
QC and MD Modelling for Predicting the Electrochemical Stability Window of Electrolytes: New Estimating Algorithm


 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Theoretical Part
2.1.1. Molecular Dynamic Simulations
2.1.2. Quantum Chemical Calculations
2.2. Experimental
3. Results
3.1. Theoretical Studies
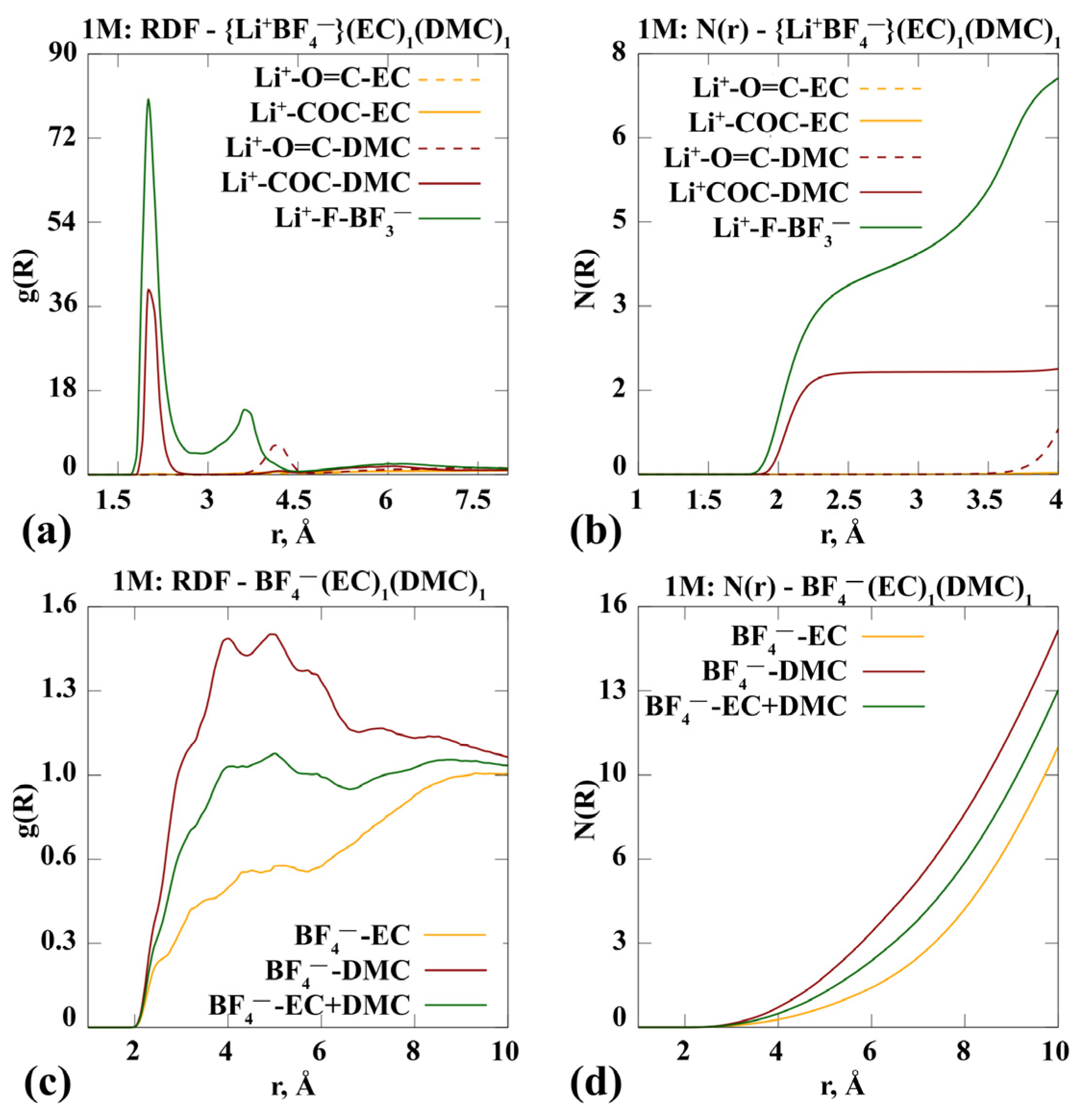
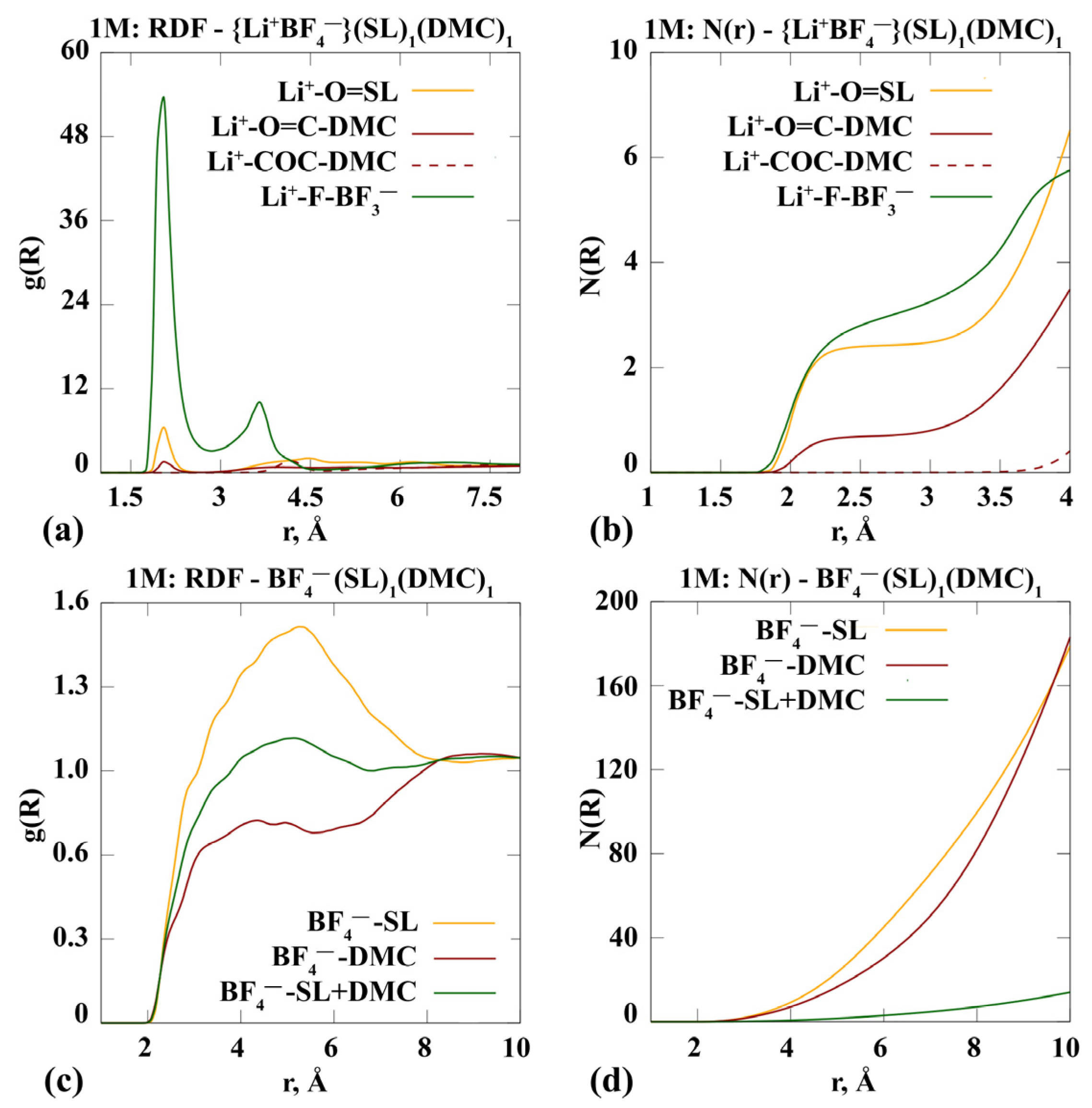
3.1.1. Molecular Dynamics
3.1.2. Quantum Chemical Calculations
- ✓ Cationic, in which solvent molecules are coordinated around a lithium cation;
- ✓ Anionic, where BF4− is also surrounded by solvent molecules and solvated ionic pairs, and where the LiBF4 salt is surrounded by EC, DMC or SL molecules.
