
Electrochemical Properties of LiFePO4 Cathodes: The Effect of Carbon Additives




Abstract
1. Introduction
2. Materials and Methods
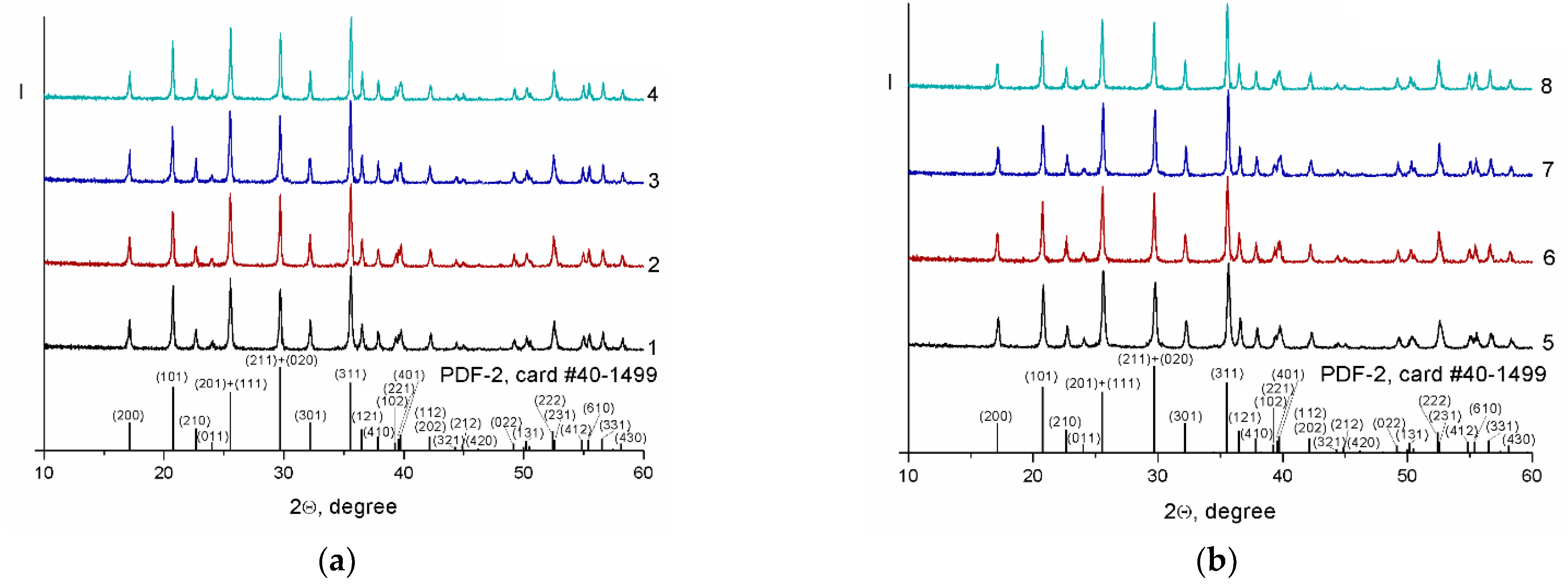
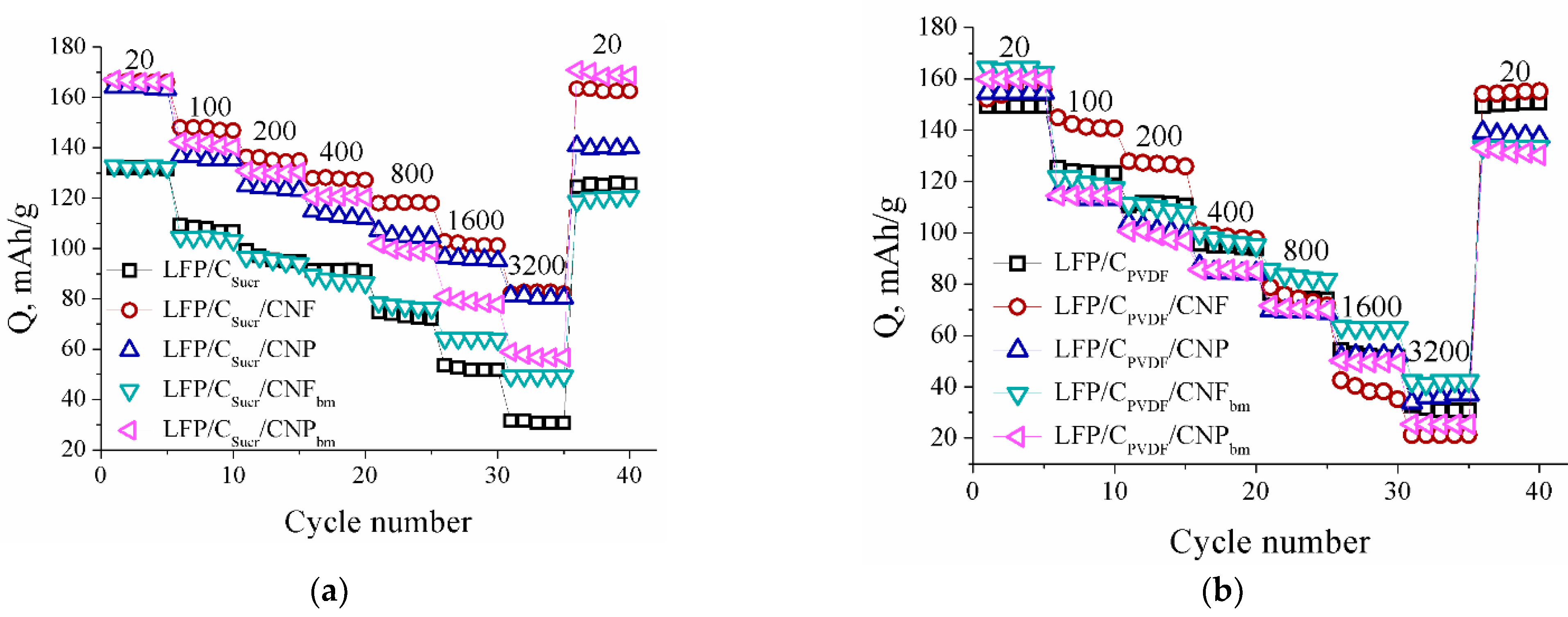
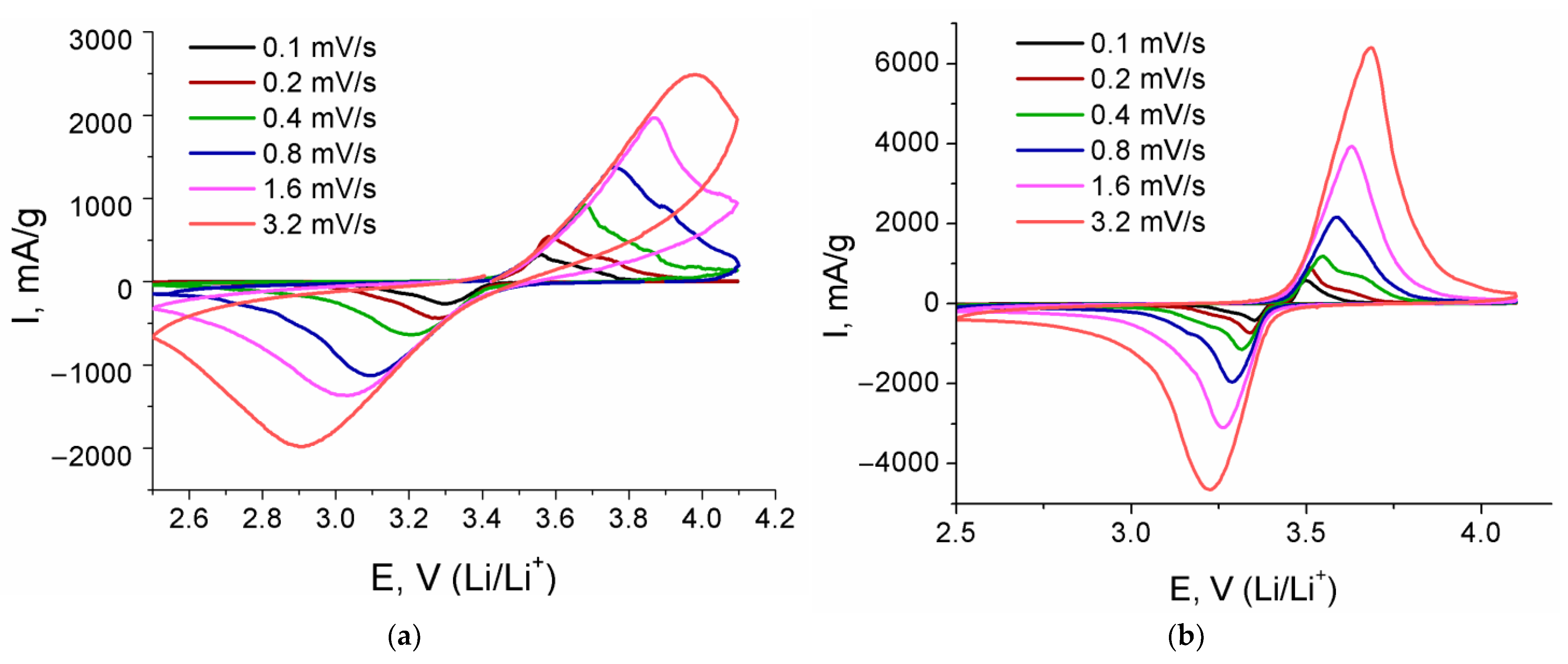
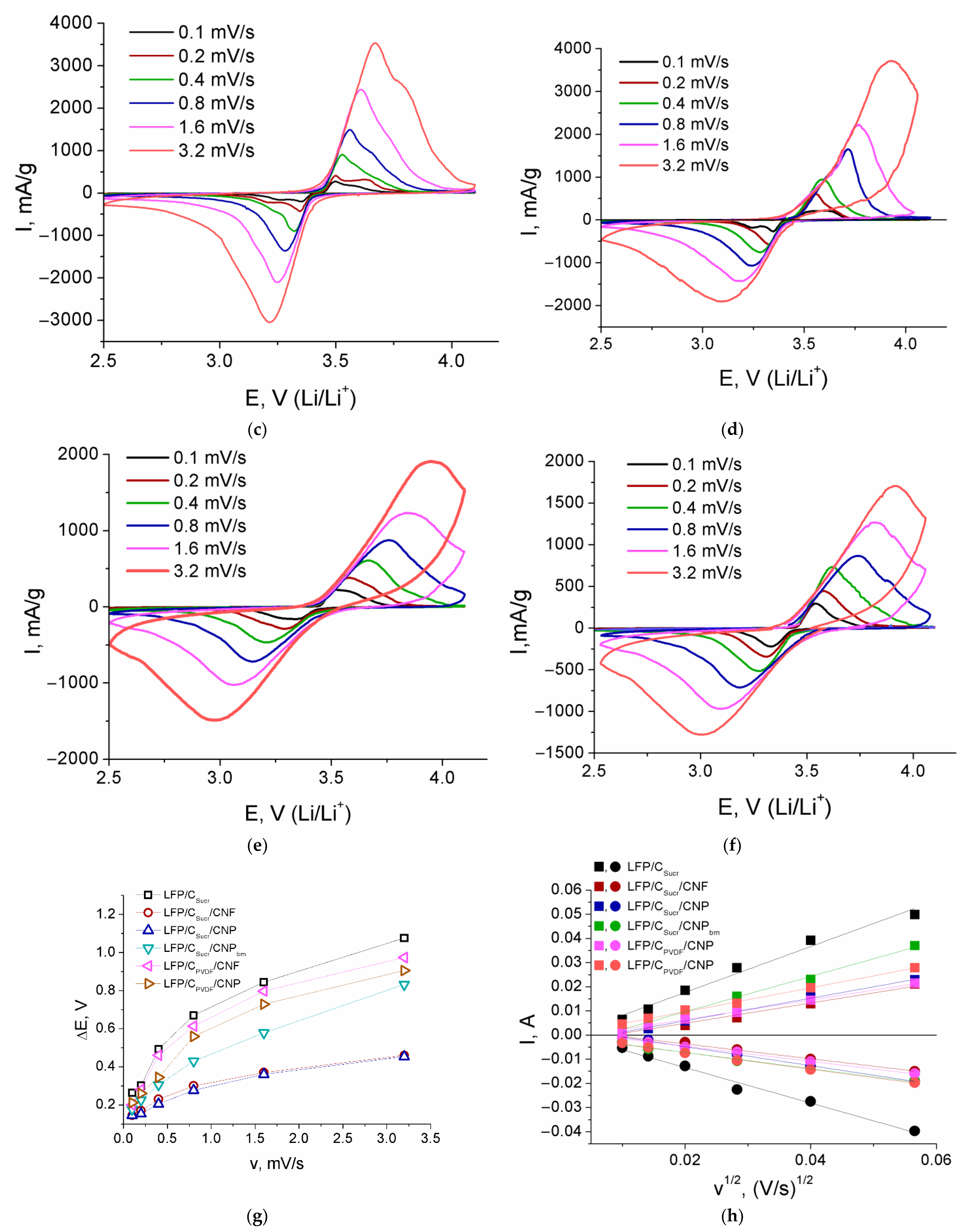
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-ion battery materials: Present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Grey, C.P.; Hall, D.S. Prospects for lithium-ion batteries and beyond—A 2030 vision. Nat. Commun. 2020, 11, 6279. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, X.; Tian, J.; Ma, X.; Wang, B.; Li, W.; Wang, Q. Adhesive nanocomposites of hypergravity induced Co3O4 nanoparticles and natural gels as Li-ion battery anode materials with high capacitance and low resistance. RSC Adv. 2017, 7, 21061–21067. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Pan, R.; Chen, Z. Modeling and State-of-Charge Prediction of Lithium-Ion Battery and Ultracapacitor Hybrids with a Co-Estimator. Energy 2017, 121, 739–750. [Google Scholar] [CrossRef]
- Duh, Y.S.; Lin, K.H.; Kao, C.S. Experimental investigation and visualization on thermal runaway of hard prismatic lithium-ion batteries used in smart phones. J. Therm. Anal. Calorim. 2018, 132, 1677–1692. [Google Scholar] [CrossRef]
- Galushkin, N.E.; Yazvinskaya, N.N.; Galushkin, D.N. Mechanism of thermal runaway in lithium-ion cells. J. Electrochem. Soc. 2018, 165, 1303–1308. [Google Scholar] [CrossRef]
- Kang, Y.; Deng, C.; Chen, Y.; Liu, X.; Liang, Z.; Li, T.; Hu, Q.; Zhao, Y. Binder-Free Electrodes and Their Application for Li-Ion Batteries. Nanoscale Res. Lett. 2020, 15, 112. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.; Xia, M.; Wei, K.; Zhang, L.; Zhang, X.; Cui, Y.; Shu, J. Issues and challenges of layered lithium nickel cobalt manganese oxides for lithium-ion batteries. J. Electroanal. Chem. 2021, 895, 115412. [Google Scholar] [CrossRef]
