
Lithium Growth on Alloying Substrates and Effect on Volumetric Expansion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Electrolyte Preparation
2.2. Fabrication of Alloying Substrates
2.3. Electrochemical Characterization
2.4. Scanning Electron Microscopy/Energy-Dispersive X-Ray Spectroscopy (SEM/EDS)
2.5. Laser Plasma Focused Ion Beam (Laser PFIB) Cross Sectional Imaging
3. Results and Discussion
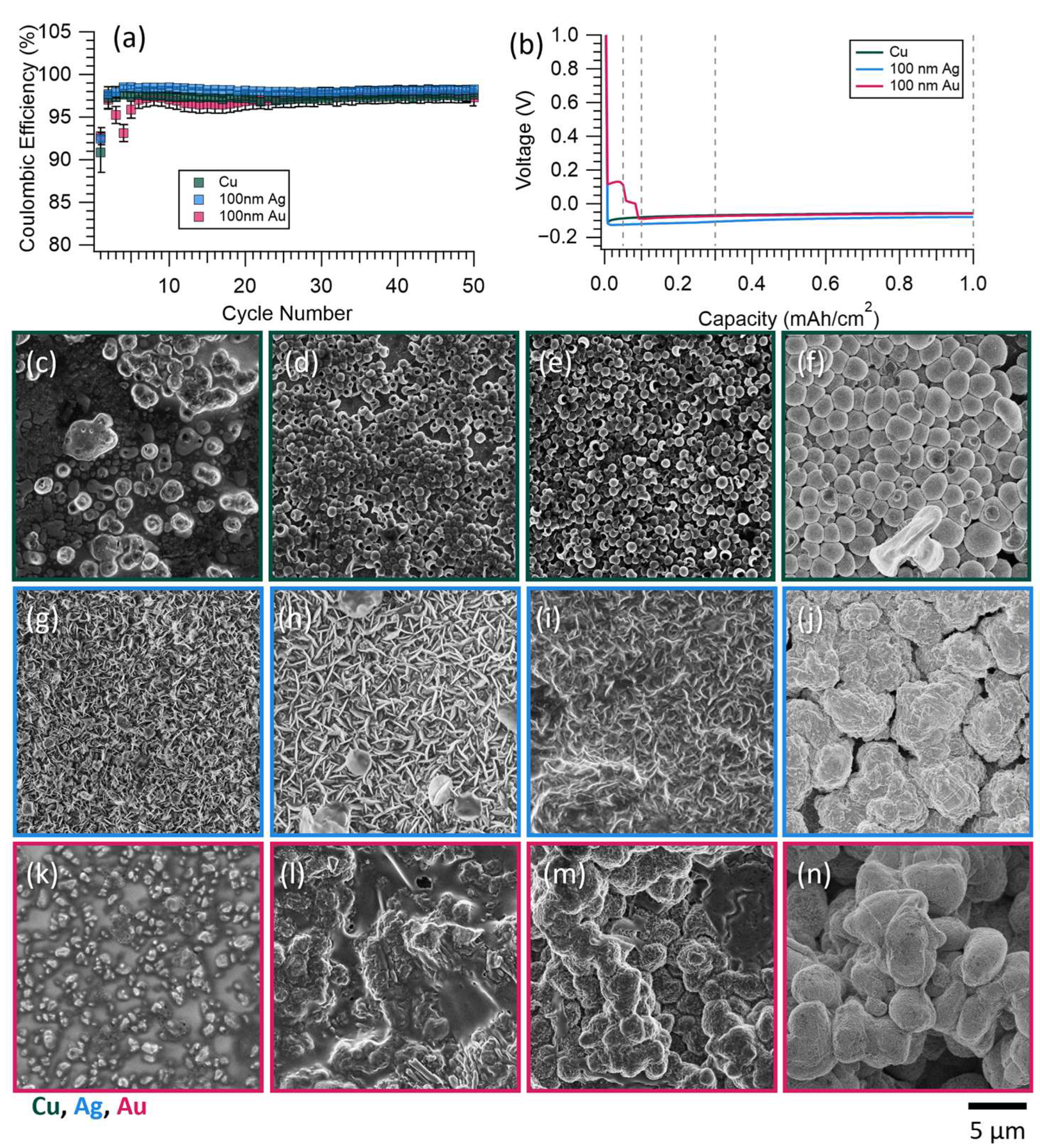
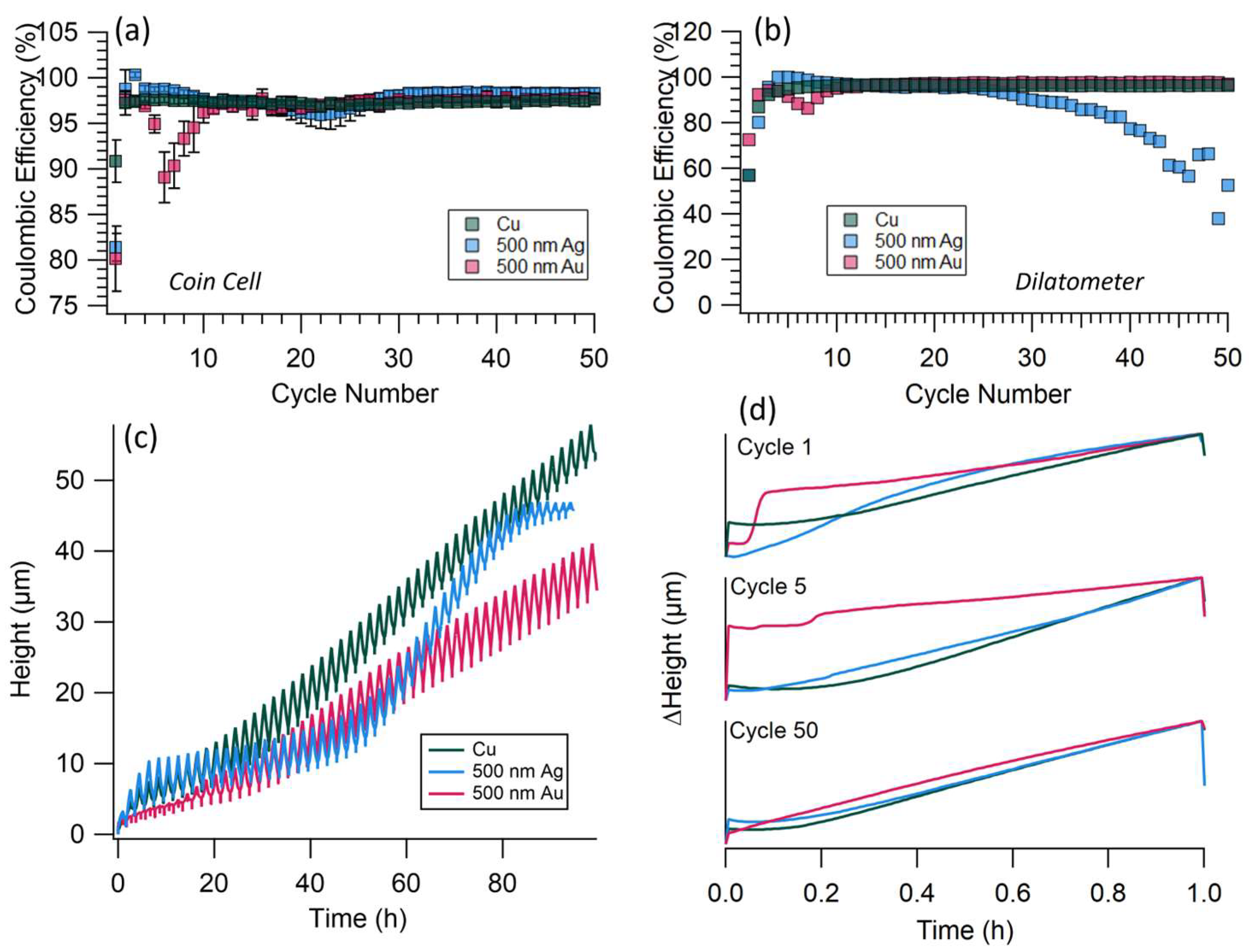
3.1. Coin Cell Results
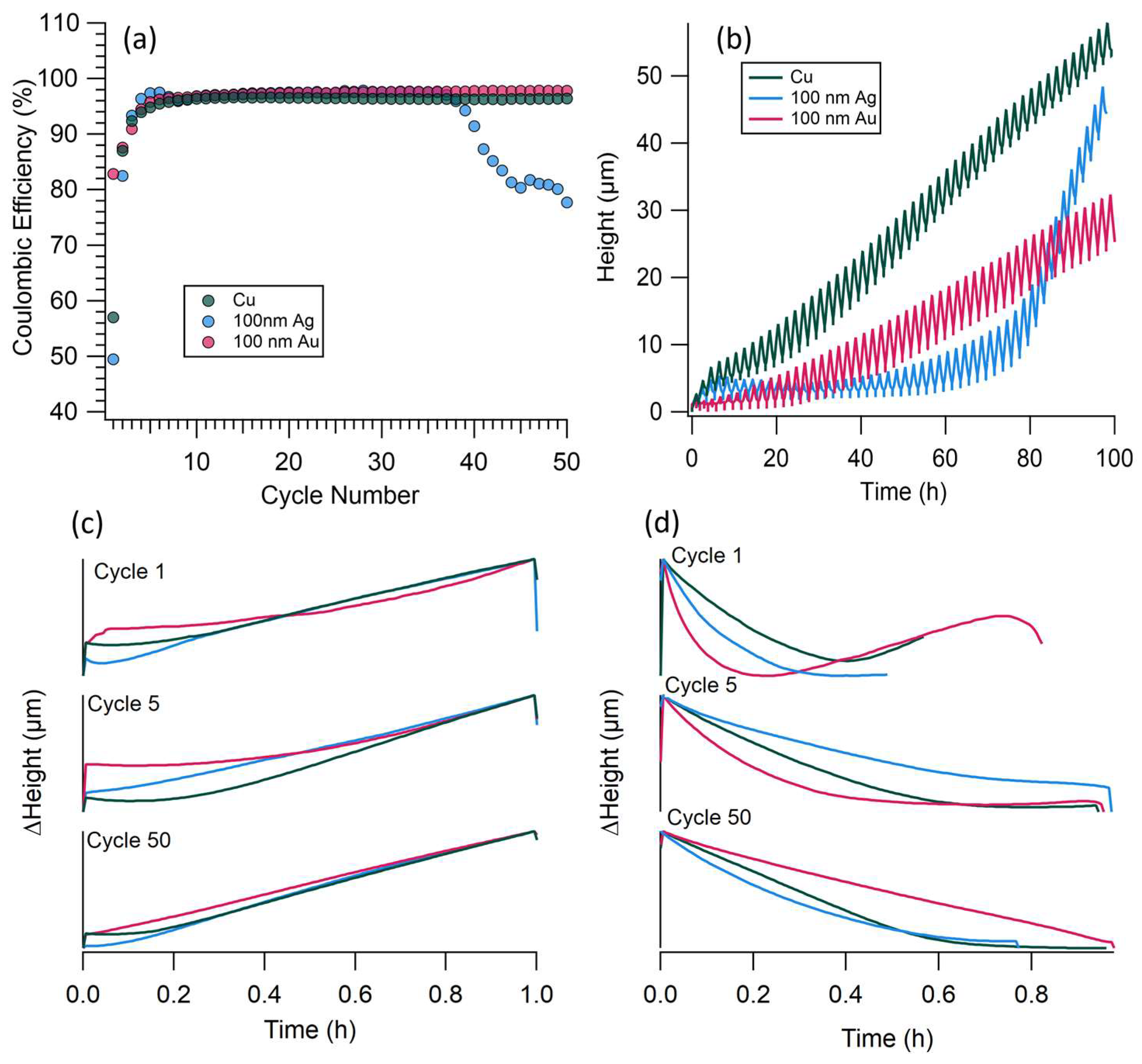
3.2. Dilatometer Results
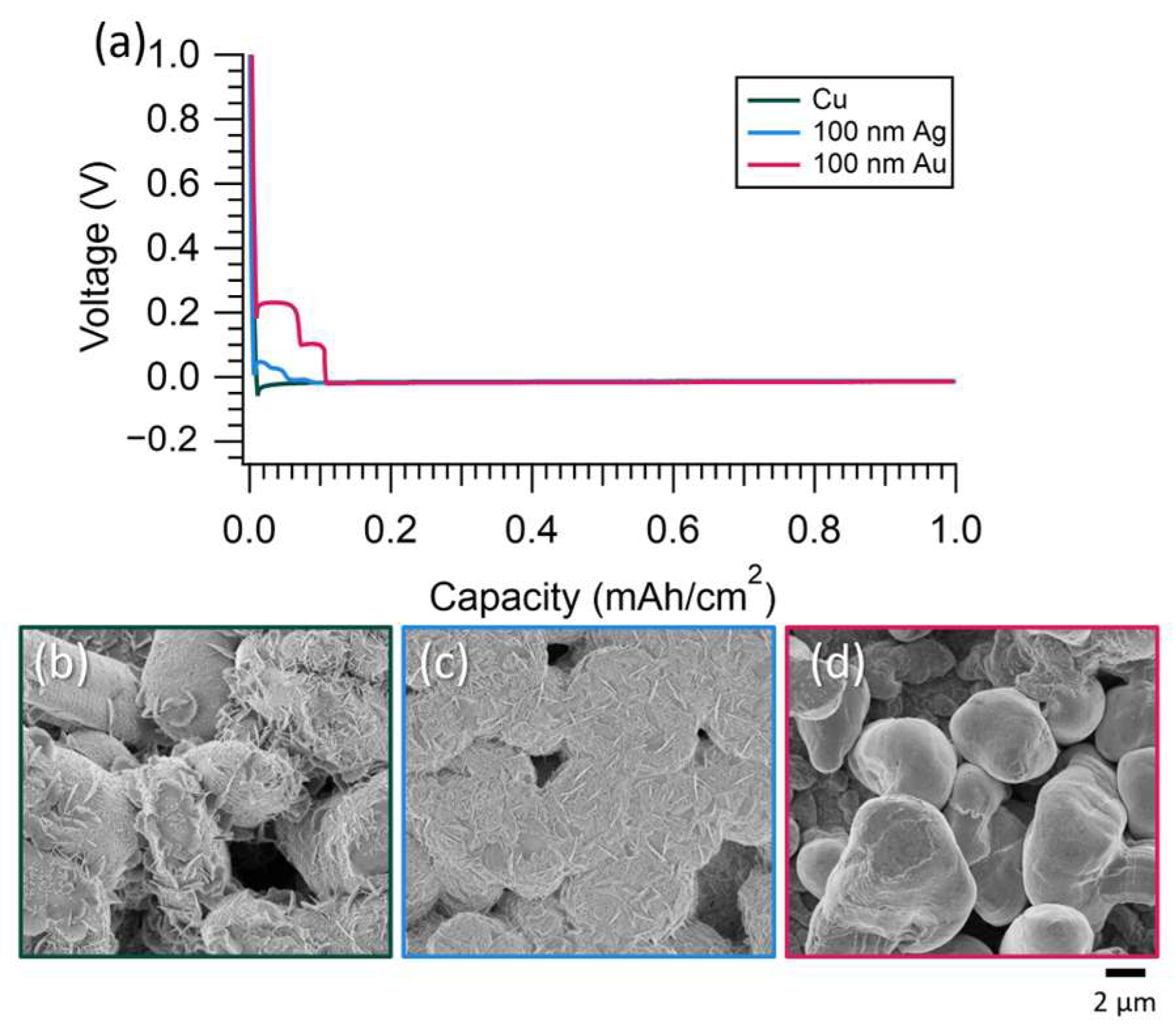
3.3. Decreased Current
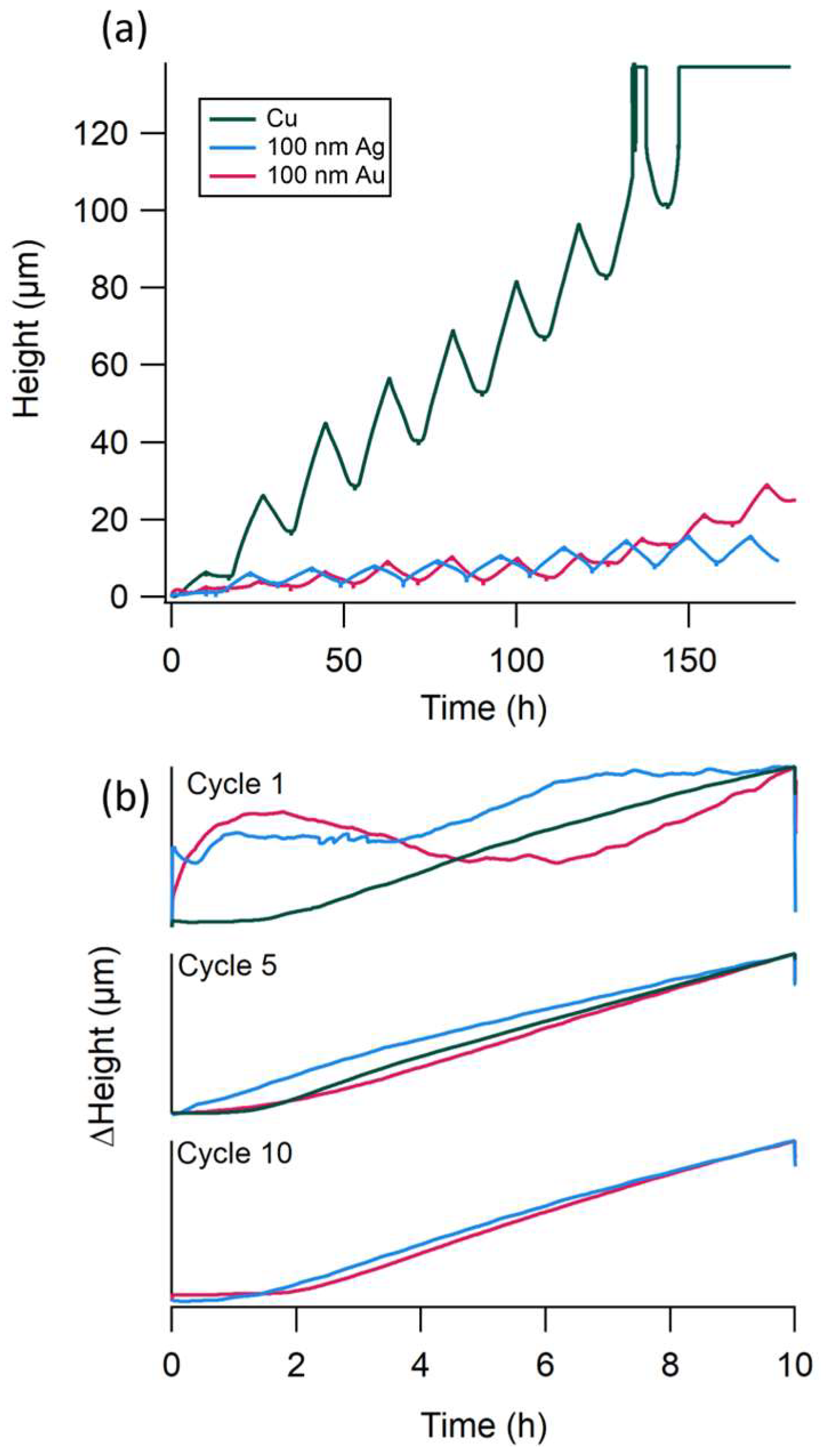
3.4. Increasing Substrate Thickness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CE | Coulombic efficiency |
| CV | Cyclic voltammogram/cyclic voltammetry |
| DME | 1,2-dimethoxyethane |
| DOL | 1,3-dioxolane |
| EDS | energy-dispersive X-ray spectroscopy |