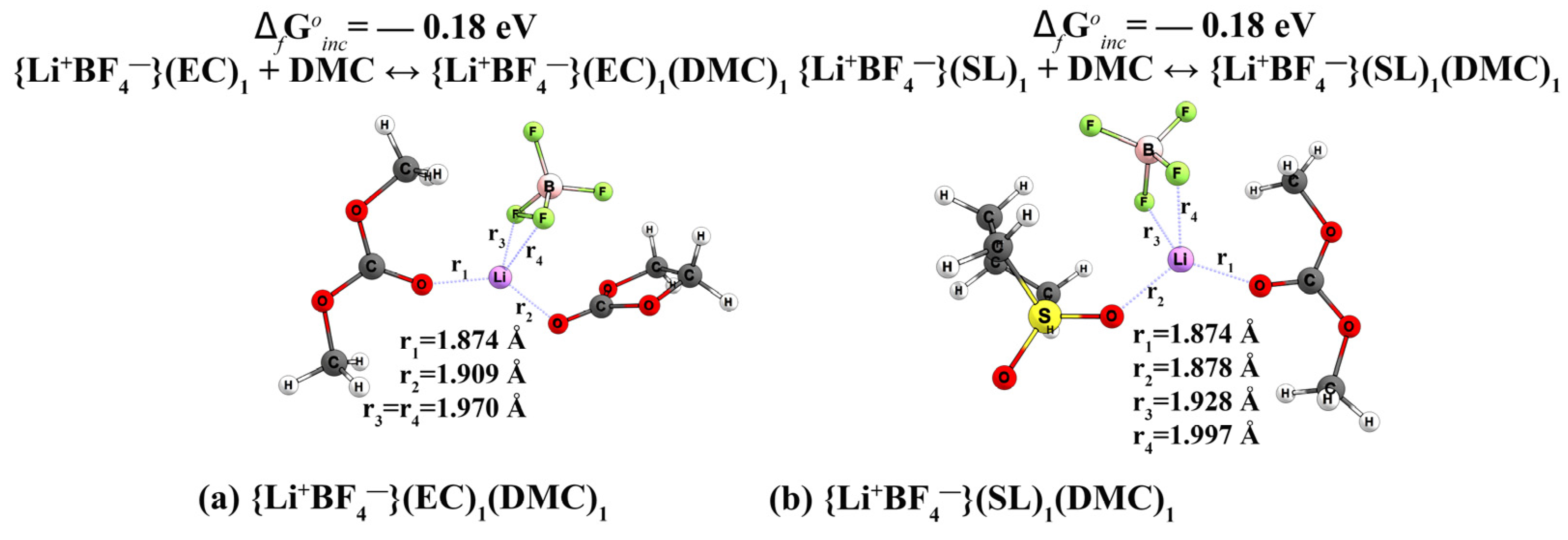
Structure and Thermodynamic Stability of Complexes
- Complexes of the 1 m {Li+BF4−} в EC/DMC (1:1)
- Complexes of the 1 m {Li+BF4−} в SL/DMC (1:1)
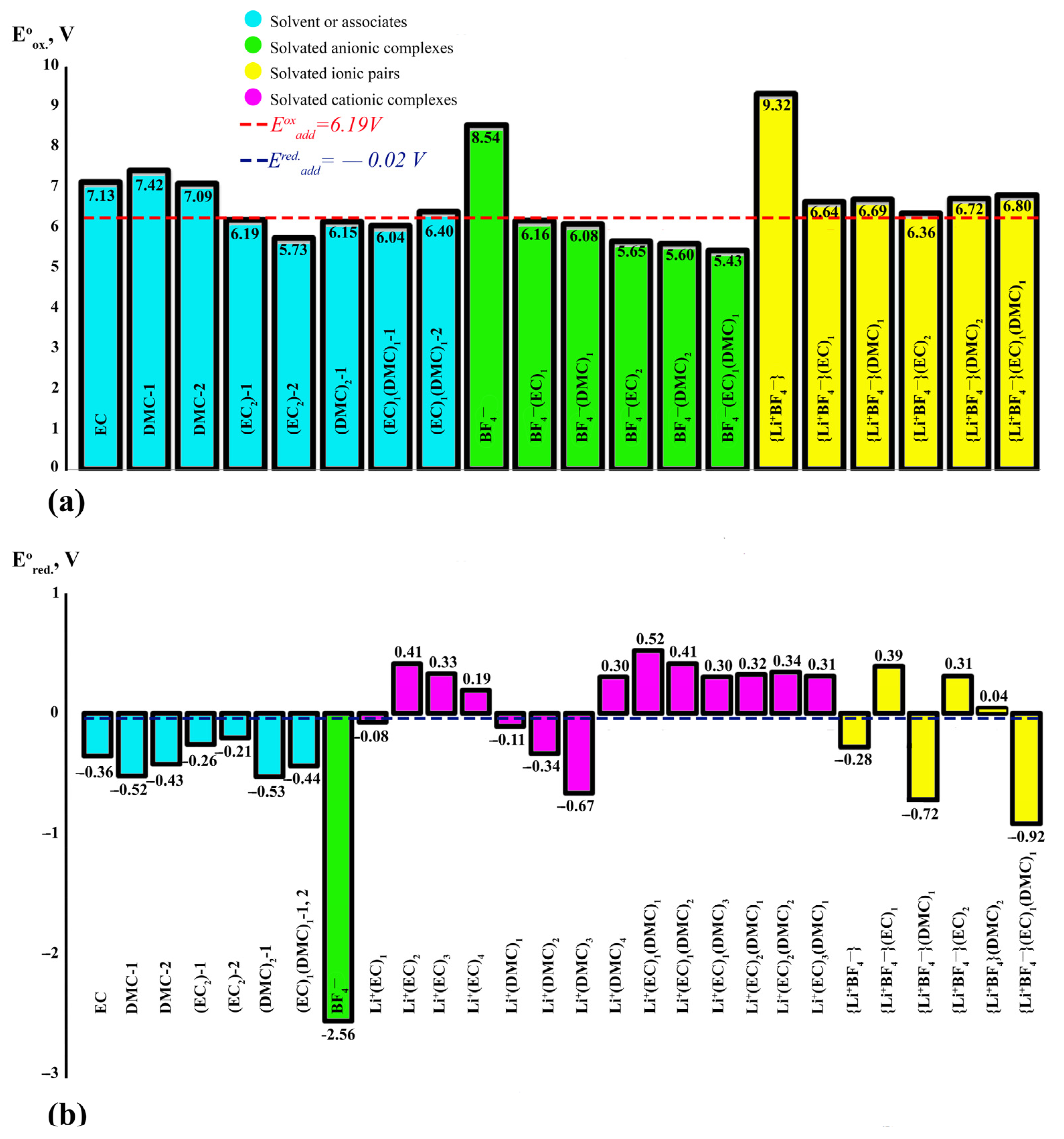
Electrochemical Stability
- Oxidation potentials
- Reduction potentials
- Oxidation potentials
- Reduction potentials
- Oxidation potentials
- Reduction potentials
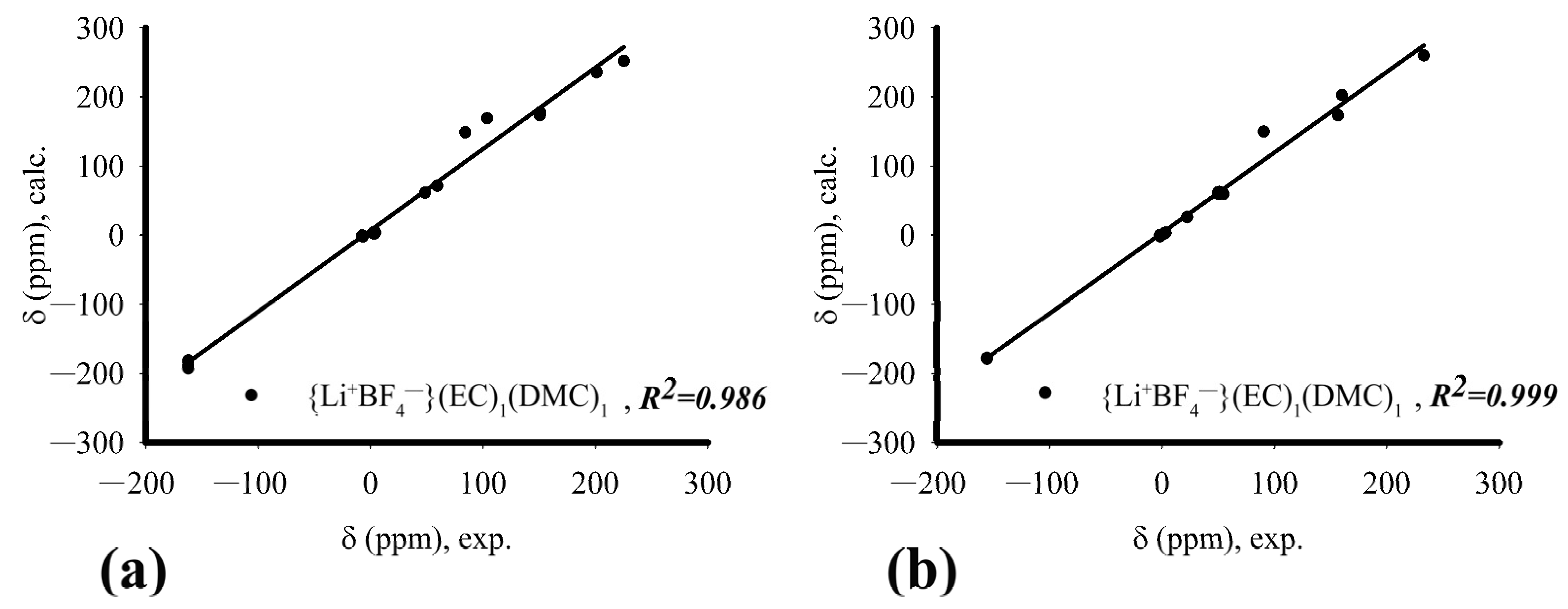
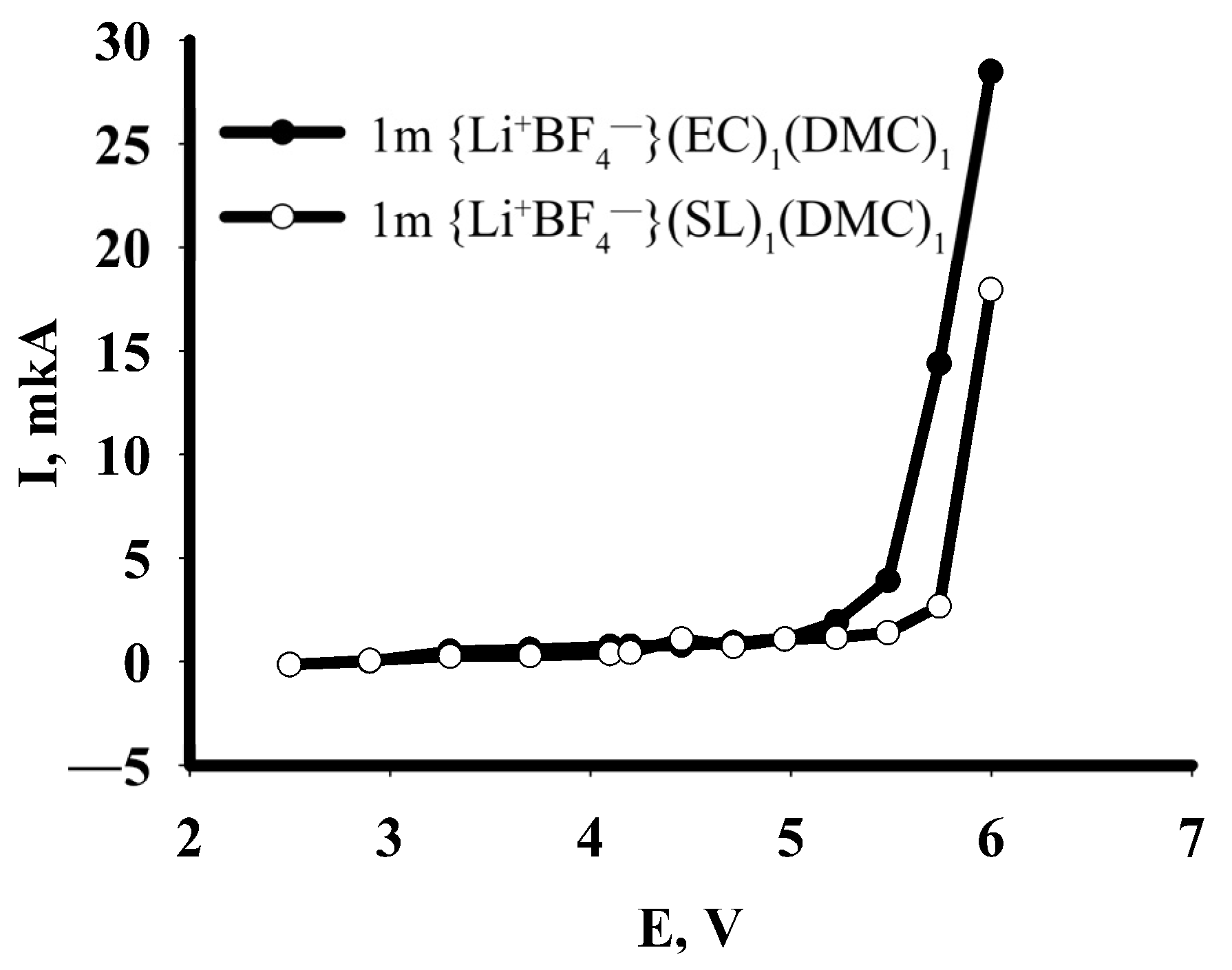
3.2. Experimental Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| ΔfGoin | incremental (stepped) Gibbs energy of formation (eV) |
| N | Maxwell–Boltzmann energy distribution |
| ΔEoxabs | adiabatic oxidation potential estimated in this work (M052X/TZVP) |
| ΔEox*abs-calc | adiabatic oxidation potential, published earlier, estimated based on quantum chemical calculations |
| ΔEox | experimentally measured value of the oxidation potential |
| ΔEredabs | adiabatic reduction potential estimated in this work |
| ΔEred*abs-calc | adiabatic reduction potential published earlier, which was estimated based on quantum chemical calculations |
| ΔEred | experimentally measured value of the reduction potential |
Abbreviations
| EC | ethylene carbonate |
| DMC | dimethyl carbonate |
| SL | sulfolane |
| LIB | lithium-ion battery |
| SEI | solid electrolyte layer |
| CEI | cathode–electrolyte interface |
| AOP | adiabatic oxidation potential |
| ARP | adiabatic reduction potential |
| OP | oxidation potential |
| RP | reduction potential |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Solvent or Complex | ΔfGoinc, eV | N, % | ΔEoxabs, V | ΔEoxabs-calc 1, V | ΔEoxabs-exp 2, V | N, % | ΔEredabs, V | ΔEredabs-calc 1, V | ΔEredabs-exp 2, V |
|---|---|---|---|---|---|---|---|---|---|---|
| Solvent or associates | ||||||||||
| 1 | EC | - | - | 7.13 | 5.58–8.50 [30,48,62,79,80] | 4.60–6.70 [57,64,82,83] | - | −0.36 | −0.32–0.90 [58,62,63] | <1 [30] 0.21 [65] 0.11, 1.94 [64] |
| 2 | DMC-1 | - | - | 7.42 | 5.62–7.60 [48,80] | 5.3–6.7 [57,58,64,82] | - | −0.52 | n/d 3 | n/d |
| 3 | DMC-2 | - | - | 7.09 | 5.62–7.13 [48,80] | n/d | - | −0.43 | n/d | n/d |
| 4 | (EC)2-1 | 0.33 | 6.5 | 6.19 | 5.94–6.00 [81] | n/d | 3.4 | −0.26 | n/d | n/d |
| 5 | (EC)2-2 | 0.25 | 6.7 | 5.73 | 5.91–5.90 [48] | 6.00–6.20 [58] | 3.5 | −0.21 | n/d | n/d |
| 6 | (DMC)2 | 0.13 | 7.0 | 6.15 | 5.84–6.13 [48] | n/d | 3.6 | −0.53 | n/d | n/d |
| 7 | (EC)1(DMC)1-1 | 0.16 | 3.5 | 6.04 | 5.95–7.70 [48] | 6.69 [84] | 2.0 | −0.44 | n/d | n/d |
| 8 | (EC)1(DMC)1-2 | 0.25 | 3.5 | 6.40 | n/d | n/d | 1.6 | −0.44 | n/d | n/d |
| Solvated anionic complexes | ||||||||||
| 9 | Non-solvent anion BF4− | - | - | 8.54 | 8.00–8.57 [48] | n/d | - | −2.56 | n/d | n/d |
| 10 | BF4− (EC)1 | 0.24 | 6.7 | 6.16 | 6.07–6.39 [48,60,79] | n/d | - | Non-optimised | n/d | n/d |
| 11 | BF4−(DMC)1 | 0.33 | 6.5 | 6.08 | 5.79–6.29 [48,79] | n/d | - | n/d | n/d | |
| 12 | BF4− (EC)2 | 0.24 | 6.7 | 5.65 | 6.30–6.46 [48] | n/d | - | n/d | n/d | |
| 13 | BF4−(DMC)2 | 0.33 | 6.5 | 5.60 | n/d | n/d | - | n/d | n/d | |
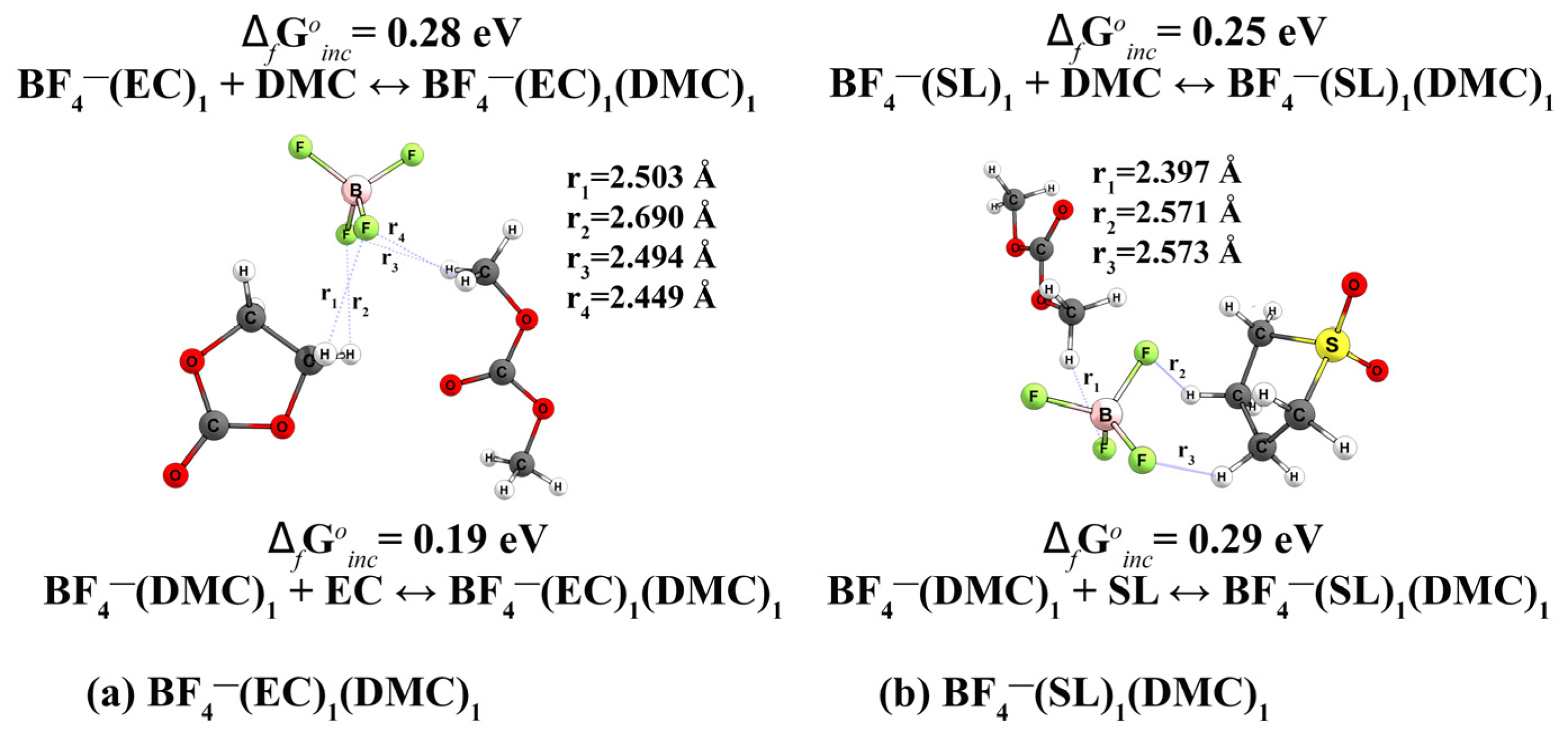
| 14 | BF4−(EC)1(DMC)1 (Reaction BF4−(EC)1 + DMC ↔ BF4−(EC)1(DMC)1) (Reaction BF4−(DMC)1 + EC ↔ BF4−(EC)1(DMC)1) | 0.28 0.19 | 6.7 | 5.43 | n/d | n/d | - | n/d | n/d | |
| Solvated cationic complexes | ||||||||||
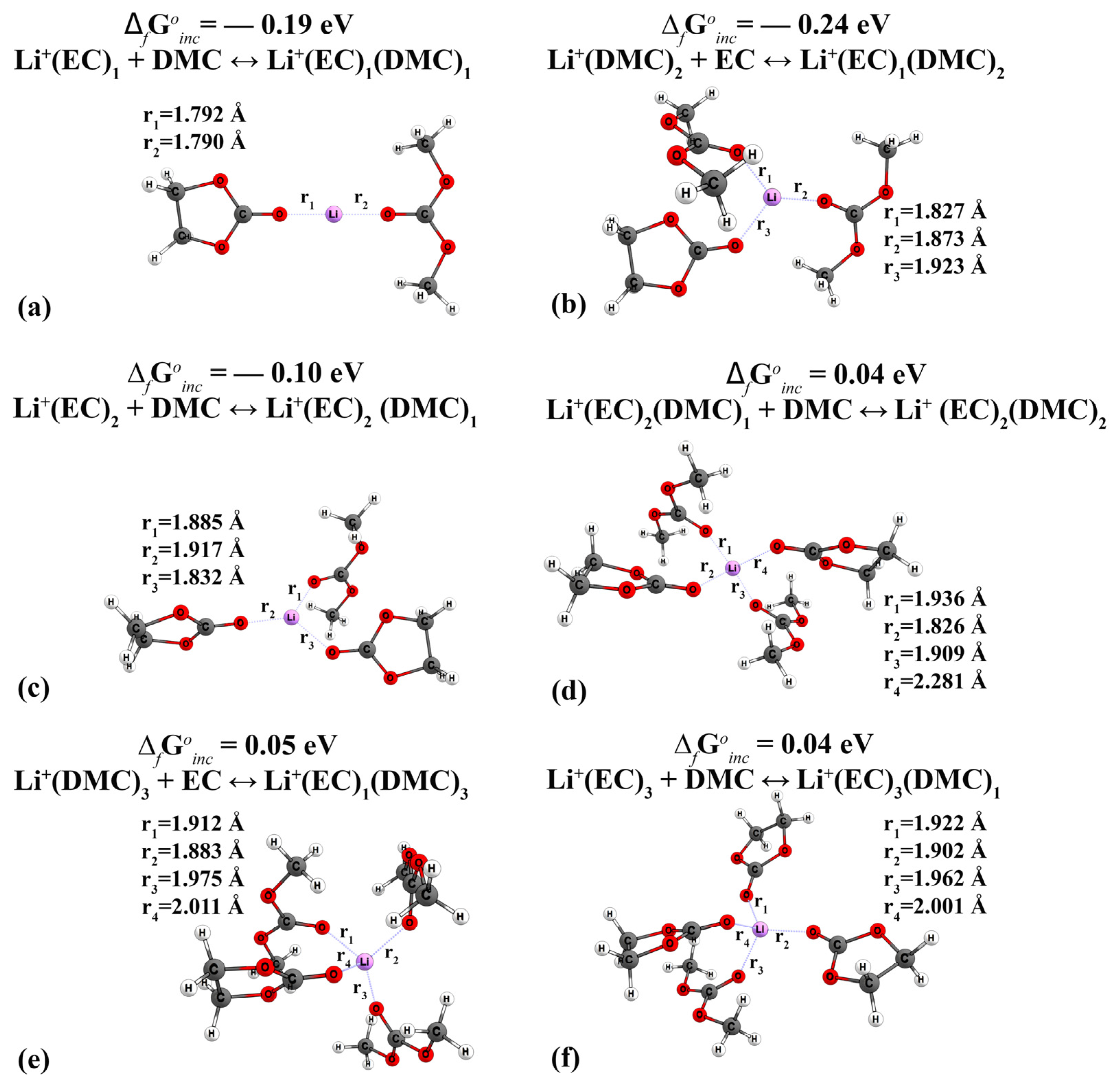
| 15 | Li+(EC)1 | −0.39 | - | - | n/d | n/d | 4.5 | −0.08 | 0.45–0.61 [27,49,60] | 0.54 [65] |
| 16 | Li+(EC)2 (Reaction Li+(EC)1 + EC ↔ Li+(EC)2) | −0.27 | - | - | n/d | n/d | 4.3 | 0.41 | n/d | n/d |
| 17 | Li+(EC)3 (Reaction Li+(EC)2 + EC ↔ Li+(EC)3) | −0.11 | - | - | n/d | n/d | 4.0 | 0.33 | n/d | n/d |
| 18 | Li+(EC)4 (Reaction Li+(EC)3 + EC ↔ Li+(EC)4) | −0.07 | - | - | n/d | n/d | 4.0 | 0.19 | n/d | 0.49 [65] |
| 19 | Li+(DMC)1 | −0.38 | - | - | n/d | n/d | 4.5 | −0.11 | 0.22–0.60 [27,60,61,66] | n/d |
| 20 | Li+(DMC)2 (Reaction Li+(DMC)1 + DMC ↔ Li+(DMC)2) | −0.13 | - | - | n/d | n/d | 4.0 | −0.34 | n/d | n/d |
| 21 | Li+(DMC)3 (Reaction Li+(DMC)2 + DMC ↔ Li+(DMC)3) | −0.21 | - | - | n/d | n/d | 4.2 | −0.67 | n/d | n/d |
| 22 | Li+(DMC)4 (Reaction Li+(DMC)3 + DMC ↔ Li+(DMC)4) | 0.07 | - | - | n/d | n/d | 3.7 | 0.30 | n/d | n/d |
| 23 | Li+(EC)1(DMC)1 (Reaction Li+(EC)1 + DMC ↔ Li+(EC)1(DMC)1) (Reaction Li+(DMC)1 + EC ↔ Li+(EC)1(DMC)1) | −0.19 −0.20 | - | - | n/d | n/d | 4.2 | 0.52 | n/d | n/d |