- Dunn, B.; Kamath, H.; Tarascon, J.M. Electrical Energy Storage for the Grid: A Battery of Choices. Science 2011, 334, 928–935. [Google Scholar] [CrossRef]
- Chen, Y.; Kang, Y.; Zhao, Y.; Wang, L.; Liu, J.; Li, Y.; Liang, Z.; He, X.; Li, X.; Tavajohi, N.; et al. A review of lithium-ion battery safety concerns: The issues, strategies, and testing standards. J. Energy Chem. 2021, 59, 83–99. [Google Scholar] [CrossRef]
- Susantyoko, R.; Alkindi, T.S.; Kanagaraj, A.B.; An, B.; Alshibli, H.; Choi, D.; Aldahmani, S.; Fadaq, H.; Almheiri, S. Performance optimization of freestanding MWCNF-LiFePO4 sheets as cathodes for improved specific capacity of lithium-ion batteries. RSC Adv. 2018, 8, 16566–16573. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, B.; Wang, Y.; Wang, F. Solvothermal synthesis of LiFePO4 nanorods as high-performance cathode materials for lithium ion batteries. Ceram. Int. 2016, 42, 10297–10303. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Q.; Liang, Q. Carbon-Coatings Improve Performance of Li-Ion Battery. Nanomaterials 2022, 12, 1936. [Google Scholar] [CrossRef] [PubMed]
- Voropaeva, D.Y.; Safronova, E.Y.; Novikova, S.A.; Yaroslavtsev, A.B. Recent progress in lithium-ion and lithium metal batteries. Mendeleev Commun. 2022, 32, 287–297. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R.; Liu, L.; Jiang, S.; Ni, L.; Bie, X.; Yang, X.; Hu, J.; Wang, Z.; Chen, H.; et al. In-situ self-polymerization restriction to form core-shell LiFePO4/C nanocomposite with ultrafast rate capability for high-power Li-ion batteries. Nano Energy 2017, 39, 346–354. [Google Scholar] [CrossRef]
