1. Introduction
Lithium-ion batteries (LIBs) have influenced the technological developments of the last 30 years, from portable electronics to electric vehicles (EVs). Regarding the latter, almost all car manufacturers offer an electric model based on different LIB chemistries [
1,
2]. Nevertheless, it is well known that many car users foster concerns regarding ‘driving range anxiety’, which is caused by the limited mileage that EVs can achieve without recharging, the availability of recharging points, and the shorter driving range under certain climatic conditions (such as low temperature) [
1]. As a result, it is necessary to develop novel materials capable of providing higher capacities at higher voltages, which translate into higher energy densities.
LiFePO
4 (LFP) is the safest, state-of-the-art cathode material for automotive applications. In fact, it has been selected by Tesla for the Model 3 [
3]. LFP can provide 170 mAh·g
−1 at an average voltage of 3.45 V vs. Li, roughly providing 586 Wh·kg
LFP−1 [
4]. In order to increase the energy density of LIBs, many EV manufacturers have chosen layered metal oxides as cathode material instead of LFP. Among these layered oxides, LiNi
xMn
yCo
zO
2 (NMC) and LiNi
xCo
yAl
zO
2 (NCA), both with x + y + z = 1, have been used in different EV models [
1,
5]. Many of these materials can provide higher capacities at higher voltages, leading to higher energy densities [
6,
7]. In particular, research associated with NMCs has pursued a decrease in cobalt content in the material replacing cobalt with nickel, which is cheaper and can lead to higher capacities [
8,
9]. In this context, NMC111 (Ni:Mn:Co = 0.33:0.33:0.33) has been subsequently replaced by NMC532, NMC622, and ultimately NMC811 [
10,
11]. Nevertheless, there is a trade-off between the high capacity resulting from high nickel content and the cycle life, as well as the thermal stability of these materials [
10,
11].
Regarding the negative electrode, graphite, which possesses a 372 mAh·g
−1 capacity and a redox potential of 0.1 V vs. Li
+/Li, has been the predominant material of the last 25 years [
12]. Only Li
4Ti
5O
12 (LTO) has questioned the supremacy of graphite, mostly for power applications [
13]. However, its low discharge capacity (175 mAh·g
−1) and high redox potential (1.55 V vs. Li
+/Li) limit its implementation in high-energy applications [
4]. Nevertheless, graphite is insufficient to achieve the highest volumetric energy density goals [
14,
15]. Thus, it has been blended with silicon oxide (SiO
x) and silicon (Si) to enhance its capacity and energy density [
12,
16]. Silicon offers an excellent capacity and works at ~0.4 V vs. Li
+/Li, which makes it an ideal candidate as an anode material [
17,
18]. Nonetheless, the drawback of this material is its low cycling stability; its immense capacity is associated with a significant volumetric expansion (+280%) that compromises the mechanical stability of Si anodes [
19,
20]. The continuous expansion/contraction cycles during the lithiation/delithiation cycles lead to the thickening of the solid electrolyte interphase (SEI) and fracture of the Si particles, causing the electric disconnection of these particles [
18,
21]. This loss of active material causes a gradual capacity decrease with the number of cycles; thus, Si displays a limited cycle life [
16,
22]. Si is usually combined with graphite in small fractions to obtain a compromise between an increased energy density and an acceptable cycle life. Nevertheless, the exploitation of silicon as an active material cannot be based on the decrease in its concentration until it is functional; it is necessary to determine the reactions occurring in the material upon lithiation/delithiation to optimise its use.
Theoretically and at high temperatures, Si is sequentially lithiated from its original phase to crystalline phases Li
12Si
7, Li
7Si
3, Li
13Si
4, and Li
22Si
5, providing a total capacity of 4200 mAh·g
−1 [
23]. At room temperature and in real LIBs, however, Si undergoes a two-phase lithiation in which the intermediate phases are amorphous [
24]. By the end of the lithiation, no Li
22Si
5 is formed; the metastable and crystalline Li
15Si
4 is the silicon phase [
23]. The capacity that can be obtained with the lithiation of Si to Li
22Si
5 is 3579 mAh·g
−1 [
25]. During subsequent delithiation, the crystalline Li
15Si
4 is removed, and an amorphous phase is obtained [
23]. Thus, analysis by means of X-ray diffraction (XRD) can provide valuable information on the degree of lithiation of Si [
26].
