1. Introduction
Carbon nanotubes (CNTs) are quasi-1D nano materials that have attracted significant attention because of remarkable properties such as high chemical stability, high electrical conductivity, strength, flexibility and impressive surface area [
1,
2,
3,
4]. All these features make CNTs very appealing for various applications such as hydrogen storage, fuel cells, supercapacitors, lithium ion batteries, stealth paints, sensors, gas and toxin detection systems, catalysts, etc. [
5,
6,
7,
8]. CNTs also may be used as an additive component to enhance properties of various materials [
9,
10,
11,
12]. Many attempts have been made to deposit metal or metal compounds into and onto CNTs. One of the most obvious routes would be melting of metal onto the CNT surface, however, most metals are not able to wet its surface [
13]. Typically three methods are in use to manufacture CNT composites: mechanical alloying of metal nanopowders with CNT in a planetary mill (and further annealing or plasma sintering), molecular level mixing (i.e., CNTs functionalization and reaction with metal salt) and electrodeposition process [
14]. The results show that pre-functionalization of CNTs with oxygen-containing functional groups gives much better interaction with the metal matrix [
15]. As-made CNTs are sparsely dispersed in every common medium therefore they have to be functionalized before metal incorporation. The simplest methodology is oxidation, which creates oxygen-containing groups being active sites for metal deposition. Many different chemical oxidation techniques of CNTs have been proposed using concentrated mineral acids additionally enhanced by oxidizing agent for example HNO
3, H
2SO
4, aqua regia, KMnO
4, OsO
4/NaIO
4 [
16,
17,
18,
19,
20,
21]. Nitric acid or combination of nitric acid and sulfuric acid are most commonly used to activate CNT surface. Here important seems to be a proper ratio of the oxidative acid mixture, which is responsible for attaching oxygen species. The disadvantage is that such approach ultimately leads to disruption of the highly-conductive sp
2 network of carbon atoms if the degree of functionalization is too high [
22]. Although oxidation of CNTs often suffer from low precision and poor control of the final material, it is the simplest and most conventional pretreatment method for preparation of high-performance CNT-metal composites.
Although Cu-CNT composites have attracted much attention recently [
23,
24], it should be pointed out that despite its high electrical conductivity Cu produces only a weak bonding with carbon matrix and combining these two material is a challenge [
15]. To achieve low-resistance ohmic contacts, and thus create a Cu-CNT composite of appreciable properties, improvement in connection between CNT and copper at the nanoscale is crucial. This is commonly handled by manufacture of CNT—copper composites by wet preparation methods, which are composed of multi component single-step or two step sensitization—activation processes in tin chloride, palladium chloride, copper salt, sodium formate, formaldehyde, ethylenediaminetetraacetic acid (EDTA) and polyethyleneglycole-containing bath [
25,
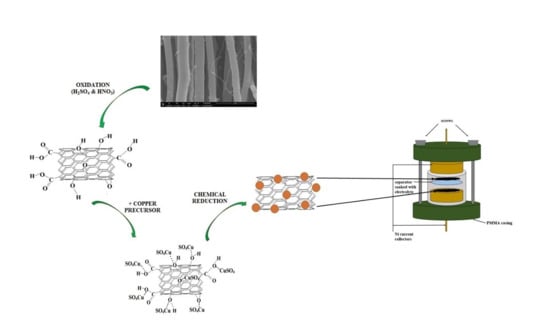
26]. Here we report facile preparation of Cu-CNT composites by CNTs acid functionalization and fast copper salt reduction. To confirm that this material may have appreciable electrical properties, we evaluated its potential for the application in supercapacitors. The product was configured in two-electrode assembly to probe its electrochemical performance.
2. Experimental
2.1. CNT Synthesis and Oxidation
Vertically-aligned carbon nanotubes were obtained in a chemical vapor deposition process from toluene (HPLC grade, Fisher Scientific, Hempton, NH, USA) as a carbon precursor and ferrocene (99.5%, Alfa Aesar, Haverhill, MA, USA) as a catalyst [
27]. Toluene was ultrasonicated with 3 wt. % ferrocene just before the process. Reaction was carried out in a horizontal furnace with three heating sections under argon atmosphere at 760 °C. After the synthesis, 0.5 g of freshly prepared CNT were mixed with 187.5 mL H
2SO
4 (>95%, Fisher Scientific) and 62.5 mL HNO
3 (70%, Fisher Scientific) in a 500 mL flask, and then ultrasonicated at 50 °C for 20 h. Next, the material was diluted with deionized water to 2 L and filtrated using polytetrafluoroethylene (PTFE) membrane filters (pore size 0.45 µm, Whatman, Maidstone, UK) on a fritted-glass funnel under vacuum and dried overnight at 110 °C. The oxidized CNTs were denoted in further experiments and analysis as CNTOX.
