an inexpensive precursor, the lignin-derived LCNFs could serve as a very good substitute for the PAN-derived PCNFs.
An additional advantage of LCNFs appears during fast charging rates. When a supercapacitor undergoes cyclic voltammetry at the scan rate of 500 mV/s, the performance of LCNFs does not fall far behind that of PCNFs. Indeed, it is only 21% for LCNFs Z1_NO_H3 and 38% for LCNFs Z1_POL_H3, in comparison to 54% for the PAN-derived carbon specimens (taking for 100% the best performance of the latter). It should be emphasized that at the fast scan rate, Z1_NO_H3 possessed a charging rate of only 21% lower than that of P1_PO_H3, even though its electrode was 3.5 times thicker than that of P1_PO_H3 and its mass was 10 times larger (cf. Table S7). In reference [32], it is noted that one of the hurdles in applying supercapacitors commercially is that the capacitance is greatly reduced by the mass load, which should be >10 mg/cm2, although it is often <3 mg/cm2 in research-scale experiments. In the present case, none of the electrodes reached such high mass loading, and yet, one could observe that Z1_NO_H3 had a much better chances of succeeding in high-material load supercapacitors than P1_PO_H3 (with the unit of coin cell capacitance of 31.8 mF in comparison to 8.3 mF). It is evident how the capacitance, not just by weight, but by area and volume, should be of dramatic importance for commercial applications. Yet, many papers do not report detailed information on either the mass and thickness of the electrodes or their real volumetric capacitance. In the present work, this important information is contained in the Supplementary Materials, where one can find the thickness and weight of the tested electrodes in Table S7, the volumetric and area capacitances in Figure 11b and Figure S21, the cyclic voltammetry results and the capacitance by unit of coin cell in Figure S22 and Table S8, and the cyclability test after 1000 cycles (Figure S23). A chart comparing advantages and disadvantages on the use of PAN and lignin as carbon precursors is provided in Table 5. The latter figure elucidates the fact that lignin-derived CNFs have several advantages over PAN-derived CNFs.
Table 5.
Comparison of the results obtained for PAN-derived and lignin-derived carbon nanofibers. The arrow directions ascertain higher or lower values.
5. Conclusions
A new method of production of carbon nanofibers (CNFs) from lignin, a renewable biopolymer, a bio-waste of the paper industry, is proposed: solution blowing, a technique much faster than electrospinning. A high yield and fast method of the preparation of precursor nanofibers, which does not require a long stabilization step, while carbonization time is short (the production time varied from 7 to 11 h). The resulting CNFs can be used as self-standing electrodes and have revealed graphitization levels comparable with those reported in the literature, with I(D)/I(G) ratios as small as 1.18. The high carbon yield of carbon nanofibers with cross-sectional diameters in the 74 nm to 307 nm range and high porosity was achieved without any need for activation agents. The effects of pre-oxidation step in air, as well as that of carbonization temperature, were evaluated in detail. In addition, the lignin-derived CNFs were compared to ordinary electrospun PAN-derived CNFs. The addition of a pore-forming additive was also investigated. The pre-oxidation (PO) step was found to be unnecessary in order to retain nanofiber shape during carbonization. In fact, PO causes fiber coalescence due to the phase separation of lignin and nylon 6 when maintained at low temperatures for 3 h, resulting in an increase in fiber diameter. Even though PO caused an increase in lignin fiber diameter, it positively affected the degree of graphitization and resulted in increased carbon content and electrical conductivity and improved mass yield.