Another key aspect in the development of high-energy LIBs is the format of the cells. There are three main categories: prismatic, cylindrical, and pouch [
4]. Cylindrical cells are the most widely implemented format [
27]. Their dense container helps to prevent deformation due to swelling in the presence of side reactions [
28]. They are defined with a numeric code (XXYYY) in which the first two numbers (XX) represent the diameter in mm, and the remaining numbers (YYY) represent the height of the cell in tenths of mm [
2] Among them, the 18650 cells are the most popular; these cells were initially manufactured by Sony for their cameras, and the length of 65 mm is due to space limitations in such a device designed to be held in the palm of a single adult hand [
29]. On the other hand, the diameter of 18 mm was selected due to safety reasons; it was determined as the maximum size to avoid thermal runaway for a cell of ~1 Ah capacity [
29]. Recently, TESLA has announced the shift to 4860 cylindrical cells, despite the safety issues that can arise [
30], which will probably have an impact on the cell size selected by other EV developers.
Even if cylindrical cells are the first option for industry, their low packing density and poor heat transport motivated battery developers to search for alternatives. Prismatic cells, with hard casings similar to those of cylindrical cells, provide safety towards swelling with increased packing density, but their energy density is ~20% lower than that of cylindrical cells [
5]. In any case, most manufacturers select this format in their EVs [
27]. In addition, it is believed that pouch cells (prismatic cells with a soft packaging) will be able to outperform their competitors, becoming the primary option in the near future [
28].
In this work, high-energy cells with 1.8 Ah capacity were assembled in two different formats to assess the impact of the cell design and casing/packaging: 18650 cylindrical and pouch. The anode consisted of a graphite/Si mix material, while the cathode comprised of an LFP/NMC532 blend. A combination of materials was utilised to increase the energy density of the electrodes through the addition of NMC532 and Si to the stable-cycle-life LFP and graphite, respectively. NMC532 was selected due to its good compromise between high capacity and stability at high voltages [
31]. Both types of cells were assembled using the same batch of electrodes and subjected to the same cycling protocols. The calendar ageing of some cells was investigated, while the cycling age of the other cells was studied using three different working voltages. Lastly, one cell was opened and characterised at the end of its cycle life.
2. Materials and Methods (Experimental)
2.1. Anode Manufacturing
The negative electrode of this work was prepared at CIDETEC’s electrode manufacturing line. The components of this electrode were nanoparticulate silicon (N-100, Tekna, Solli, Oslo) and graphite (MEG-2C, SGL Carbon, Meitingen, Germany) as anode active materials, Super C45 carbon (Imerys, Paris, France) as the conductive additive, carboxymethyl cellulose (CMC, Wallocel DOW, Midland, MI, USA) as the dispersant and binder, and styrene butadiene rubber (SBR, JSRmicro, Leuven, Belgium) as the co-binder. These components were mixed in a weight ratio of [Si/Gr/C45/CMC/SBR] = 10.4/74.6/5/5/5. The experiments performed to define the anode formulation are shown in
Figure S1.
The components were water-processed in a planetary mixer. In addition, the procedure was adapted to eliminate agglomerates via pre-dispersion of Si in the CMC solution and addition of the solids (C45 and graphite) at different steps and the SBR latex at the end. Unexpected low slurry pH was measured (pH ~3), which could affect the polymer (CMC and especially the SBR) conformation. Thus, the slurry pH was adjusted to pH = 6–7 by addition of ammonia (NH4OH). Finally, a mirror-like wet coating with minimal fish-eye spots, straight edges, two-side alignment within <1 mm, and targeted loading (2.54 mAh/cm2) of 3.7 mg/cm2 within 0.3 mg/cm2 deviation between faces was achieved.