2.2. Preparation of Cu-CNT Composites
20 mg of as-oxidized CNTs was ultrasonically re-dispersed in 10 mL of distilled water, and then suspension was combined with metal precursor copper (II) sulfate pentahydrate (Fisher Scientific, Hempton, NH, USA). The mixture was ultrasonicated overnight. It was subsequently treated with 2 mL hydrazine hydrate (78–82%, Sigma-Aldrich, St Louis, MO, USA). Color change from black to brown was immediately observed, which indicated formation of metallic copper. Product was filtrated on PTFE membrane filter and obtained slurry (denoted as Cu-CNT) was put into the oven (120 °C) to remove the solvent.
2.3. Characterization and Electrochemical Testing
The materials were qualitatively analyzed using infra-red spectrometer (Bruker Optics, IFS 66/s Billerica, MA, USA) and Raman spectroscope (Renishaw, inVia, λ = 633 nm laser Wotton under Edge, UK). Carbon amount in a primary material and oxidized product, copper load in composites and impurities content were quantitatively evaluated by EDX analysis (EDX, Bruker Quantax coupled with Nova NanoSEM, Goettingem, Germany). All materials were additionally tested by thermogravimetry and calorimetry (Mettler Toledo, TGA/DSC Columbus, OH, USA). Morphology was examined by means of Scanning Electron Microscopy (SEM, FEI Nova NanoSEM). Electrochemical experiments were carried out using two-electrode system. The working electrode materials were placed on electrochemical nickel current collectors to form films and separated with membrane (Whatman) soaked with 1 M Na2SO4. Electrodes, current collectors and separator were pressed with four screws in a poly (methyl methacrylate) casing. Cyclic voltammetry (CV) and galvanostatic charge/discharge (GC) characteristics were performed with Autolab PGSTAT 30 workstation.
3. Results
Microstructure of neat CNTs and oxidized form (CNTOX) were analyzed using SEM to find changes in structure produced during acid treatment. As-made material was composed of vertically-aligned CNTs tens of µm long (
Figure 1a,b). EDX study confirmed about 98 wt% carbon and residual Fe catalyst (2 wt%) (
Figure 2a).
Acid treatment induced defects, shortened CNTs and increased densification degree of the material (
Figure 1c,d). Moreover, the entangled CNTOX material was no longer anisotropic. Presence of the oxygen in CNTOX was confirmed by EDX analysis as shown in
Figure 2b.
Acid treatment introduced ca. 37 wt% oxygen. Additionally, 11 wt% of sulfur, which is caused by contamination of oxidized CNT with strongly adsorbed sulfur-based chemical groups (most probably sulfate) introduced by sulfuric acid (could not be removed by the work-up despite copious amounts of distilled water used for filtration), was detected.
SEM images of Cu-CNT composite revealed presence of copper in the carbon matrix (
Figure 3). CNTs were covered by two types of particles, which were equally distributed: very fine Cu particles (
Figure 3b) and regions with bigger agglomerates (
Figure 3c) containing copper, sulfur and oxygen (probably unreduced copper sulfate or some copper oxide species).
EDX analysis showed 79 wt% Cu and 13 wt% C with the rest attributed to the residual oxygen groups from CNT functionalization and Cu precursor (
Figure 4a).
Raman spectroscopy evaluated changes in the chemical structure of the material in the course of functionalization and Cu deposition (
Figure 4b). The higher the ratio of the D to G band intensity the more disordered structure of the analyzed material. The D band located around 1350 cm
−1 is a consequence of structure disorder by the introduction of sp
3 hybridized carbon atoms, whereas G band around 1600 cm
−1 determines carbon domain with sp
2 hybridization. Ordered structure of as-synthesized CNTs had relatively low D/G ratio around 0.36. Acid treated CNTs had plethora of oxygen-bearing functional groups and were partially unzipped, much more corrugated and consequently more disordered. This resulted in an increase in D band intensity to 0.92. Oxidized CNT matrix coated with copper precursor and reduced using hydrazine was even more affected because of chemical reduction which caused elimination of some carbon atoms from CNT structure together with the oxygen. Consequently, the D/G ratio for the Cu-CNTOX composite was as high as 0.99.