| Laser PFIB | laser plasma focused ion beam |
| LiTFSI | lithium bis(trifluoromethane)sulfonimide |
| SEI | solid electrolyte interphase |
| SEM | scanning electron microscopy |
References
- Wang, S.; Ren, D.; Xu, C.; Han, X.; Liu, X.; Lu, L.; Ouyang, M. Lithium plating induced volume expansion overshoot of lithium-ion batteries: Experimental analysis and modeling. J. Power Sources 2024, 593, 233946. [Google Scholar] [CrossRef]
- Merrill, L.C.; Gannon, R.N.; Jungjohann, K.L.; Randolph, S.J.; Goriparti, S.; Zavadil, K.R.; Johnson, D.C.; Harrison, K.L. Evaluation of Lithium Metal Anode Volumetric Expansion through Laser Plasma Focused Ion Beam Cross-Sectional Imaging. J. Electrochem. Soc. 2023, 170, 080527. [Google Scholar] [CrossRef]
- Harrison, K.L.; Goriparti, S.; Merrill, L.C.; Long, D.M.; Warren, B.; Roberts, S.A.; Perdue, B.R.; Casias, Z.; Cuillier, P.; Boyce, B.L.; et al. Effects of Applied Interfacial Pressure on Li-Metal Cycling Performance and Morphology in 4 M LiFSI in DME. ACS Appl. Mater. Interfaces 2021, 13, 31668–31679. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.L.; Merrill, L.C.; Long, D.M.; Randolph, S.J.; Goriparti, S.; Cristian, J.; Warren, B.; Roberts, S.A.; Harris, S.J.; Perry, D.L.; et al. Cryogenic electron microscopy reveals that applied pressure promotes short circuits in Li batteries. iScience 2021, 24, 103394. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Yu, Y.; Shen, C.; Zhou, C.; Dong, C.; Zhao, T.; Xu, X. One-Pot Preparation of Lithium Compensation Layer, Lithiophilic Layer, and Artificial Solid Electrolyte Interphase for Lean-Lithium Metal Anode. ACS Appl. Mater. Interfaces 2022, 14, 19437–19447. [Google Scholar] [CrossRef]
- Liu, W.; Liu, P.; Mitlin, D. Review of Emerging Concepts in SEI Analysis and Artificial SEI Membranes for Lithium, Sodium, and Potassium Metal Battery Anodes. Adv. Energy Mater. 2020, 10, 2002297. [Google Scholar] [CrossRef]
- Kang, D.; Xiao, M.; Lemmon, J.P. Artificial Solid-Electrolyte Interphase for Lithium Metal Batteries. Batter. Supercaps 2020, 4, 445–455. [Google Scholar] [CrossRef]
- Zhang, D.; Dai, A.; Fan, B.; Li, Y.; Shen, K.; Xiao, T.; Hou, G.; Cao, H.; Tao, X.; Tang, Y. Three-Dimensional Ordered Macro/Mesoporous Cu/Zn as a Lithiophilic Current Collector for Dendrite-Free Lithium Metal Anode. ACS Appl. Mater. Interfaces 2020, 12, 31542–31551. [Google Scholar] [CrossRef]
- Stan, M.C.; Becking, J.; Kolesnikov, A.; Wankmiller, B.; Frerichs, J.E.; Hansen, M.R.; Bieker, P.; Kolek, M.; Winter, M. Sputter coating of lithium metal electrodes with lithiophilic metals for homogeneous and reversible lithium electrodeposition and electrodissolution. Mater. Today 2020, 39, 137–145. [Google Scholar] [CrossRef]