Overall, 75 m was produced in two different coating widths (130 and 205 mm, onto 250 mm-width and 10 μm thick Cu foil, Schlenk, Roth, Germany) for each of the cell formats (cylindrical and pouch cells, respectively).
The anodes with 205 mm width for soft packaged pouch cells did not need slitting. Electrodes were die-cut directly (four anodes on 14 cm wide sheet) after calendering, for the stacked design of 100 × 61 mm coated area, by CIDETEC. The anode rolls manufactured in 130 mm width coating were slit by CEA for cylindrical cells. To limit waste, CEA used a lab slitting equipment to slit the anode coating.
The calendering step for the anodes was aimed at an expected optimum porosity of 32% (1.41 g/cm3). Control of flexibility performed by bending test (no damage when the electrode was wound on mandrels with decreasing diameter) revealed no cracks on the 2 mm diameter mandrel. This coupled with the 90° peel test strength (67 ± 2 N/m) provided satisfactory mechanical results with very high adhesion to the Cu current collector.
2.2. Cathode Manufacturing
The positive electrode in the current work was developed in CEA and then upscaled, adapting viscosity with coating equipment capability for 50 L of slurry and a coating machine with an oven of 5 m length (Megtec, De Pere, WI, USA). The positive electrode consisted of LiFePO4 (LFP, beLife, Dnipropetrovsk, Ukraine) and LiNi0.5Mn0.3Co0.2O2 (NMC532) as active materials, Super C65 carbon black (Imerys, Paris, France) as the conductive additive, and polyvinylidene fluoride (PVDF, Solvay Solef® 5130, Brussels, Belgium) as the binder. The weight ratio of these components was [LFP/NMC/C45/PVDF] = 45.25/45.25/5/4.5. Finally, CEA coated 380 m of two-sided electrode from the slurry onto an aluminium current collector of 20 µm thickness and a width of 30 cm (Hydro, Oslo, Norway). The loading of this coating was 14.4 mg/cm2 (2.3 mAh/cm2). During the slitting step, the electrode width was adjusted by cutting the coils. Then, the cathode was calendered to 36% porosity (2.3 g/cm3). After calendering, a control of flexibility (satisfactory at 4 mm diameter bending) and adhesion strength (260 ± 21 N/m) was applied.
2.3. Cell Manufacturing
In order to compare the two cell designs, both the cylindrical and the pouch cells consisting of the same components (except the separator which was specific to the assembly) were conditioned with the same protocol. The separator was a tri-layer Celgard 2325 grade (Charlotte, NC, USA) for the cylindrical hard-case cells, while the stacked soft packaging cells were assembled with a modified Celgard ECT-2015 grade (same thickness) suitable for the specific lamination/winding process on the cell-assembly line. The electrolyte was composed of ethylene carbonate and dimethyl carbonate (EC:DMC) in a volume proportion of 1:1 with 1 M lithium hexafluorophosphate (LiPF6) and a blend of additives: 10% fluoroethylene carbonate (FEC), 2% lithium bis(trifluoromethanesulfonyl)imide (LiTFSI), and 2% vinylene carbonate (VC). The electrolyte was purchased from UBE Industries.
2.3.1. Assembly of 18650 Cylindrical Cells
Cell assembly was performed on semiautomatic winding equipment inside a dry room with a dew point of −40 °C. Each cell consisted of a double-side coated 55 mm wide cathode and a 57 mm wide anode with two 60 mm wide separators. Electrodes and separators were wound around a mandrel, and the resultant jellyroll was dried in a vacuum oven overnight. After welding of the tabs on the bottom and the cap for the anode and cathode, respectively, and grooving, the cells were placed in an Ar-filled glove box for electrolyte filling and crimping. A picture of the components used for the assembly of 18650 cells is shown in
Figure S2.
2.3.2. Assembly of Pouch Cells
Electrodes were cut to size in a semiautomatic die-cutting unit (MTI Corp., Richmond, CA, USA) to 14 cm sheets from the electrode rolls. The cathodes and anodes were cut to different sizes (10 cm × 6.1 cm and 9.8 × 5.9 cm for the anodes and cathodes, respectively). Pictures of the die-cutting unit, a schematic representation of the cells, and a picture of the final cell are shown in
Figure S3.