| 24 | Li+(EC)1(DMC)2 (Reaction Li+(DMC)2 + EC ↔ Li+(EC)1(DMC)2) (Reaction Li+(EC)1(DMC)1 + DMC ↔ Li+(EC)1(DMC)2) | −0.24 −0.17 | - | - | n/d | n/d | 4.2 | 0.41 | n/d | n/d |
| 25 | Li+(EC)1(DMC)3 (Reaction Li+(DMC)3 + EC ↔ Li+(EC)1(DMC)3) (Reaction Li+(EC)1(DMC)2 + DMC ↔ Li+(EC)1(DMC)3) | 0.05 0.07 | - | - | n/d | n/d | 3.7 | 0.30 | n/d | n/d |
| 26 | Li+(EC)2(DMC)1 (Reaction Li+(EC)2 + DMC ↔ Li+(EC)2 (DMC)1) (Reaction Li+(EC)1(DMC)1 + EC ↔ Li+(EC)2 (DMC)1) | −0.10 −0.18 | - | - | n/d | n/d | 4.1 | 0.32 | n/d | n/d |
| 27 | Li+(EC)2(DMC)2 (Reaction Li+(EC)2(DMC)1 + DMC ↔ Li+ (EC)2(DMC)2) (Reaction Li+ (EC)1(DMC)2 + EC ↔ Li+ (EC)2(DMC)2) | 0.04 0.03 | - | - | n/d | n/d | 3.8 | 0.34 | n/d | n/d |
| 28 | Li+(EC)3(DMC)1 (Reaction Li+(EC)3 + DMC ↔ Li+(EC)3(DMC)1) (Reaction Li+(EC)2DMC + EC ↔ Li+(EC)3(DMC)) | 0.04 0.03 | - | - | n/d | n/d | 3.8 | 0.31 | n/d | n/d |
| Solvated ionic pairs | ||||||||||
| 29 | Non-solvent ion pair {Li+BF4−} | −0.51 | - | 9.32 | - | 5.80 [91] | - | −0.28 | n/d | n/d |
| 30 | {Li+BF4−}(EC)1 | −0.19 | 8.0 | 6.64 | 6.64–8.74 [48] | n/d | 4.4 | 0.39 | n/d | n/d |
| 31 | {Li+BF4−}(DMC)1 | −0.26 | 8.2 | 6.69 | - | n/d | 4.5 | −0.72 | n/d | n/d |
| 32 | {Li+BF4−}(EC)2 (Reaction {Li+BF4−}(EC)1 + EC ↔ {Li+BF4−}(EC)2) | −0.06 | 7.6 | 6.36 | 6.60–6.72 [48] | n/d | 4.1 | 0.31 | n/d | n/d |
| 33 | {Li+BF4−}(DMC)2 (Reaction {Li+BF4−}(DMC)1 +DMC ↔ {Li+BF4−}(DMC)2) | −0.12 | 7.8 | 6.72 | n/d | n/d | 4.2 | 0.04 | n/d | n/d |
| 34 | {Li+BF4−}(EC)1(DMC)1 (Reaction {Li+BF4−}(EC)1+DMC ↔ {Li+BF4−}(EC)1(DMC)1) (Reaction {Li+BF4−}(DMC)1 +EC ↔ {Li+BF4−}(EC)1(DMC)1) | −0.18 −0.11 | 7.9 | 6.80 | n/d | n/d | 4.3 | −0.92 | n/d | n/d |
| All system | ||||||||||
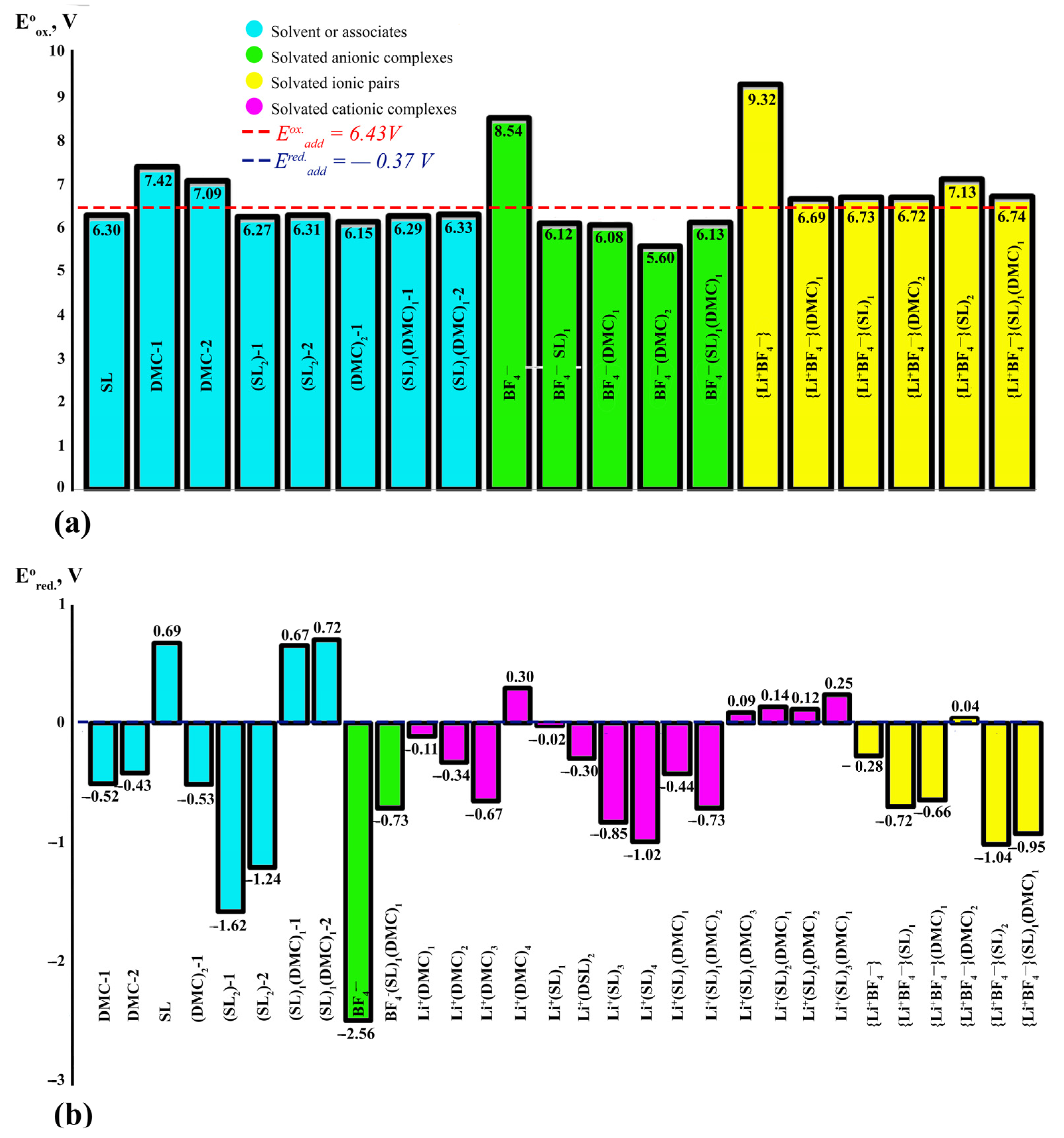
| Eox additive (calculation) = 6.19 V | Ered additive (calculation) = −0.02 V | |||||||||
| № | Solvent or Complex | ΔfGoinc, eV | N, % | ΔEoxabs, V | ΔEoxabs-calc, V | ΔEoxabs-exp, V | N, % | ΔEredabs, V | ΔEredabs-calc, V | ΔEredabs-exp, V |
|---|---|---|---|---|---|---|---|---|---|---|
| Solvent or associates | ||||||||||
| 1 | DMC-1 | - | - | 7.42 | 5.62–7.60 [48,80] | 5.3–6.7 [57,58,64,82] | - | −0.52 | n/d | n/d |
| 2 | DMC-2 | - | - | 7.09 | 5.62–7.13 [48,80] | n/d | - | −0.43 | n/d | n/d |
| 3 | SL | - | - | 6.30 | 6.64–6.74 [48] | 4.81 [91] 5.80 [83] | - | 0.69 | n/d | 0.40 [85] |
| 4 | (DMC)2 | 0.13 | 6.2 | 6.15 | 5.84–6.13 [48] | n/d | 3.5 | −0.53 | n/d | n/d |
| 5 | (SL)2-1 | 0.29 | 5.8 | 6.27 | n/d | n/d | 3.3 | −1.62 | n/d | n/d |
| 6 | (SL)2-2 | 0.29 | 5.8 | 6.31 | n/d | n/d | 3.3 | −1.24 | n/d | n/d |
| 7 | (SL)1(DMC)1-1 | 0.28 | 5.9 | 6.29 | n/d | 6.56 [64,84] 4.50 [64] | 3.3 | 0.67 | n/d | 0.10, 1.39 [64] |
| 8 | (SL)1(DMC)1-2 | 0.32 | 5.8 | 6.33 | n/d | n/d | 3.3 | 0.72 | n/d | n/d |
| Solvated anionic complexes | ||||||||||
| 9 | Non-solvent anion BF4− | - | - | 8.54 | 8.00–8.57 [48] | n/d | - | −2.56 | n/d | n/d |
| 10 | BF4−(DMC)1 | 0.33 | 5.7 | 6.08 | 5.79–6.29 [48,79] | n/d | - | Non-optimised | n/d | n/d |
| 11 | BF4− (SL)1 | 0.37 | 5.6 | 6.12 | 6.49, 5.68 [48,91] | 5.80 [92] | - | n/d | n/d | |
| 12 | BF4−(DMC)2 | 0.33 | 5.7 | 5.60 | n/d | n/d | - | n/d | n/d | |
| 13 | BF4− (SL)2 | 0.56 | 5.2 | 6.38 | n/d | n/d | - | n/d | n/d | |
| 14 | BF4−(SL)1(DMC)1 (Reaction BF4−(SL)1 + DMC ↔ BF4−(SL)1(DMC)1) (Reaction BF4−(DMC)1 + SL ↔ BF4−(SL)1(DMC)1) | 0.25 0.29 | 5.9 | 6.13 | n/d | n/d | - | −0.73 | n/d | n/d |
| Solvated cationic complexes | ||||||||||
| 15 | Li+(DMC)1 | −0.38 | - | - | n/d | n/d | 4.3 | −0.11 | n/d | n/d |
| 16 | Li+(DMC)2 (Reaction Li+(DMC)1 + DMC ↔ Li+(DMC)2) | −0.13 | - | - | n/d | n/d | 3.9 | −0.34 | n/d | n/d |
| 17 | Li+(DMC)3 (Reaction Li+(DMC)2 + DMC ↔ Li+(DMC)3) | −0.21 | - | - | n/d | n/d | 4.0 | −0.67 | n/d | n/d |
| 18 | Li+(DMC)4 (Reaction Li+(DMC)3 + DMC ↔ Li+(DMC)4) | 0.07 | - | - | n/d | n/d | 3.6 | 0.30 | n/d | n/d |
| 19 | Li+(SL)1 | −0.38 | - | - | n/d | n/d | 4.3 | −0.02 | n/d | n/d |
| 20 | Li+(SL)2 (Reaction Li+(SL)1 + SL ↔ Li+(SL)2) | −0.20 | - | - | n/d | n/d | 4.0 | −0.30 | n/d | n/d |
| 21 | Li+(SL)3 (Reaction Li+(SL)2 + SL ↔ Li+(SL)3) | −0.26 | - | - | n/d | n/d | 4.1 | −0.85 | n/d | n/d |
| 22 | Li+(SL)4 (Reaction Li+(SL)3 + SL ↔ Li+(SL)4) | −0.14 | - | - | n/d | n/d | 3.9 | −1.02 | n/d | n/d |
| 23 | Li+(SL)1(DMC)1 (Reaction Li+(SL)1 + DMC ↔ Li+(SL)1(DMC)1) Reaction Li+(DMC)1 + SL ↔ Li+(SL)1(DMC)1) | −0.24 −0.24 | - | - | n/d | n/d | 4.1 | −0.44 | n/d | n/d |