Qualitative analysis was performed using FTIR (
Figure 5). In comparison to the as-made CNT the oxidized form had several signals with three of them most important: at 1750 cm
−1 assigned to the carboxylic groups, 1620 cm
−1 to hydroxyl groups and around 1220 cm
−1 probably from sulfates embedded during acid treatment. Because of reduction of copper precursor and oxidized CNT matrix by hydrazine hydrate most of signals vanished in Cu-CNT. Only presence of the residual hydroxyl groups can be observed. Additionally signals located at 2200–2000 cm
−1 ascribed to carbon structure were present in all three spectra, however, in the Cu-CNT composite they had the smallest intensities probably because of high copper loading. Not observable in this figure because of spectra overlapping was a broad and small signal located at 3500–3000 cm
−1 from stretching vibration of C–OH groups and water.
TGA and DSC curves recorded for all tested materials were presented in
Figure 6. In CNT only one gravimetric change can be found that was assigned to combustion of carbon honeycomb matrix that was satisfied by the presence of intensive exothermic signal in DSC curve. For the CNTOX matrix thermogravimetric curve was composed of several steps related to removal of residual solvent around 100 °C, water of hydration starting from 220 °C, gradual elimination of the oxygen containing groups up to 540 °C and decomposition of carbon matrix close to 640 °C. In both CNT and CNTOX weight loss was 100%, which satisfied absence of any significant metal impurities, with a strong exothermic signal attributed to carbon combustion. In the copper—CNT composite carbon matrix weight loss was observed at lower temperature than in CNT and CNTOX that could have been caused by catalytic effect of copper. In the temperature range 410 °C–700 °C there was also oxidation of the residual CuSO
4 (left after hydrazine reduction) to copper sulfate hydroxide hydrate and copper sulfate hydroxide, but their DSC signal was much lower in comparison to dominant carbon decomposition. As the amount of carbon with respect to copper in the composite was smaller the TG curve step was also not so significant. Final weight of the sample was about 70–75% of the initial—because carbon loss was equilibrated by copper oxidation compounds.
As obtained Cu-CNT composite was electrochemically tested using two electrode symmetric cell as a potential material for supercapacitor electrodes. Results were compared to the as-made CNT as well as CNTOX.
Figure 7a presents CV curves obtained for tested materials during 1000 charge discharge cycles at scan rate 500 mV/s in the potential window 0–1 V. In case of CNT curves were overlaid with the
x-axis as the current intensity was very low of the order of 0.00001 A. In contrary both CNTOX and Cu-CNT curves had much higher value of current intensity ca. 0.001 A. The more ideal box-like shape was obtained for Cu-CNT whereas for CNTOX more asymmetric curves with a strong oxidizing peak near 0.5V, particularly in the first few cycles, were registered. That was caused by irreversible redox reactions of electrode material with an electrolyte. Capacity loss after 1000 cycles was as high as 5% for CNT, 30.4% for CNTOX and 16% for Cu-CNT. However, it should be emphasized that the difference between first and last step in Cu-CNT was as high as 48% (different shape for three initial cycles), which is a consequence of chemical reaction of electrolyte and active species.
The real specific capacity was calculated from the galvanostatic charge/discharge curves (
Figure 7b). Curves recorded for all three types of materials were very similar, however, the least symmetric shape was found for copper-based composite. This was caused by reactions of copper, oxygen and residual sulfur present within material with an electrolyte. Consequently, the concept of Cu incorporation onto CNT may be a proper route to enhance electrical and capacity properties of CNT, but more efforts should be done to obtain material of high purity that will limit effect of undesirable side reactions. In case of sparsely conductive CNTOX, relatively high specific capacity and shape of galvanostatic curves was obtained. The oxygen containing groups are responsible for pseudocapacitance that enhances specific capacity, however, very often these species cause self-discharge of electrode material. Measurements were performed in a broad range of current density from 0.02 A/g to 7 A/g. The calculated specific capacity was 1.2 F/g, 25 F/g and 46 F/g for CNT, CNTOX and Cu-CNT, respectively.
It was relatively easy to charge and discharge CNT with resulting in proper triangular shape of galvanostatic curve, similarly for CNTOX, while for Cu-CNT problems with complete discharge were found. This showed that incorporation of copper copper-bearing species strongly limited pores accessibility and induced some irreversible redox reactions. Although materials morphology was changed the net specific capacity of the composite was enhanced based on pseudocapacitive effect.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