The stacked soft-packaging cell was designed comprising eight cathodes and nine anodes per cell. The assembly was carried out in a dry room (dew point −50 °C) by manual stacking of the electrodes after vacuum-drying at 120–140 °C for 12 h. The process, using a guiding tool to guarantee stack alignment, is depicted by the photographic sequence in
Figure S4.
Electrode flanges (tabs) were ultrasonically welded to terminal tabs (100 μm thick Al (+) and Ni-plated Cu (−)) and then placed between two half-shells of aluminium laminated foil (ALF) pouch material (without depth-forming) and heat sealed on three sides before the filling step.
The cells were filled with 11.5 g (9 mL) of electrolyte, and the remaining side was thermally sealed under −850 mbar using a vacuum chamber sealer. The cells were then ready to be formed (see
Section 2.4) under external pressure applied by sandwiching the cell between two stainless-steel plates.
After this formation, the cells were degassed and finally sealed under full vacuum for grading characterisation.
2.4. Electrochemical Tests
2.4.1. Conditioning (Formation)
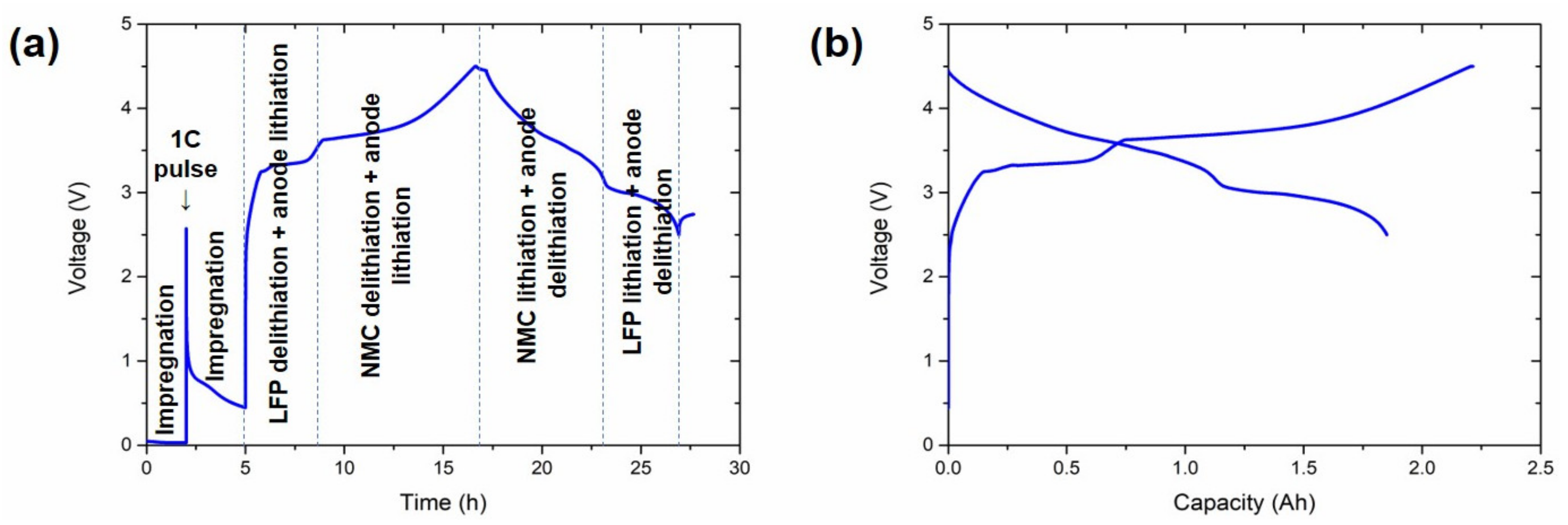
The conditioning experiments were performed inside a temperature chamber set at 45 °C. After a resting period of 2 h, a 1 C pre-charge pulse of 10 s was applied, which was followed by a 3 h rest period for the impregnation of the electrolyte. After that, a C/10 constant current cycle between 4.5 and 2.5 V was conducted, including a constant voltage step by the end of the charge at 4.5 V until the current decreased to C/20. Afterwards, the cells were removed from the chamber, waiting until their temperature dropped below 30 °C. After this formation, the pouch cells were degassed and finally sealed under full vacuum for grading characterisation.