| 24 | Li+(SL)1(DMC)2 (Reaction Li+(DMC)2 + SL ↔ Li+(SL)1(DMC)2) (Reaction Li+(SL)1(DMC)1 + DMC ↔ Li+(SL)1(DMC)2 | −0.24 −0.14 | - | - | n/d | n/d | 4.0 | −0.73 | n/d | n/d |
| 25 | Li+(SL)1(DMC)3 (Reaction Li+(DMC)3 + SL ↔ Li+(SL)1(DMC)3) (Reaction Li+ (SL)1(DMC)2 + DMC ↔ Li+(SL)1(DMC)3) | −0.11 −0.08 | - | - | n/d | n/d | 0.09 | 0.09 | n/d | n/d |
| 26 | Li+(SL)2(DMC)1 (Reaction Li+(SL)2 + DMC ↔ Li+(SL)2(DMC)1) (Reaction Li+(SL)(DMC)1 + SL ↔ Li+(SL)2(DMC)1) | −0.31 −0.26 | - | - | n/d | n/d | 0.14 | 0.14 | n/d | n/d |
| 27 | Li+(SL)2(DMC)2 (Reaction Li+(SL)2(DMC) + DMC ↔ Li+(SL)2(DMC)2) (Reaction Li+(SL)(DMC)2 + SL ↔ Li+(SL)2(DMC)2) | −0.03 −0.16 | - | - | n/d | n/d | 0.12 | 0.12 | n/d | n/d |
| 28 | Li+(SL)3(DMC)1 (Reaction Li+(SL)3 + DMC ↔ Li+(SL)3(DMC)1) (Reaction Li+(SL)2(DMC)1 + SL ↔ Li+(SL)3(DMC)1) | −0.02 0.04 | - | - | n/d | n/d | 0.25 | 0.25 | n/d | n/d |
| Solvated ionic pairs | ||||||||||
| 29 | Non-solvent ion pair {Li+BF4−} | - | - | 9.32 | n/d | 5.80 [92] | - | −0.28 | n/d | n/d |
| 30 | {Li+BF4−}(DMC)1 | −0.26 | 7.3 | 6.69 | n/d | n/d | 4.1 | −0.72 | n/d | n/d |
| 31 | {Li+BF4−}(SL)1 | −0.25 | 7.3 | 6.73 | n/d | n/d | 4.1 | −0.66 | n/d | n/d |
| 32 | {Li+BF4−}(DMC)2 (Reaction {Li+BF4−}(DMC)1 +DMC ↔ {Li+BF4−}(DMC)2) | −0.12 | 6.9 | 6.72 | n/d | n/d | 3.9 | 0.04 | n/d | n/d |
| 33 | {Li+BF4−}(SL)2 (Reaction {Li+BF4−}(SL)1 + EC ↔ {Li+BF4−}(SL)2 | −0.22 | 7.2 | 7.13 | n/d | n/d | 4.1 | −1.04 | n/d | n/d |
| 34 | {Li+BF4−}(SL)1(DMC)1 (Reaction {Li+BF4−}(SL)1+DMC ↔ {Li+BF4−}(SL)1(DMC)1 (Reaction {Li+BF4−}(DMC)1 +SL ↔ {Li+BF4−}(SL)1(DMC)1 | −0.18 −0.16 | 7.0 | 6.74 | n/d | n/d | 4.0 | −0.95 | n/d | n/d |
| All system | ||||||||||
| Eox °additive (calculation) = 6.43 V | Ered°additive (calculation) = −0.37 V | |||||||||
References
- Choi, J.; Aurbach, D. Promise and Reality of Post-Lithium-Ion Batteries with High Energy Densities. Nat. Rev. Mater. 2016, 1, 16013. [Google Scholar] [CrossRef]
- Guo, K.; Qi, S.; Wang, H.; Huang, J.; Wu, M.; Yang, Y.; Li, X.; Ren, Y.; Ma, J. High-Voltage Electrolyte Chemistry for Lithium Batteries. Small Sci. 2022, 2. [Google Scholar] [CrossRef]
- Xiang, J.; Wei, Y.; Zhong, Y.; Yang, Y.; Cheng, H.; Yuan, L.; Xu, H.; Huang, Y. Building Practical High-voltage Cathode Materials for Lithium-ion Batteries. Adv. Mater. 2022, 2200912. [Google Scholar] [CrossRef]
- Lee, W.; Muhammad, S.; Sergey, C.; Lee, H.; Yoon, J.; Kang, Y.-M.; Yoon, W.-S. Advances in the Cathode Materials for Making a Breakthrough in the Li Rechargeable Batteries. Angew. Chem. 2019, 132. [Google Scholar] [CrossRef]
- Jung, S.K.; Kim, H.; Cho, M.G.; Cho, S.P.; Lee, B.; Kim, H.; Park, Y.U.; Hong, J.; Park, K.Y.; Yoon, G.; et al. Lithium-free transition metal monoxides for positive electrodes in lithium-ion batteries. Nat. Energy 2017, 2, 16208. [Google Scholar] [CrossRef]
- Wu, J.; Tsai, C.-J. Qualitative modeling of the electrolyte oxidation in long-term cycling of LiCoPO4 for high-voltage lithium-ion batteries. Electrochim. Acta 2021, 368, 137585. [Google Scholar] [CrossRef]
- Sreedeep, S.; Natarajan, S.; Aravindan, V. Recent advancements in LiCoPO4 cathodes using electrolyte additives. Curr. Opin. Electrochem. 2022, 31, 100868. [Google Scholar] [CrossRef]
- Han, J.-G.; Kim, K.; Lee, Y.; Choi, N.-S. Scavenging Materials to Stabilize LiPF6 -Containing Carbonate-Based Electrolytes for Li-Ion Batteries. Adv. Mater. 2019, 31, 1804822. [Google Scholar] [CrossRef]
- Xu, K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 2004, 104, 4303–4417. [Google Scholar] [CrossRef]
- Xu, K. Electrolytes and Interphases in Li-Ion Batteries and Beyond. Chem. Rev. 2014, 114, 11503–11618. [Google Scholar] [CrossRef]
- Aurbach, D.; Markovsky, B.; Salitra, G.; Markevich, E.; Talyossef, Y.; Koltypin, M.; Nazar, L.F.; Ellis, B.L.; Kovacheva, D.A. Review on electrode–electrolyte solution interactions, related to cathode materials for Li-ion batteries. J. Power Sources 2007, 165, 491–499. [Google Scholar] [CrossRef]
- Fan, X.; Wang, C. High-voltage liquid electrolytes for Li batteries: Progress and perspectives. Chem. Soc. Rev. 2021, 50, 10486–10566. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Petibon, R.; Xiong, D.; Ma, L.; Dahn, J. Enabling linear alkyl carbonate electrolytes for high voltage Li-ion cells. J. Power Sources 2016, 328, 124–135. [Google Scholar] [CrossRef]