- Li, Q.Y.; Zheng, F.H.; Huang, Y.G.; Zhang, X.H.; Wu, Q.; Fu, D.J.; Zhang, J.J.; Yin, J.C.; Wang, H.Q. Surfactants assisted synthesis of nano-LiFePO4/C composite as cathode materials for lithium-ion batteries. J. Mater. Chem. A 2015, 3, 2025–2035. [Google Scholar] [CrossRef]
- Stenina, I.A.; Yaroslavtsev, A.B. Nanomaterials for lithium-ion batteries and hydrogen energy. Pure Appl. Chem. 2017, 89, 1185–1194. [Google Scholar] [CrossRef]
- Saroha, R.; Panwar, A.K.; Sharma, Y.; Tyagi, P.K.; Ghosh, S. Development of surface functionalized ZnO-doped LiFePO4/C composites as alternative cathode material for lithium ion batteries. Appl. Surf. Sci. 2017, 394, 25–36. [Google Scholar] [CrossRef]
- Li, X.; Yu, L.; Cui, Y.; Li, A.; Shao, H.; Shao, Z.; Zhang, W.; Shao, Z. Enhanced properties of LiFePO4/C cathode materials co-doped with V and F ions via high-temperature ball milling route. Int. J. Hydrogen Energy 2019, 44, 27204–27213. [Google Scholar] [CrossRef]
- Harrison, K.L.; Bridges, C.A.; Paranthaman, M.P.; Segre, C.U.; Katsoudas, J.; Maroni, V.A.; Idrobo, J.C.; Goodenough, J.B.; Manthiram, A. Temperature dependence of aliovalent-vanadium doping in LiFePO4 cathodes. Chem. Mater. 2013, 25, 768–781. [Google Scholar] [CrossRef]
- Yu, Z.; Jiang, L. Olivine LiFePO4 nanocrystals grown on nitrogen-doped grapheme sheets as high-rate cathode for lithium-ion batteries. Solid State Ion. 2018, 325, 12–16. [Google Scholar] [CrossRef]
- Zhan, T.T.; Jiang, W.F.; Li, C.; Luo, X.D.; Lin, G.; Li, Y.W.; Xiao, S.H. High performed composites of LiFePO4/3DG/C based on FePO4 by hydrothermal method. Electrochim. Acta 2017, 246, 322–328. [Google Scholar] [CrossRef]
- Yaroslavtsev, A.B.; Stenina, I.A. Carbon coating of electrode materials for lithium-ion batteries. Surf. Innov. 2021, 9, 92–110. [Google Scholar] [CrossRef]