Surface area analysis revealed that the carbonization at high temperatures of the nanofibers derived from PAN, lignin, or lignin/ZnCl2 yields specimens with a high surface area in the range of 1435–3640 m2/g, with mesopores of 2–50 nm, micropores < 2 nm, and even ultra-micropores of <1 nm. The large specific surface area (SSA) values were associated with high-temperature carbonization, high carbon content, high weight loss, and pre-oxidation in air. The PAN_PO_H3 specimen revealed the high contribution of pores < 2 nm and pores in the 2–5 nm range to the SSA value. On the other hand, the lignin-derived carbon nanofibers acquired a major part of their SSA from micropores and ultra-micropores. It was shown that the pre-oxidation step (PO) causes an increase in mesoporosity in all groups of specimens. The addition of zinc chloride provided Z1_NO_H3 specimens with up to 88.9% of its SSA values associated from pores of <2 nm. It should be emphasized that fibers did not undergo any activation process, such as the impregnation by KOH, and still, the lignin-derived carbon nanofibers (LCNFs) and PAN-derived carbon nanofibers (PCNFs) revealed the SSA values that were much higher than those reported in the literature.
The electrochemical results obtained using supercapacitor prototypes demonstrated that at slow scan rates, the P1_POL_H3 specimen reached it maximum capacitance of 26 F/g at the scan rate of 30 mV/s, although this value was reduced to 13.8 F/g at 500 mV/s. Similarly, for the Z1_NO_H3 specimen, the maximum capacitance was 11.13 F/g at 200 mV/s, which was only slightly reduced to 10.9 F/g at 500 mV/s. The specimens with a higher packing density also revealed an improved capacitance at higher scan rates, as well as higher volumetric capacitance. Such phenomena were attributed to an improved electrolyte ion mobility due to a shorter diffusion path.
The lignin-derived carbon nanofibers possessed specific capacitance of about one-half that of the PAN-derived specimens at 50 mV/s. However, the difference was only 21% at 500 mV/s. Hence, the LCNFs performance does not fall far behind that of PCNFs. Considering the choice of lignin as a carbon nanofiber precursor, one should consider that, despite lower SSA value and specific capacitance, lignin guarantees a much higher mass yield, which is four to five times higher than that of PAN. The latter, together with a higher packing density, a lower price, an improved performance at the fast scan rates, and a much lower environmental impact, suggests that lignin holds great promise as a raw material for supercapacitor electrodes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/batteries9120566/s1. References [64,65] are cited in the supplementary materials.
Author Contributions
Conceptualization, Y.J.D. and A.L.Y.; Methodology, Y.J.D. and V.D.S.; Software, Y.J.D. and V.D.S.; Validation, A.L.Y.; Formal analysis, Y.J.D. and V.D.S.; Investigation, Y.J.D.; Resources, B.P.; Data curation, V.D.S.; Writing—original draft, Y.J.D., V.D.S., E.S.M. and A.L.Y.; Writing—review & editing, Y.J.D., V.D.S., B.P., E.S.M. and A.L.Y.; Supervision, A.L.Y.; Project administration, A.L.Y.; Funding acquisition, B.P. and A.L.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Nonwovens Institute, grant no. 18 231. V. D. Silva and E. S. Medeiros thank the Brazilian funding agencies (CAPES and CNPQ) for their support. V. D. Silva thanks CNPQ (Process No. 202290/2020 4) and CAPES (finance code 001).