- Park, J.B.; Choi, C.; Yu, S.; Chung, K.Y.; Kim, D.-W. Porous Lithiophilic Li–Si Alloy-Type Interfacial Framework via Self-Discharge Mechanism for Stable Lithium Metal Anode with Superior Rate. Adv. Energy Mater. 2021, 11, 2101544. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, G.; Liu, Z.; Weng, S.; Li, X.; Wang, X.; Gao, Y.; Wang, Z.; Chen, L. Phase Diagram Determined Lithium Plating/Stripping Behaviors on Lithiophilic Surfaces. ACS Energy Lett. 2021, 6, 4118–4126. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, W.; Ming, H.; Cao, G.; Fan, L.Z.; Zhang, H. Chemical Energy Release Driven Lithiophilic Layer on 1 m2 Commercial Brass Mesh toward Highly Stable Lithium Metal Batteries. Nano Lett. 2019, 19, 1832–1837. [Google Scholar] [CrossRef]
- Zhao, F.; Zhou, X.; Deng, W.; Liu, Z. Entrapping lithium deposition in lithiophilic reservoir constructed by vertically aligned ZnO nanosheets for dendrite-free Li metal anodes. Nano Energy 2019, 62, 55–63. [Google Scholar] [CrossRef]
- Chen, X.-R.; Chen, X.; Yan, C.; Zhang, X.-Q.; Zhang, Q.; Huang, J.-Q. Role of Lithiophilic Metal Sites in Lithium Metal Anodes. Energy Fuels 2021, 35, 12746–12752. [Google Scholar] [CrossRef]
- Cao, X.; Ren, X.; Zou, L.; Engelhard, M.; Huang, W.; Wang, H.; Matthews, B.E.; Lee, H.; Niu, C.; Arey, B.W.; et al. Monolithic solid–electrolyte interphases formed in fluorinated orthoformate-based electrolytes minimize Li depletion and pulverization. Nat. Energy 2019, 4, 796–805. [Google Scholar] [CrossRef]
- Zheng, J.; Kim, M.S.; Tu, Z.; Choudhury, S.; Tang, T.; Archer, L.A. Regulating electrodeposition of lithium: Towards commercially relevant secondary Li metal batteries. Chem. Soc. Rev. 2020, 49, 2701–2750. [Google Scholar] [CrossRef]
- Merrill, L.C.; Long, D.M.; Small, K.A.; Jungjohann, K.L.; Leung, K.; Bassett, K.L.; Harrison, K.L. Role of Coatings as Artificial Solid Electrolyte Interphases on Lithium Metal Self-Discharge. J. Phys. Chem. C. 2022, 126, 17490–17501. [Google Scholar] [CrossRef]