2.4.2. Calendar Ageing
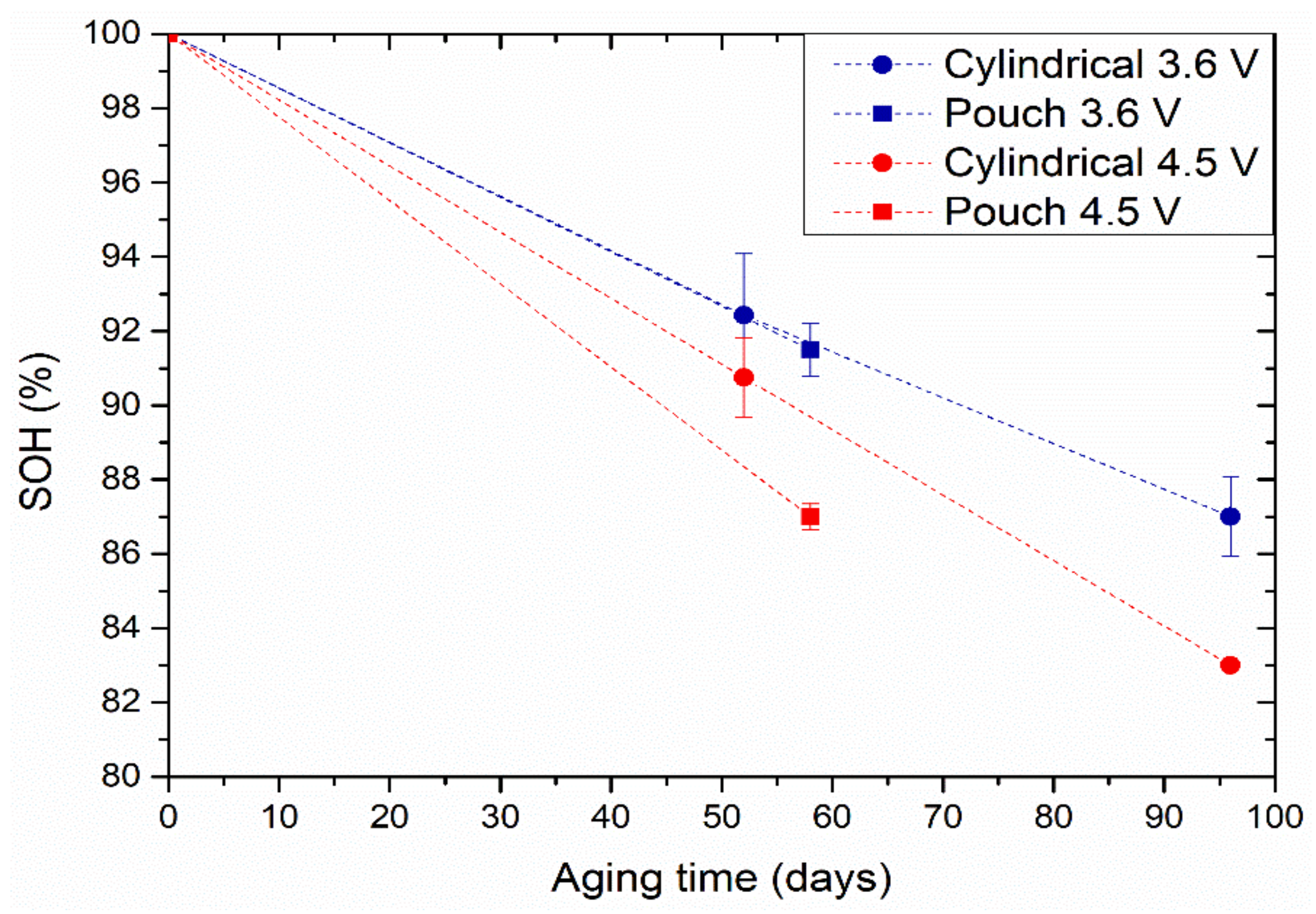
After conditioning, four cells per format were charged to 3.6 V (two cells per format) and 4.5 V (two cells per format) and stored at 25 °C for 58 days (for the pouch cells) and 96 days (cylindrical cells). The cells were kept at an open-circuit voltage state. The capacity evolution during calendar ageing was determined after two consecutive cycles with a 0.3 C charge and discharge current rate.