- Nagasubramanian, G.; Orendorff, C.J. Hydrofluoroether electrolytes for lithium-ion batteries: Reduced gas decomposition and nonflammable. J. Power Sources 2011, 196, 8604–8609. [Google Scholar] [CrossRef]
- Achiha, T.; Nakajima, T.; Ohzawa, Y.; Koh, M.; Yamauchi, A.; Kagawa, M.; Aoyama, H. Thermal Stability and Electrochemical Properties of Fluorine Compounds as Nonflammable Solvents for Lithium-Ion Batteries. J. Electrochem. Soc. 2010, 157, A707. [Google Scholar] [CrossRef]
- Nanbu, N.; Takimoto, K.; Takehara, M.; Ue, M.; Sasaki, Y. Electrochemical properties of fluoropropylene carbonate and its application to lithium-ion batteries. Electrochem. Commun. 2008, 10, 783–786. [Google Scholar] [CrossRef]
- Lee, S.H.; Hwang, J.-Y.; Park, S.-J.; Park, G.-T.; Sun, Y.-K. Adiponitrile (C6H8N2): A New Bi-Functional Additive for High-Performance Li-Metal Batteries. Adv. Funct. Mater. 2019, 29, 1902496. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, K.; Ding, F.; Li, W.; Liu, X.; Zhang, J. Safety-Reinforced Succinonitrile-Based Electrolyte with Interfacial Stability for High-Performance Lithium Batteries. ACS Appl. Mater. Interfaces 2017, 9, 29820–29828. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, K.; Ding, F.; Li, W.; Liu, X.; Zhang, J. Enhancing the high voltage interface compatibility of LiNi0.5Co0.2Mn0.3O2 in the succinonitrile-based electrolyte. Electrochim. Acta 2019, 298, 818–826. [Google Scholar] [CrossRef]
- Hofmann, A.; Schulz, M.; Indris, S.; Heinzmann, R.; Hanemann, T. Mixtures of Ionic Liquid and Sulfolane as Electrolytes for Li-Ion Batteries. Electrochim. Acta 2014, 147, 704–711. [Google Scholar] [CrossRef]
- Xue, L.; Ueno, K.; Lee, S.-Y.; Angell, C.A. Enhanced performance of sulfone-based electrolytes at lithium ion battery electrodes, including the LiNi0.5Mn1.5O4 high voltage cathode. J. Power Sources 2014, 262, 123–128. [Google Scholar] [CrossRef]
- Sun, X.; Angell, C.A. Doped sulfone electrolytes for high voltage Li-ion cell applications. Electrochem. Commun. 2009, 11, 1418–1421. [Google Scholar] [CrossRef]
- Zhang, T.; Paillard, E. Recent advances toward high voltage, EC-free electrolytes for graphite-based Li-ion battery. Front. Chem. Sci. Eng. 2018, 12, 577–591. [Google Scholar] [CrossRef]
- Zuo, X.; Fan, C.; Liu, J.; Xiao, X.; Wu, J.; Nan, J. Lithium Tetrafluoroborate as an Electrolyte Additive to Improve the High Voltage Performance of Lithium-Ion Battery. J. Electrochem. Soc. 2013, 160, A1199–A1204. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, S.; Jow, T.R.; Xu, W.; Angell, C.A. LiBOB as Salt for Lithium-Ion Batteries: A Possible Solution for High Temperature Operation. Electrochem. Solid-State Lett. 2002, 5, A26. [Google Scholar] [CrossRef]
- Schedlbauer, T.; Krüger, S.; Schmitz, R.; Schmitz, R.W.; Schreiner, C.; Gores, H.J.; Passerini, S.; Winter, M. Lithium difluoro(oxalato)borate: A promising salt for lithium metal based secondary batteries? Electrochim. Acta 2013, 92, 102–107. [Google Scholar] [CrossRef]
- Delp, S.A.; Borodin, O.; Olguin, M.; Eisner, C.G.; Allen, J.L.; Jow, T.R. Importance of Reduction and Oxidation Stability of High Voltage Electrolytes and Additives. Electrochim. Acta 2016, 209, 498–510. [Google Scholar] [CrossRef]
- Ravdel, B.; Abraham, K.M.; Gitzendanner, R.; DiCarlo, J.; Lucht, B.; Campion, C. Thermal stability of lithium-ion battery electrolytes. J. Power Sources 2003, 119–121, 805–810. [Google Scholar] [CrossRef]
- Ellis, L.; Hill, I.; Gering, K.; Dahn, J. Synergistic Effect of LiPF6 and LiBF4 as Electrolyte Salts in Lithium-Ion Cells. J. Electrochem. Soc. 2017, 164, A2426–A2433. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.; Yang, L.; Lu, H.; Liu, H.; Zheng, B. Exploring the redox decomposition of ethylene carbonate–propylene carbonate in Li-ion batteries. Mater. Adv. 2021, 2, 1747–1751. [Google Scholar] [CrossRef]
- Doi, T.; Shimizu, Y.; Hashinokuchi, M.; Inaba, M. LiBF4 -Based Concentrated Electrolyte Solutions for Suppression of Electrolyte Decomposition and Rapid Lithium-Ion Transfer at LiNi0.5Mn1.5O4 /Electrolyte Interface. J. Electrochem. Soc. 2016, 163, A2211–A2215. [Google Scholar] [CrossRef]
- Doi, T.; Shimizu, Y.; Hashinokuchi, M.; Inaba, M. Dilution of Highly Concentrated LiBF4 /Propylene Carbonate Electrolyte Solution with Fluoroalkyl Ethers for 5-V LiNi0.5Mn1.5O4 Positive Electrodes. J. Electrochem. Soc. 2017, 164, A6412–A6416. [Google Scholar] [CrossRef]