Data Availability Statement
Data available upon reasonable request to corresponding authors. The data are not publicly available due to privacy.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aykut, S.C.; Castro, M. Globalising the Climate; Aykut, S.C., Foyer, J., Morena, E., Eds.; Routledge: London, UK, 2017; ISBN 9781315560595. [Google Scholar]
- Wang, Y.; Song, Y.; Xia, Y. Electrochemical Capacitors: Mechanism, Materials, Systems, Characterization and Applications. Chem. Soc. Rev. 2016, 45, 5925–5950. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, L.M.; Cesar, R.; Moreira, C.M.R.; Santos, J.H.M.; De Souza, L.G.; Pires, B.M.; Vicentini, R.; Nunes, W.; Zanin, H. Reviewing the Fundamentals of Supercapacitors and the Difficulties Involving the Analysis of the Electrochemical Findings Obtained for Porous Electrode Materials. Energy Storage Mat. 2020, 27, 555–590. [Google Scholar] [CrossRef]
- Zhang, Q.; Uchaker, E.; Candelaria, S.L.; Cao, G. Nanomaterials for Energy Conversion and Storage. Chem. Soc. Rev. 2013, 42, 3127. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Chang, D.W.; Baek, J.-B.; Lu, W. Carbon Nanomaterials for Advanced Energy Conversion and Storage. Small 2012, 8, 1130–1166. [Google Scholar] [CrossRef]
- Miao, F.; Shao, C.; Li, X.; Wang, K.; Lu, N.; Liu, Y. Three-Dimensional Freestanding Hierarchically Porous Carbon Materials as Binder-Free Electrodes for Supercapacitors: High Capacitive Property and Long-Term Cycling Stability. J. Mater. Chem. A Mater. 2016, 4, 5623–5631. [Google Scholar] [CrossRef]
- Silva, V.D.; Dias, Y.J.; Macedo, D.A.; Medeiros, E.S.; Yarin, A.L. Freestanding, Flexible, Ultrahigh-Surface Area Carbon Nanofibers by Supersonic Solution Blowing: Promising Electrodes for Supercapacitors. Appl. Mater. Today 2023, 31, 101776. [Google Scholar] [CrossRef]
- Collins, M.N.; Nechifor, M.; Tanasă, F.; Zănoagă, M.; McLoughlin, A.; Stróżyk, M.A.; Culebras, M.; Teacă, C.-A. Valorization of Lignin in Polymer and Composite Systems for Advanced Engineering Applications—A Review. Int. J. Biol. Macromol. 2019, 131, 828–849. [Google Scholar] [CrossRef]
- Wang, S.; Bai, J.; Innocent, M.T.; Wang, Q.; Xiang, H.; Tang, J.; Zhu, M. Lignin-Based Carbon Fibers: Formation, Modification and Potential Applications. Green Energy Environ. 2022, 7, 578–605. [Google Scholar] [CrossRef]
- Xuan, D.; Liu, J.; Wang, D.; Lu, Z.; Liu, Q.; Liu, Y.; Li, S.; Zheng, Z. Facile Preparation of Low-Cost and Cross-Linked Carbon Nanofibers Derived from PAN/PMMA/Lignin as Supercapacitor Electrodes. Energy Fuels 2021, 35, 796–805. [Google Scholar] [CrossRef]
- Fang, W.; Yang, S.; Wang, X.-L.; Yuan, T.-Q.; Sun, R.-C. Manufacture and Application of Lignin-Based Carbon Fibers (LCFs) and Lignin-Based Carbon Nanofibers (LCNFs). Green Chem. 2017, 19, 1794–1827. [Google Scholar] [CrossRef]
- Hu, S.; Hsieh, Y.-L. Ultrafine Microporous and Mesoporous Activated Carbon Fibers from Alkali Lignin. J. Mater. Chem. A Mater. 2013, 1, 11279. [Google Scholar] [CrossRef]