- He, Z.; Jiang, Y.; Zhu, J.; Wang, H.; Li, Y.; Zhou, H.; Meng, W.; Dai, L.; Wang, L. N-doped carbon coated LiTi2(PO4)3 as superior anode using PANi as carbon and nitrogen bi-sources for aqueous lithium ion battery. Electrochim. Acta 2018, 279, 279–288. [Google Scholar] [CrossRef]
- Lei, C.; Han, F.; Li, D.; Li, W.-C.; Sun, Q.; Zhang, X.-Q.; Lu, A.-H. Dopamine as the coating agent and carbon precursor for the fabrication of N-doped carbon coated Fe3O4 composites as superior lithium ion anodes. Nanoscale 2013, 5, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, I.; Sharma, P.; Nebhani, L. Polybenzoxazine-an enticing precursor for engineering heteroatom-doped porous carbon materials with applications beyond energy, environment and catalysis. Mater. Today Chem. 2022, 23, 100734. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, Z.; Fan, C.; Liu, Z.; Han, S.; Liu, J. Construction and Theoretical Calculation of an Ultra-High-Performance LiVPO4F/C Cathode by B-Doped Pyrolytic Carbon from Poly(vinylidene Fluoride). ACS Appl. Mater. Interfaces 2021, 13, 15190–15204. [Google Scholar] [CrossRef] [PubMed]
- Stenina, I.A.; Kulova, T.L.; Skundin, A.M.; Yaroslavtsev, A.B. Effects of carbon coating from sucrose and PVDF on electrochemical performance of Li4Ti5O12/C composites in different potential ranges. J. Solid State Electrochem. 2018, 22, 2631–2639. [Google Scholar] [CrossRef]
- Wang, X.; Feng, Z.; Hou, X.; Liu, L.; He, M.; He, X.; Huang, J.; Wen, Z. Fluorine doped carbon coating of LiFePO4 as a cathode material for lithium-ion batteries. Chem. Eng. J. 2020, 379, 122371. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, H.; Chen, Y.; Wang, W.; Ye, Y.; Zheng, C.; Deng, P.; Shi, Z. A three-dimensional LiFePO4/carbon nanotubes/graphene composite as a cathode material for lithium-ion batteries with superior high-rate performance. J. Alloys Compd. 2015, 626, 280–286. [Google Scholar] [CrossRef]