2.4.3. Electrochemical Ageing
The cycling ageing tests were applied on cylindrical and pouch cells. Cycle ageing was performed with a current of 0.3 C for both charging and discharging within three different voltage windows: (i) between the minimum and maximum voltage limits (2.5–4.5 V), which includes the transition between LFP and NMC and between two intermediate voltages, (ii) 3.6–4.5 V, and (iii) 2.5–4.5 V to investigate the ageing degradation on the different LFP and NMC voltage working range. Each test was carried out on two cells from the same batch to ensure the test result repeatability. Electrochemical tests were performed with a Basytec Cell Test System potentiostat at 25 °C ± 1 °C (at CIDETEC facilities, Donostia-San Sebastian, Spain), a Maccor cycler S4000 (at Helmholtz Institute Ulm, Ulm, Germany), and a PEC SBT0550 battery cycler (at CEA, Grenoble, France).
2.5. Post-Mortem Characterisation
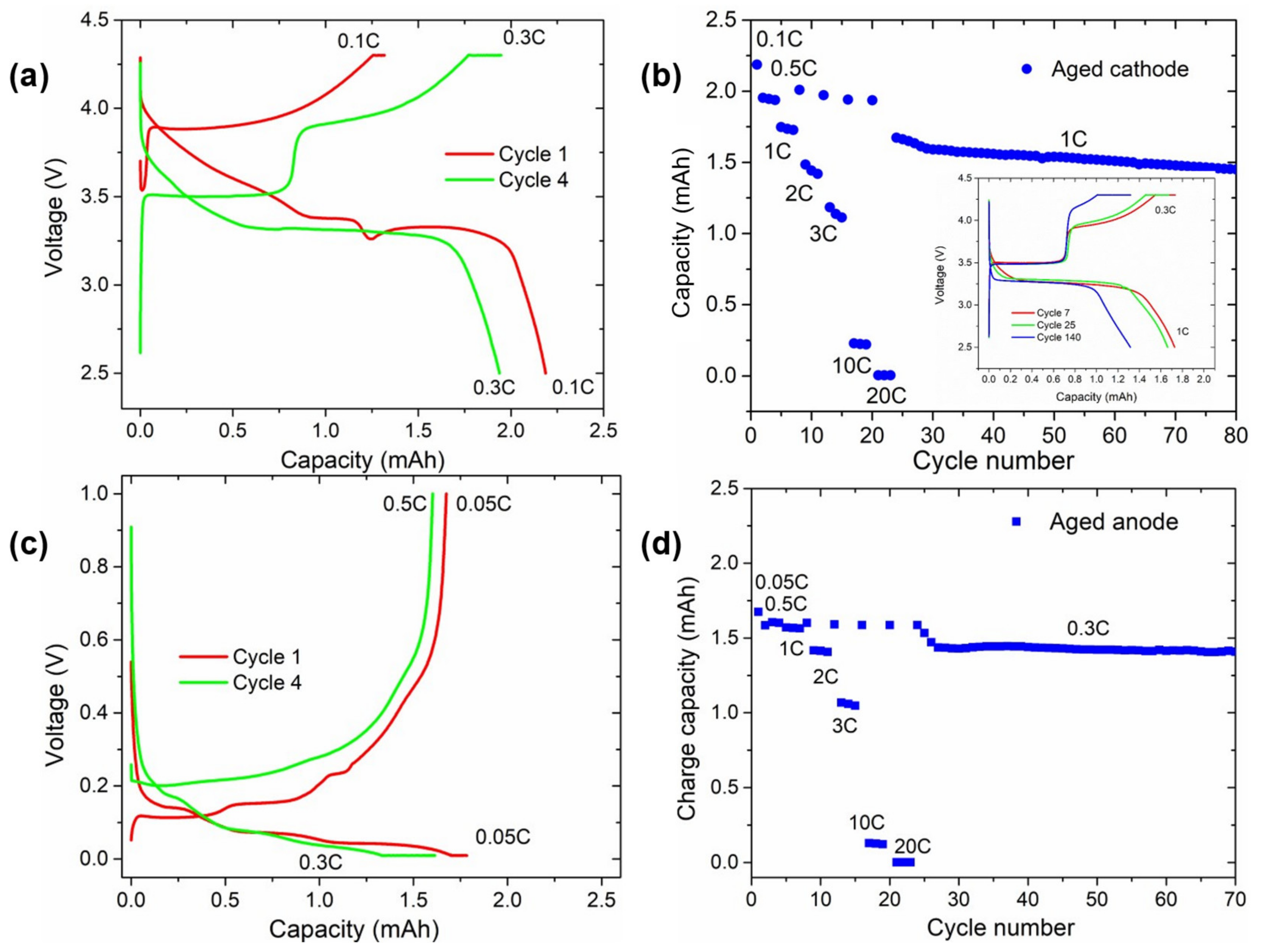
One cylindrical cell was opened to conduct post-mortem characterisation of its electrodes. This cell was previously cycled at 25 °C and within the voltage range 2.5 V–4.5 V until it reached 70% state of health (SOH) after 44 cycles. The cell was then fully discharged (0% state of charge, SOC) and introduced in an argon-filled glove box (MBraun, München, Germany) with O2 and H2O concentration below 1 ppm, respectively. The venting was pierced to evaluate the internal pressure, the free electrolyte was recovered by the venting, and the cell case was cut. Afterward, the electrode roll was extracted and unwound. The positive and negative electrodes were separated, and samples for post-mortem and extended electrochemical analyses were cut out from