- Cao, Q.; Zhu, M.; Chen, J.; Song, Y.; Li, Y.; Zhou, J. Novel Lignin-Cellulose-Based Carbon Nanofibers as High-Performance Supercapacitors. ACS Appl. Mater. Interfaces 2020, 12, 1210–1221. [Google Scholar] [CrossRef]
- Schlee, P.; Hosseinaei, O.; Baker, D.; Landmér, A.; Tomani, P.; Mostazo-López, M.J.; Cazorla-Amorós, D.; Herou, S.; Titirici, M.-M. From Waste to Wealth: From Kraft Lignin to Free-Standing Supercapacitors. Carbon 2019, 145, 470–480. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, H.; Cao, Q.; Zheng, H.; Xu, D.; Guo, H.; Wang, S.; Li, Y.; Zhou, J. Electrospun Lignin-Based Carbon Nanofibers as Supercapacitor Electrodes. ACS Sustain. Chem. Eng. 2020, 8, 12831–12841. [Google Scholar] [CrossRef]
- Landers, J.; Gor, G.Y.; Neimark, A.V. Density Functional Theory Methods for Characterization of Porous Materials. Colloids Surf. A Physicochem. Eng. Asp. 2013, 437, 3–32. [Google Scholar] [CrossRef]
- Chen, T.; Dai, L. Flexible Supercapacitors Based on Carbon Nanomaterials. J. Mater. Chem. A Mater. 2014, 2, 10756. [Google Scholar] [CrossRef]
- Nataraj, S.K.; Yang, K.S.; Aminabhavi, T.M. Polyacrylonitrile-Based Nanofibers—A State-of-the-Art Review. Prog. Polym. Sci. 2012, 37, 487–513. [Google Scholar] [CrossRef]
- Liu, Y.; Chae, H.G.; Kumar, S. Gel-Spun Carbon Nanotubes/Polyacrylonitrile Composite Fibers. Part II: Stabilization Reaction Kinetics and Effect of Gas Environment. Carbon 2011, 49, 4477–4486. [Google Scholar] [CrossRef]
- Pan, Z.; Yang, J.; Kong, J.; Loh, X.J.; Wang, J.; Liu, Z. “Porous and Yet Dense” Electrodes for High-Volumetric-Performance Electrochemical Capacitors: Principles, Advances, and Challenges. Adv. Sci. 2022, 9, 2103953. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, E.S.; Glenn, G.M.; Klamczynski, A.P.; Orts, W.J.; Mattoso, L.H.C. Solution Blow Spinning: A New Method to Produce Micro- and Nanofibers from Polymer Solutions. J. Appl. Polym. Sci. 2009, 113, 2322–2330. [Google Scholar] [CrossRef]
- Inagaki, M.; Yang, Y.; Kang, F. Carbon Nanofibers Prepared via Electrospinning. Adv. Mater. 2012, 24, 2547–2566. [Google Scholar] [CrossRef]
- Jia, C.; Li, L.; Song, J.; Li, Z.; Wu, H. Mass Production of Ultrafine Fibers by a Versatile Solution Blow Spinning Method. Acc. Mater. Res. 2021, 2, 432–446. [Google Scholar] [CrossRef]
- Ju, Y.-W.; Choi, G.-R.; Jung, H.-R.; Lee, W.-J. Electrochemical Properties of Electrospun PAN/MWCNT Carbon Nanofibers Electrodes Coated with Polypyrrole. Electrochim. Acta 2008, 53, 5796–5803. [Google Scholar] [CrossRef]
- Liu, S.; Song, C.; Zhang, W.; Zhang, T.; Shao, W.; Weng, Z.; Yao, M.; Huang, H.; Jian, X.; Hu, F. Optimizing Solvated Anion Accessibility and Pseudocapacitive Charge Storage in Carbon Cathode toward High-Energy Sodium-Ion Capacitor. Chem. Eng. J. 2022, 450, 138103. [Google Scholar] [CrossRef]
- Lai, C.; Zhou, Z.; Zhang, L.; Wang, X.; Zhou, Q.; Zhao, Y.; Wang, Y.; Wu, X.-F.; Zhu, Z.; Fong, H. Free-Standing and Mechanically Flexible Mats Consisting of Electrospun Carbon Nanofibers Made from a Natural Product of Alkali Lignin as Binder-Free Electrodes for High-Performance Supercapacitors. J. Power Sources 2014, 247, 134–141. [Google Scholar] [CrossRef]