- Luo, G.-Y.; Gu, Y.-J.; Liu, Y.; Chen, Z.-L.; Huo, Y.-l.; Wu, F.-Z.; Mai, Y.; Dai, X.-Y.; Deng, Y. Electrochemical performance of in situ LiFePO4 modified by N-doped graphene for Li-ion batteries. Ceram. Int. 2021, 47, 11332–11339. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, L. Outstanding Li-storage performance of LiFePO4@MWCNFs cathode material with 3D network structure for lithium-ion batteries. J. Phys. Chem. Solid. 2018, 116, 216–221. [Google Scholar] [CrossRef]
- Kim, M.-S.; Lee, G.-W.; Lee, S.-W.; Jeong, J.H.; Mhamane, D.; Roh, K.C.; Kim, K.-B. Synthesis of LiFePO4/graphene microspheres while avoiding restacking of graphene sheet’s for high-rate lithium-ion batteries. J. Ind. Eng. Chem. 2017, 52, 251–259. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Zhang, H.; Liang, F.; Yao, Y.; Zhang, K. Freeze drying under vacuum assisted synthesis of LiFePO4@MWCNFs composite with phytic acid as phosphorus source for advanced Li-storage. Vacuum 2021, 193, 110541. [Google Scholar] [CrossRef]
- Deng, F.; Zeng, X.R.; Zou, J.Z.; Huang, J.F.; Sheng, H.C.; Xiong, X.B.; Qian, H.X.; Li, X.H. Synthesis of LiFePO4 in situ vapor-grown carbon fiber (VGCF) composite cathode material via microwave pyrolysis chemical vapor deposition. Chin. Sci. Bull. 2011, 56, 1832–1835. [Google Scholar] [CrossRef][Green Version]
- Stenina, I.A.; Minakova, P.V.; Kulova, T.L.; Desyatov, A.V.; Yaroslavtsev, A.B. LiFePO4/carbon nanomaterial composites for cathodes of high-power lithium ion batteries. Inorg. Mater. 2021, 57, 620–628. [Google Scholar] [CrossRef]
- Li, W.; Garg, A.; Le, M.L.P.; Ruhatiya, C.; Gao, L.; Tran, V.M. Electrochemical performance investigation of LiFePO4/C0.15-x (x=0.05, 0.1, 0.15 CNTs) electrodes at various calcination temperatures: Experimental and Intelligent Modelling approach. Electrochim. Acta 2020, 330, 135314. [Google Scholar] [CrossRef]