the middle part of the recovered sheets (avoiding the external parts of the electrodes). These samples were rinsed with DMC solvent; rinsing baths of solvent were used, in which each sample was soaked for approximately 30 s. Pristine samples were studied in parallel.
Scanning electron microscopy (SEM) imaging and energy-dispersive X-ray spectroscopy (EDX) was performed on a carbon-sputtered sample using a JSM 7600F (JEOL, Tokyo, Japan). The crystallographic analysis of the different samples was performed by means of powder XRD, using a Bruker D8 Discover diffractometer (Cu Kα radiation, λ = 0.154 nm, Billerica, MA, USA) equipped with a LynxEye PSD detector (Stockholm, Sweden). The diffractograms were recorded between 2θ = 10° and 80° at 0.003°·s−1. XRD and SEM analyses were performed using an inert transfer chamber to protect the sample from the external atmosphere.
Lastly, some samples were also used to assemble half coin cells (HCCs, CR2032 configuration) using lithium metal (Rockwood Lithium, 500 μm thick, Frankfurt, Germany) as the counter electrode. Electrodes of 1.13 cm2 were punched and assembled in an argon-filled glove box (H2O < 0.1 ppm, O2 < 0.1 ppm) versus lithium, using Whatman, GF/D separator (Maidstone, UK), and 120 μL of 1 M of LiPF6 in EC:DMC (1:1) + 10% FEC, 2% LiTFSI, and 2% VC electrolyte. These cells were subjected to two cycles at C/20 followed by a rate capability test and 150 cycles at C/3 (for the half coin cells with recovered negative electrode) or 1 C (for the half coin cells with recovered positive electrode). The tests were conducted at 20 °C. The potential windows for positive and negative electrode HCCs were 4.3 V–2.6 V and 1.0 V–10 mV, respectively.