- Ding, R.; Wu, H.; Thunga, M.; Bowler, N.; Kessler, M.R. Processing and Characterization of Low-Cost Electrospun Carbon Fibers from Organosolv Lignin/Polyacrylonitrile Blends. Carbon 2016, 100, 126–136. [Google Scholar] [CrossRef]
- Rahaman, M.S.A.; Ismail, A.F.; Mustafa, A. A Review of Heat Treatment on Polyacrylonitrile Fiber. Polym. Degrad. Stab. 2007, 92, 1421–1432. [Google Scholar] [CrossRef]
- Zhang, W.; Yin, J.; Lin, Z.; Lin, H.; Lu, H.; Wang, Y.; Huang, W. Facile Preparation of 3D Hierarchical Porous Carbon from Lignin for the Anode Material in Lithium Ion Battery with High Rate Performance. Electrochim. Acta 2015, 176, 1136–1142. [Google Scholar] [CrossRef]
- Kopeć, M.; Lamson, M.; Yuan, R.; Tang, C.; Kruk, M.; Zhong, M.; Matyjaszewski, K.; Kowalewski, T. Polyacrylonitrile-Derived Nanostructured Carbon Materials. Prog. Polym. Sci. 2019, 92, 89–134. [Google Scholar] [CrossRef]
- Jiang, Q.; Pang, X.; Geng, S.; Zhao, Y.; Wang, X.; Qin, H.; Liu, B.; Zhou, J.; Zhou, T. Simultaneous Cross-Linking and Pore-Forming Electrospun Carbon Nanofibers towards High Capacitive Performance. Appl. Surf. Sci. 2019, 479, 128–136. [Google Scholar] [CrossRef]
- Yarin, A.L.; Pourdeyhimi, B.; Ramakrishna, S. Fundamentals and Applications of Micro- and Nanofibers; Cambridge University Press: Cambridge, UK, 2014; ISBN 9781107060296. [Google Scholar]
- Qu, W.; Yang, J.; Sun, X.; Bai, X.; Jin, H.; Zhang, M. Towards Producing High-Quality Lignin-Based Carbon Fibers: A Review of Crucial Factors Affecting Lignin Properties and Conversion Techniques. Int. J. Biol. Macromol. 2021, 189, 768–784. [Google Scholar] [CrossRef]
- de Gonzaga, L.A.C.; Martins, M.C.F.; Correa, A.C.; Facchinatto, W.M.; da Silva, C.M.P.; Colnago, L.A.; Mattoso, L.H.C. Production of Carbon Nanofibers from PAN and Lignin by Solution Blow Spinning. J. Polym. Res. 2021, 28, 237. [Google Scholar] [CrossRef]
- Cho, M.; Ko, F.K.; Renneckar, S. Impact of Thermal Oxidative Stabilization on the Performance of Lignin-Based Carbon Nanofiber Mats. ACS Omega 2019, 4, 5345–5355. [Google Scholar] [CrossRef]
- Thunga, M.; Chen, K.; Grewell, D.; Kessler, M.R. Bio-Renewable Precursor Fibers from Lignin/Polylactide Blends for Conversion to Carbon Fibers. Carbon 2014, 68, 159–166. [Google Scholar] [CrossRef]
- Sallem-Idrissi, N.; Sclavons, M.; Debecker, D.P.; Devaux, J. Miscible Raw Lignin/Nylon 6 Blends: Thermal and Mechanical Performances. J. Appl. Polym. Sci. 2016, 133, 1–10. [Google Scholar] [CrossRef]
- Ago, M.; Borghei, M.; Haataja, J.S.; Rojas, O.J. Mesoporous Carbon Soft-Templated from Lignin Nanofiber Networks: Microphase Separation Boosts Supercapacitance in Conductive Electrodes. RSC Adv. 2016, 6, 85802–85810. [Google Scholar] [CrossRef]
- Moezzi, A.; Cortie, M.; McDonagh, A. Transformation of Zinc Hydroxide Chloride Monohydrate to Crystalline Zinc Oxide. Dalton Trans. 2016, 45, 7385–7390. [Google Scholar] [CrossRef]
- Hayashi, J.; Muroyama, K.; Gomes, V.G.; Watkinson, A.P. Fractal Dimensions of Activated Carbons Prepared from Lignin by Chemical Activation. Carbon 2002, 40, 630–632. [Google Scholar] [CrossRef]