- Wang, B.; Liu, T.; Liu, A.; Liu, G.; Wang, L.; Gao, T.; Wang, D.; Zhao, X.S. A Hierarchical Porous C@LiFePO4/Carbon Nanotubes Microsphere Composite for High-Rate Lithium-Ion Batteries: Combined Experimental and Theoretical Study. Adv. Energy Mater. 2016, 6, 1600426. [Google Scholar] [CrossRef]
- Sun, X.; Li, J.; Shi, C.; Wang, Z.; Liu, E.; He, C.; Du, X.; Zhao, N. Enhanced electrochemical performance of LiFePO4 cathode with in-situ chemical vapor deposition synthesized carbon nanotubes as conductor. J. Power Sources. 2012, 220, 264–268. [Google Scholar] [CrossRef]
- Xu, J.; Chen, G.; Li, X. Electrochemical performance of LiFePO4 cathode material coated with multi-wall carbon nanotubes. Mater. Chem. Phys. 2009, 118, 9–11. [Google Scholar] [CrossRef]
- Yang, J.; Wang, J.; Li, X.; Wang, D.; Liu, J.; Liang, G.; Gauthier, M.; Li, Y.; Geng, D.; Li, R.; et al. Hierarchically porous LiFePO4/nitrogen-doped carbon nanotubes composite as a cathode for lithium ion batteries. J. Mater. Chem. 2012, 22, 7537–7543. [Google Scholar] [CrossRef]
- Shih, J.-Y.; Lin, G.-Y.; Li, Y.-J.J.; Hung, T.-F.; Jose, R.; Karuppiah, C.; Yang, C.-C. Operando investigation on the fast two-phase transition kinetics of LiFePO4/C composite cathodes with carbon additives for lithium-ion batteries. Electrochim. Acta 2022, 419, 140356. [Google Scholar] [CrossRef]
- Rajoba, S.J.; Jadhav, L.D.; Kalubarme, R.S.; Patil, P.S.; Varma, S.; Wani, B.N. Electrochemical performance of LiFePO4/GO composite for Li-ion batteries. Ceram. Int. 2018, 44, 6886–6893. [Google Scholar] [CrossRef]