- Ma, T.; Xu, G.-L.; Li, Y.; Wang, L.; He, X.; Zheng, J.; Liu, J.; Engelhard, M.H.; Zapol, P.; Curtiss, L.A.; et al. Revisiting the Corrosion of the Aluminum Current Collector in Lithium-Ion Batteries. J. Phys. Chem. Lett. 2017, 8, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Porcher, W.; Paillard, E. Towards practical sulfolane based electrolytes: Choice of Li salt for graphite electrode operation. J. Power Sources 2018, 395, 212–220. [Google Scholar] [CrossRef]
- Liu, T.; Han, X.; Zhang, Z.; Chen, Z.; Wang, P.; Han, P.; Ding, N.; Cui, G. A high concentration electrolyte enables superior cycleability and rate capability for high voltage dual graphite battery. J. Power Sources 2019, 437, 226942. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, J.; Yang, Q.; Wang, S.; Wang, W.; Li, B. Stable Cycling of High-Voltage Lithium-Metal Batteries Enabled by High-Concentration FEC-Based Electrolyte. ACS Appl. Mater. Interfaces 2020, 12, 22901–22909. [Google Scholar] [CrossRef]
- Yamada, Y.; Wang, J.; Ko, S.; Watanabe, E.; Yamada, A. Advances and issues in developing salt-concentrated battery electrolytes. Nat. Energy 2019, 4, 269–280. [Google Scholar] [CrossRef]
- Sheina, L.V.; Kuz’mina, E.V.; Karaseva, E.V.; Gallyamov, A.G.; Prosochkina, T.R.; Kolosnitsyn, V.S. Thermochemical and Electrochemical Stability of Electrolyte Systems based on Sulfolane. Russ. J. Appl. Chem. 2018, 91, 1427–1433. [Google Scholar] [CrossRef]
- Etacheri, V.; Marom, R.; Elazari, R.; Salitra, G.; Aurbach, D. Challenges in the development of advanced Li-ion batteries: A review. Energy Environ. Sci. 2011, 4, 3243–3262. [Google Scholar] [CrossRef]
- Yamada, Y.; Yamada, A. Review—Superconcentrated Electrolytes for Lithium Batteries. J. Electrochem. Soc. 2015, 162, A2406–A2423. [Google Scholar] [CrossRef]
- Xing, L.; Tu, W.; Vatamanu, J.; Liu, Q.; Huang, W.; Wang, Y.; Zhou, H.; Zeng, R.-H.; Li, W. On anodic stability and decomposition mechanism of sulfolane in high-voltage lithium ion battery. Electrochim. Acta 2014, 133, 117–122. [Google Scholar] [CrossRef]
- Wang, X.; Xue, W.; Hu, K.; Li, Y.; Li, Y.; Huang, R. Adiponitrile as Lithium-Ion Battery Electrolyte Additive: A Positive and Peculiar Effect on High-Voltage Systems. ACS Appl. Energy Mater. 2018, 1, 5347–5354. [Google Scholar] [CrossRef]
- Raberg, J.; Vatamanu, J.; Harris, S.; van Oversteeg, C.; Ramos, A.; Borodin, O.; Cuk, T. Probing Electric Double Layer Composition via in-Situ Vibrational Spectroscopy and Molecular Simulations. J. Phys. Chem. Lett. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Ueno, K.; Watanabe, D.; Ugata, Y.; Matsumae, Y.; Liu, J.; Thomas, M.L.; Dokko, K.; Watanabe, M. Sulfolane-Based Highly Concentrated Electrolytes of Lithium Bis(trifluoromethanesulfonyl)amide: Ionic Transport, Li-Ion Coordination, and Li–S Battery Performance. J. Phys. Chem. C 2019, 123, 14229–14238. [Google Scholar] [CrossRef]
- Lin, F.-W.; Tran, N.T.T.; Hsu, W.-D. Effect of 1,3-Propane Sultone on the Formation of Solid Electrolyte Interphase at Li-Ion Battery Anode Surface: A First-Principles Study. ACS Omega 2020, 5, 13541–13547. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.; Liu, Z.; Zheng, B. Interaction Mechanisms between Lithium Polysulfides/Sulfide and Small Organic Molecules. ACS Omega 2021, 6, 4995–5000. [Google Scholar] [CrossRef]
- Jiang, Z.; Rappe, A.M. Structure, Diffusion, and Stability of Lithium Salts in Aprotic Dimethyl Sulfoxide and Acetonitrile Electrolytes. J. Phys. Chem. C 2022, 126, 10266–10272. [Google Scholar] [CrossRef]
- Borodin, O.; Behl, W.; Jow, T.R. Oxidative Stability and Initial Decomposition Reactions of Carbonate, Sulfone, and Alkyl Phosphate-Based Electrolytes. J. Phys. Chem. C 2013, 117, 8661–8682. [Google Scholar] [CrossRef]
- Borodin, O. Challenges with Prediction of Battery Electrolyte Electrochemical Stability Window and Guiding the Electrode—Electrolyte Stabilization. Curr. Opin. Electrochem. 2018, 13. [Google Scholar] [CrossRef]
- Xu, K.; Zhuang, G.V.; Allen, J.L.; Lee, U.; Zhang, S.S.; Ross, P.N.; Jow, T.R. Syntheses and Characterization of Lithium Alkyl Mono- and Dicarbonates as Components of Surface Films in Li-Ion Batteries. J. Phys. Chem. B 2006, 110, 7708–7719. [Google Scholar] [CrossRef]