2.6. Three-Electrode Cells
Three-electrode cells were assembled to monitor the potential of each of the electrodes upon galvanostatic cycling. Three-electrode Swagelok cells were assembled in an MBraun argon-filled glove box with oxygen and water contents below 1 ppm. Lithium metal foil (Rockwood Lithium, Frankfurt, Germany) was used as reference electrode along with glass fibre separators (Whatman, Cytiva, Maidstone, UK), soaked with 1 M LiPF6 in EC:DMC (1:1) + 10% FEC, 2% LiTFSI, and 2% VC electrolyte.
4. Discussion
Overall, the performance of pouch cells was lower than that of cylindrical cells in cycling and calendar ageing tests. This can be attributed to two different factors. On the one hand, the hard casing of cylindrical cells can prevent excessive volumetric expansion. Berckmans et al. evidenced that the application of external pressure can provide higher capacity and lower cell resistance with extended cycle life [
33]. They attributed this improvement to the limitation of the swelling of the Si particles. The coulombic efficiency was higher for cylindrical cells, evidencing the lower occurrence of irreversible reactions. In addition, calendar ageing experiments revealed that hard cylindrical casing was more efficient in avoiding capacity loss than soft pouch packaging. This could be associated with the easier deformability of the latter format, which could allow side reactions such as gassing. Although the pouch format could improve the packaging density of the cells, the energy and number of cycles that we could obtain from these cells remain unsatisfactory; hence, additional development is required, such as control of the optimum external pressure.
Furthermore, the electrochemical and post-mortem characterisation of the cells further support that the origin of the capacity fade of the cells could be attributed to the loss of cyclable Si. As discussed in
Section 1 and evidenced by the three-electrode cell, the lithiation of bulk crystalline silicon proceeded via a two-phase reaction (ca. 0.17 V) and involved the formation of amorphous Li
ySi. The complete lithiation resulted in crystalline Li
15Si
4. Reversing the process, amorphous Li
zSi was formed via a two-phase reaction (ca. 0.45 V), followed by the complete delithation (via a solid solution mechanism) of amorphous silicon. This phase could be relithiated to amorphous Li
xSi; while charging below 0.07 V vs Li
+/Li, the crystalline Li
15Si
4 was formed again. Rhodes et al. [
34] experimentally showed that the potential regions where extensive fracturing of Si takes place correspond to the two-phase reactions of Li
15Si
4. Therefore, the faster decay of the cells cycled down to 2.5 V, corresponding to extensive formation and dealloying of Li
15Si
4, could be partially attributed to the silicon containing anode. The three-electrode experiment showed a progressive loss of Si activity upon cycling at C/3 between 2.5 and 4.5 V, most probably due to the typical fracturing upon large volume expansion/contraction. On the contrary, when the discharge cut-off was limited to 3.6 V, the capacity retention was higher than when using 2.5 V as the cut-off limit. This was associated with silicon utilisation, with a lower working voltage of the cell resulting in higher silicon utilisation. Furthermore, it was observed that cycling in voltage windows of 2.5–3.6 V and 2.5–4.5 V had similar ageing effects. As a result, it appears that the low cut-off voltage was associated with a full silicon reaction that accelerated the capacity fade in both cylindrical and pouch cell formats.
The hypothesis of silicon governing the capacity fade of the cells was further confirmed by the post-mortem characterisation. On the one hand, disappearance of the Si signal from the XRD pattern of the post-mortem negative electrode indicated the degradation of this material, likely pulverised and disconnected from the anode. In addition, assembly of HCCs with aged electrodes suggested that the remaining active materials continued being electrochemically active, while Si was the sole unrecoverable material.
Consequently, it is necessary to study the stabilisation of silicon in the repetitive volumetric expansion/contraction during lithiation/delithiation cycles. Many efforts have been directed toward developing silicon/carbon composites that can buffer these volumetric changes, enhancing the cycle performance of the cells [
18,
35]. The use of silicon oxide (SiO
x) instead of Si can also be a good alternative; even if its capacity is lower (1200–1500 mAh·g
−1) [
36,
37], it undergoes a lower volumetric expansion and can be added in higher fractions in the anode formulation [
38].


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}