- Ren, X.; Li, Z.; Cao, J.; Tian, S.; Zhang, K.; Guo, J.; Wen, L.; Liang, G. Enhanced rate performance of the mortar-like LiFePO4/C composites combined with the evenly coated of carbon aerogel. J. Alloys Compd. 2021, 867, 158776. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composite | Crystallite Size ± 1, nm | Carbon Content, ±0.1 wt.% | Electronic Conductivity, S/cm | Specific Surface Area, m2/g |
|---|---|---|---|---|
| LFP/CSucr | 75 | 4.7 | 4.6 × 10–7 | 35 |
| LFP/CPVDF | 69 | 7.5 | 8.6 × 10–5 | 54 |
| LFP/CSucr/CNF | 60 | 12.1 | 1.3 × 10–2 | 65 |
| LFP/CSucr/CNP | 73 | 12.5 | 3.6 × 10–4 | 91 |
| LFP/CSucr/CNFbm | 58 | 12.5 | 3.2 × 10–3 | 48 |
| LFP/CSucr/CNPbm | 68 | 12.8 | 9.7 × 10–3 | 64 |
| LFP/CPVDF/CNF | 67 | 15.2 | 1.2 × 10–2 | 71 |
| LFP/CPVDF/CNP | 68 | 15.7 | 9.2 × 10–4 | 92 |
| LFP/CPVDF/CNFbm | 61 | 15.1 | 4.6 × 10–3 | 68 |
| LFP/CPVDF/CNPbm | 64 | 15.5 | 1.2 × 10–3 | 90 |
| Carbon Additive/ its Content | Preparation Method/ Calcination Conditions | Capacity, mAh/g (C Rate) | Ref. |
|---|---|---|---|
| CNTs/10 wt.% | Hydrothermal/800 °C, 6 h, N2 | 160 (0.1 C) | [37] |
| CNTs/6 wt.% | Hydrothermal/600 °C, 6 h, Ar/H2 | 155 (0.2 C), 126 (5 C) | [38] |
| CNTs/13 wt.% | Chemical vapor deposition/675 °C, 30 min, C2H2/Ar | 161 (0.1 C)/119 (5 C) | [39] |
| MWCNTs/5 wt.% | Hydrothermal/700 °C, 6 h, Ar | 139 (0.3 C)/102 (3 C) | [40] |
| N-doped CNTs/11 wt.% | Sol–gel/700 °C, 10 h, Ar | 142 (0.1 C)/82 (5 C) | [41] |
| MWCNTs/4.5 wt.% | Spray drying/700 °C, 10 h, Ar/H2 | 157 (0.2 C), 131 (5 C) | [32] |
| Graphene/4 wt.% | Solid-state reaction/650 °C, 10 h, Ar | 165 (0.2 C), 124 (5 C) | [30] |
| Porous graphene oxide/1 wt.% | Spray drying/700 °C, 10 h, Ar/H2 | 151 (0.1 C), 126 (5 C) | [42] |
| Graphene oxide/4 wt.% | solution combustion/700 °C, 5 h, N2 | 163 (0.1 C), 60 (2 C) | [43] |
| Vapor-grown carbon fiber/14.5 wt.% | Microwave pyrolysis chemical vapor deposition/800 °C, 10 min, propylene | 148 (0.1 C), 144 (0.5 C) | [35] |