- Watanabe, Y.; Kinoshita, S.-i.; Wada, S.; Hoshino, K.; Morimoto, H.; Tobishima, S.-I. Electrochemical properties and lithium ion solvation behavior of sulfone--ester mixed electrolytes for high-voltage rechargeable lithium cells. J. Power Sources 2008, 179, 770–779. [Google Scholar] [CrossRef]
- Nazri, M. Liquid electrolytes: Some theoretical and practical aspects. In Lithium Batteries; Springer: Boston, MA, USA, 2003; pp. 509–529. [Google Scholar] [CrossRef]
- Abouimrane, A.; Belharouak, I.; Amine, K. Sulfone-based electrolytes for high-voltage Li-ion batteries. Electrochem. Commun. 2009, 11, 1073–1076. [Google Scholar] [CrossRef]
- Li, S.; Zhao, D.; Wang, P.; Cui, X.; Tang, F. Electrochemical effect and mechanism of adiponitrile additive for high-voltage electrolyte. Electrochim. Acta 2016, 222, 668–677. [Google Scholar] [CrossRef]
- Lu, Y.; Xu, S.; Shu, J.B.; Aladat, W.I.A.; Archer, L.A. High voltage LIB cathodes enabled by salt-reinforced liquid electrolytes. Electrochem. Commun. 2015, 51, 23–26. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models; John Wiley and Sons: West Sussex, UK; New York, NY, USA, 2002. [Google Scholar] [CrossRef]
- Abe, K.; Ushigoe, Y.; Yoshitake, H.; Yoshio, M. Functional electrolytes: Novel type additives for cathode materials, providing high cycleability performance. J. Power Sources 2006, 153, 328–335. [Google Scholar] [CrossRef]
- Abe, K.; Hattori, T.; Kawabe, K.; Ushigoe, Y.; Yoshitake, H. Functional Electrolytes Triple-Bonded Compound as an Additive for Negative Electrode. J. Electrochem. Soc. 2007, 154. [Google Scholar] [CrossRef]
- Curtiss, L.; Redfern, P.; Raghavachari, K. Gaussian-4 theory using reduced order perturbation theory. J. Chem. Phys. 2007, 127, 124105. [Google Scholar] [CrossRef]
- Borodin, O.; Olguin, M.; Spear, C.; Leiter, K.W.; Knap, J. Towards high throughput screening of electrochemical stability of battery electrolytes. Nanotechnology 2015, 26. [Google Scholar] [CrossRef]
- Borodin, O.; Ren, X.; Vatamanu, J.; von Wald Cresce, A.; Knap, J.; Xu, K. Modeling Insight into Battery Electrolyte Electrochemical Stability and Interfacial Structure. Acc. Chem. Res. 2017, 50, 2886–2894. [Google Scholar] [CrossRef]
- Han, Y.-K.; Lee, K.; Yoo, J.; Huh, Y.S. Virtual screening of borate derivatives as high-performance additives in lithium-ion batteries. Theor. Chem. Acc. 2014, 133. [Google Scholar] [CrossRef]
- Han, Y.-K.; Yoo, J.; Yim, T. Computational screening of phosphite derivatives as high-performance additives in high-voltage Li-ion batteries. RSC Adv. 2017, 7, 20049–20056. [Google Scholar] [CrossRef]
- Xu, K.; Ding, S.P.; Jow, T.R. Toward Reliable Values of Electrochemical Stability Limits for Electrolytes. J. Electrochem. Soc. 1999, 146, 4172–4178. [Google Scholar] [CrossRef]
- Hou, T.; Yang, G.; Rajput, N.N.; Self, J.; Park, S.-W.; Nanda, J.; Persson, K.A. The influence of FEC on the solvation structure and reduction reaction of LiPF6/EC electrolytes and its implication for solid electrolyte interphase formation. Nano Energy 2019, 64, 103881. [Google Scholar] [CrossRef]
- Borodin, O.; Olguin, M.; Spear, C.; Leiter, K.; Knap, J.; Yushin, G.; Childs, A.; Xu, K. (Invited) Challenges with Quantum Chemistry-Based Screening of Electrochemical Stability of Lithium Battery Electrolytes. ECS Trans. 2015, 69, 113–123. [Google Scholar] [CrossRef]
- Sanginov, E.A.; Borisevich, S.S.; Kayumov, R.R.; Istomina, A.S.; Evshchik, E.Y.; Reznitskikh, O.G.; Yaroslavtseva, T.V.; Melnikova, T.I.; Dobrovolsky, Y.A.; Bushkova, O.V. Lithiated Nafion plasticised by a mixture of ethylene carbonate and sulfolane. Electrochim. Acta 2021, 373, 137914. [Google Scholar] [CrossRef]
- Shaw, D.E. Schrödinger: Desmond Molecular Dynamics System: 2021-4; DE Shaw Research: New York, NY, USA; Maestro-Desmond Interoperability Tools Schrödinger: New York, NY, USA, 2021. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Ikeguchi, M. Partial rigid-body dynamics in NPT, NPAT and NPγT ensembles for proteins and membranes. J. Comput. Chem. 2004, 25, 529–541. [Google Scholar] [CrossRef]
- Tildesley, D.; Allen, M. Computer Simulation of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar] [CrossRef]
- March, N.H.; Tosi, M.P. Atomic Dynamics in Liquids; Courier Corporation: North Chelmsford, MA, USA, 1991. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Borodin, O.; Jow, T.R. Quantum Chemistry Study of the Oxidation-Induced Decomposition of Tetramethylene Sulfone (TMS) Dimer and TMS/BF4-. ECS Trans. 2013, 50, 391–398. [Google Scholar] [CrossRef]