- Kim, C.; Ngoc, B.T.N.; Yang, K.S.; Kojima, M.; Kim, Y.A.; Kim, Y.J.; Endo, M.; Yang, S.C. Self-Sustained Thin Webs Consisting of Porous Carbon Nanofibers for Supercapacitors via the Electrospinning of Polyacrylonitrile Solutions Containing Zinc Chloride. Adv. Mater. 2007, 19, 2341–2346. [Google Scholar] [CrossRef]
- Yun, S.I.; Kim, S.H.; Kim, D.W.; Kim, Y.A.; Kim, B.-H. Facile Preparation and Capacitive Properties of Low-Cost Carbon Nanofibers with ZnO Derived from Lignin and Pitch as Supercapacitor Electrodes. Carbon 2019, 149, 637–645. [Google Scholar] [CrossRef]
- Kim, C.; Park, S.-H.; Cho, J.-I.; Lee, D.-Y.; Park, T.-J.; Lee, W.-J.; Yang, K.-S. Raman Spectroscopic Evaluation of Polyacrylonitrile-Based Carbon Nanofibers Prepared by Electrospinning. J. Raman Spectrosc. 2004, 35, 928–933. [Google Scholar] [CrossRef]
- Singh, M.; Gupta, A.; Sundriyal, S.; Jain, K.; Dhakate, S.R. Kraft Lignin-Derived Free-Standing Carbon Nanofibers Mat for High-Performance All-Solid-State Supercapacitor. Mater. Chem. Phys. 2021, 264, 124454. [Google Scholar] [CrossRef]
- Hérou, S.; Crespo Ribadeneyra, M.; Schlee, P.; Luo, H.; Cristian Tanase, L.; Roβberg, C.; Titirici, M. The Impact of Having an Oxygen-Rich Microporous Surface in Carbon Electrodes for High-Power Aqueous Supercapacitors. J. Energy Chem. 2021, 53, 36–48. [Google Scholar] [CrossRef]
- Esmeryan, K.D.; Castano, C.E.; Bressler, A.H.; Abolghasemibizaki, M.; Fergusson, C.P.; Roberts, A.; Mohammadi, R. Kinetically Driven Graphite-like to Diamond-like Carbon Transformation in Low Temperature Laminar Diffusion Flames. Diam. Relat. Mater. 2017, 75, 58–68. [Google Scholar] [CrossRef]
- Choi, D.; Kil, H.-S.; Lee, S. Fabrication of Low-Cost Carbon Fibers Using Economical Precursors and Advanced Processing Technologies. Carbon 2019, 142, 610–649. [Google Scholar] [CrossRef]
- Liang, J.; Zhao, H.; Yue, L.; Fan, G.; Li, T.; Lu, S.; Chen, G.; Gao, S.; Asiri, A.M.; Sun, X. Recent Advances in Electrospun Nanofibers for Supercapacitors. J. Mater. Chem. A Mater. 2020, 8, 16747–16789. [Google Scholar] [CrossRef]
- Kakoria, A.; Sinha-Ray, S. A Review on Biopolymer-Based Fibers via Electrospinning and Solution Blowing and Their Applications. Fibers 2018, 6, 45. [Google Scholar] [CrossRef]
- Jeon, J.; Zhang, L.; Lutkenhaus, J.L.; Laskar, D.D.; Lemmon, J.P.; Choi, D.; Nandasiri, M.I.; Hashmi, A.; Xu, J.; Motkuri, R.K.; et al. Controlling Porosity in Lignin-Derived Nanoporous Carbon for Supercapacitor Applications. ChemSusChem 2015, 8, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Azwar, E.; Wan Mahari, W.A.; Chuah, J.H.; Vo, D.-V.N.; Ma, N.L.; Lam, W.H.; Lam, S.S. Transformation of Biomass into Carbon Nanofiber for Supercapacitor Application—A Review. Int. J. Hydrogen Energy 2018, 43, 20811–20821. [Google Scholar] [CrossRef]
- Kolbasov, A.; Sinha-Ray, S.; Joijode, A.S.; Hassan, M.A.; Brown, D.; Maze, B.; Pourdeyhimi, B.; Yarin, A.L. Industrial Scale Solution Blowing of Soy Protein Nanofibers. Ind. Eng. Chem. Res. 2015, 55, 323–333. [Google Scholar] [CrossRef]