| Carbon aerogel/2.6 wt.% | Spray drying/720 °C, 5 h, N2 | 152 (0.2 C), 134 (5 C) | [44] |
| F-doped coating/10 wt.% | Solid-state reaction/600 °C, 8 h, Ar | 145 (0.1 C), 113 (5 C) | [29] |
| Carbon nanofibers/10 wt.% | Sol–gel/600 °C, 10 h, Ar | 169 (0.1 C), 121 (5 C) | This work |
| Sample | Redox Process | Slope of the Dependence of Ip vs. v1/2 | DLi, cm2/s |
|---|---|---|---|
| LFP/CSucr | Reduction | −0.734 | 2.1 × 10–12 |
| Oxidation | 0.950 | 3.6 × 10–12 | |
| LFP/CSucr/CNF | Reduction | −0.308 | 2.1 × 10–11 |
| Oxidation | 0.419 | 4.1 × 10–11 | |
| LFP/CSucr/CNP | Reduction | −0.393 | 1.4 × 10–11 |
| Oxidation | 0.469 | 2.1 × 10–11 | |
| LFP/CSucr/CNPbm | Reduction | −0.336 | 2.8 × 10–12 |
| Oxidation | 0.679 | 1.1 × 10–11 | |
| LFP/CPVDF/CNF | Reduction | −0.315 | 8.1 × 10–12 |
| Oxidation | 0.407 | 1.5 × 10–11 | |
| LFP/CPVDF/CNP | Reduction | −0.349 | 1.8 × 10–12 |
| Oxidation | 0.493 | 3.4 × 10–12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stenina, I.; Minakova, P.; Kulova, T.; Yaroslavtsev, A. Electrochemical Properties of LiFePO4 Cathodes: The Effect of Carbon Additives. Batteries 2022, 8, 111. https://doi.org/10.3390/batteries8090111
Stenina I, Minakova P, Kulova T, Yaroslavtsev A. Electrochemical Properties of LiFePO4 Cathodes: The Effect of Carbon Additives. Batteries. 2022; 8(9):111. https://doi.org/10.3390/batteries8090111
Chicago/Turabian StyleStenina, Irina, Polina Minakova, Tatiana Kulova, and Andrey Yaroslavtsev. 2022. "Electrochemical Properties of LiFePO4 Cathodes: The Effect of Carbon Additives" Batteries 8, no. 9: 111. https://doi.org/10.3390/batteries8090111
APA StyleStenina, I., Minakova, P., Kulova, T., & Yaroslavtsev, A. (2022). Electrochemical Properties of LiFePO4 Cathodes: The Effect of Carbon Additives. Batteries, 8(9), 111. https://doi.org/10.3390/batteries8090111