- Zhang, X.; Pugh, J.K.; Ross, P.N.J. Computation of Thermodynamic Oxidation Potentials of Organic Solvents Using Density Functional Theory. J. Electrochem. Soc. 2001, 148. [Google Scholar] [CrossRef]
- Xing, L.; Borodin, O. Oxidation induced decomposition of ethylene carbonate from DFT calculations—Importance of explicitly treating surrounding solvent. Phys. Chem. Chem. Phys. 2012, 14, 12838–12843. [Google Scholar] [CrossRef] [PubMed]
- Azcarate, I.; Yin, W.; Méthivier, C.; Ribot, F.; Laberty-Robert, C.; Grimaud, A. Assessing the Oxidation Behavior of EC:DMC Based Electrolyte on Non- Catalytically Active Surface. J. Electrochem. Soc. 2020, 167, 080530. [Google Scholar] [CrossRef]
- Abe, K.; Miyoshi, K.; Hattori, T.; Ushigoe, Y.; Yoshitake, H. Functional electrolytes: Synergetic effect of electrolyte additives for lithium-ion battery. J. Power Sources 2008, 184, 449–455. [Google Scholar] [CrossRef]
- Ue, M.; Takeda, M.; Takehara, M.; Mori, S. Electrochemical Properties of Quaternary Ammonium Salts for Electrochemical Capacitors. J. Electrochem. Soc. 1997, 144, 2684–2688. [Google Scholar] [CrossRef]
- Alvarado, J.; Schroeder, M.; Zhang, M.; Borodin, O.; Gobrogge, E.; Olguin, M.; Ding, M.; Gobet, M.; Greenbaum, S.; Meng, Y.; et al. A carbonate-free, sulfone-based electrolyte for high-voltage Li-ion batteries. Mater. Today 2018, 21, 341–353. [Google Scholar] [CrossRef]
- Nie, M.; Lucht, B.L. Role of Lithium Salt on Solid Electrolyte Interface (SEI) Formation and Structure in Lithium Ion Batteries. J. Electrochem. Soc. 2014, 161, A1001. [Google Scholar] [CrossRef]
- Agubra, V.A.; Fergus, J.W. The formation and stability of the solid electrolyte interface on the graphite anode. J. Power Sources 2014, 268, 153–162. [Google Scholar] [CrossRef]
- Li, R.; Kamali, A.R. Molten salt assisted conversion of corn lignocellulosic waste into carbon nanostructures with enhanced Li-ion storage performance. Chem. Eng. Sci. 2023, 265, 118222. [Google Scholar] [CrossRef]
- Kamali, A.R.; Haghighat-Shishavan, S.; Nazarian-Samani, M.; Rezaei, A.; Kim, K.-B. Ultra-fast shock-wave combustion synthesis of nanostructured silicon from sand with excellent Li storage performance. Sustain. Energy Fuels 2019, 3, 1396–1405. [Google Scholar] [CrossRef]
- Zhu, W.; Kamali, A.R. Green preparation of nanostructured β-MoO3/hexagonal-shaped MoS2/graphene with enhanced lithium-ion storage performance. J. Alloys Compd. 2023, 932, 167724. [Google Scholar] [CrossRef]
- Kolosnitsyn, V.S.; Sheina, L.V.; Mochalov, S.E. Physicochemical and electrochemical properties of sulfolane solutions of lithium salts. Russ. J. Electrochem. 2008, 44, 575–578. [Google Scholar] [CrossRef]
- Borodin, O.; Jow, T.R. Quantum Chemistry Studies of the Oxidative Stability of Carbonate, Sulfone and Sulfonate-Based Electrolytes Doped with BF4-, PF6- Anions. ECS Trans. 2011, 33, 77–84. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobrovolsky, Y.A.; Ilyina, M.G.; Evshchik, E.Y.; Khamitov, E.M.; Chernyak, A.V.; Shikhovtseva, A.V.; Melnikova, T.I.; Bushkova, O.V.; Borisevich, S.S. QC and MD Modelling for Predicting the Electrochemical Stability Window of Electrolytes: New Estimating Algorithm. Batteries 2022, 8, 292. https://doi.org/10.3390/batteries8120292
Dobrovolsky YA, Ilyina MG, Evshchik EY, Khamitov EM, Chernyak AV, Shikhovtseva AV, Melnikova TI, Bushkova OV, Borisevich SS. QC and MD Modelling for Predicting the Electrochemical Stability Window of Electrolytes: New Estimating Algorithm. Batteries. 2022; 8(12):292. https://doi.org/10.3390/batteries8120292
Chicago/Turabian StyleDobrovolsky, Yuri A., Margarita G. Ilyina, Elizaveta Y. Evshchik, Edward M. Khamitov, Alexander V. Chernyak, Anna V. Shikhovtseva, Tatiana I. Melnikova, Olga V. Bushkova, and Sophia S. Borisevich. 2022. "QC and MD Modelling for Predicting the Electrochemical Stability Window of Electrolytes: New Estimating Algorithm" Batteries 8, no. 12: 292. https://doi.org/10.3390/batteries8120292
APA StyleDobrovolsky, Y. A., Ilyina, M. G., Evshchik, E. Y., Khamitov, E. M., Chernyak, A. V., Shikhovtseva, A. V., Melnikova, T. I., Bushkova, O. V., & Borisevich, S. S. (2022). QC and MD Modelling for Predicting the Electrochemical Stability Window of Electrolytes: New Estimating Algorithm. Batteries, 8(12), 292. https://doi.org/10.3390/batteries8120292