- Tuinstra, F.; Koenig, J.L. Raman Spectrum of Graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef]
- Dadol, G.C.; Kilic, A.; Tijing, L.D.; Lim, K.J.A.; Cabatingan, L.K.; Tan, N.P.B.; Stojanovska, E.; Polat, Y. Solution Blow Spinning (SBS) and SBS-Spun Nanofibers: Materials, Methods, and Applications. Mater. Today Commun. 2020, 25, 101656. [Google Scholar] [CrossRef]
- Cai, Y.; Huang, F.; Wei, Q.; Song, L.; Hu, Y.; Ye, Y.; Xu, Y.; Gao, W. Structure, Morphology, Thermal Stability and Carbonization Mechanism Studies of Electrospun PA6/Fe-OMT Nanocomposite Fibers. Polym. Degrad. Stab. 2008, 93, 2180–2185. [Google Scholar] [CrossRef]
- Cho, M.; Karaaslan, M.A.; Renneckar, S.; Ko, F. Enhancement of the Mechanical Properties of Electrospun Lignin-Based Nanofibers by Heat Treatment. J. Mater. Sci. 2017, 52, 9602–9614. [Google Scholar] [CrossRef]
- Perera Jayawickramage, R.A.; Ferraris, J.P. High Performance Supercapacitors Using Lignin Based Electrospun Carbon Nanofiber Electrodes in Ionic Liquid Electrolytes. Nanotechnology 2019, 30, 155402. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Luo, J.; Chen, L. Facile Preparation of Self-Standing Hierarchical Porous Nitrogen-Doped Carbon Fibers for Supercapacitors from Plant Protein–Lignin Electrospun Fibers. ACS Omega 2018, 3, 4647–4656. [Google Scholar] [CrossRef]
- Mathis, T.S.; Kurra, N.; Wang, X.; Pinto, D.; Simon, P.; Gogotsi, Y. Energy Storage Data Reporting in Perspective—Guidelines for Interpreting the Performance of Electrochemical Energy Storage Systems. Adv. Energy Mater. 2019, 9, 1902007. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, W.; Alhebshi, N.A.; Salah, N.; Alshareef, H.N. Synthesis Strategies of Porous Carbon for Supercapacitor Applications. Small Methods 2020, 4, 1900853. [Google Scholar] [CrossRef]
- Santos, J.P.A.; Pinzón, M.J.; Santos, É.A.; Vicentini, R.; Pagan, C.J.B.; Da Silva, L.M.; Zanin, H. Boosting Energy-Storage Capability in Carbon-Based Supercapacitors Using Low-Temperature Water-in-Salt Electrolytes. J. Energy Chem. 2022, 70, 521–530. [Google Scholar] [CrossRef]
- Vicentini, R.; Aguiar, J.P.; Beraldo, R.; Venâncio, R.; Rufino, F.; Da Silva, L.M.; Zanin, H. Ragone Plots for Electrochemical Double-Layer Capacitors. Batter. Supercaps 2021, 4, 1291–1303. [Google Scholar] [CrossRef]
- Real, C.G.; Vicentini, R.; Nunes, W.G.; Pascon, A.M.; Campos, F.A.; Da Silva, L.M.; Freitas, R.G.; Zanin, H. Analyses of Dispersive Effects and the Distributed Capacitance in the Time and Frequency Domains of Activated Carbon Nanofiber Electrodes as Symmetric Supercapacitors. Electrochim. Acta 2022, 402, 139299. [Google Scholar] [CrossRef]
- Chen, X.; Wang, X.; Fang, D. A review on C1s XPS-spectra for some kinds of carbon materials. Fuller. Nanotub. Carbon Nanostructures 2020, 28, 1048–1058. [Google Scholar] [CrossRef]
- Chae, J.S.; Kang, W.; Roh, K.C. sp2–sp3 Hybrid Porous Carbon Materials Applied for Supercapacitors. Energies 2021, 14, 5990. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).