- Meyerson, M.L.; Maraschky, A.M.; Watt, J.; Small, L.J. Fast cycling of “anode-less”, redox-mediated Li-S flow batteries. J. Energy Storage 2023, 72, 108767. [Google Scholar] [CrossRef]
- Kreissl, J.J.A.; Petit, J.; Oppermann, R.; Cop, P.; Gerber, T.; Joos, M.; Abert, M.; Tubke, J.; Miyazaki, K.; Abe, T.; et al. Electrochemical Lithiation/Delithiation of ZnO in 3D-Structured Electrodes: Elucidating the Mechanism and the Solid Electrolyte Interphase Formation. ACS Appl. Mater. Interfaces 2021, 13, 35625–35638. [Google Scholar] [CrossRef]
- Choudhury, S.; Tu, Z.; Stalin, S.; Vu, D.; Fawole, K.; Gunceler, D.; Sundararaman, R.; Archer, L.A. Electroless Formation of Hybrid Lithium Anodes for Fast Interfacial Ion Transport. Angew. Chem. Int. Ed. Engl. 2017, 56, 13070–13077. [Google Scholar] [CrossRef]
- Gu, X.; Dong, J.; Lai, C. Li-containing alloys beneficial for stabilizing lithium anode: A review. Eng. Rep. 2020, 3, e12339. [Google Scholar] [CrossRef]
- Lin, C.F.; Qi, Y.; Gregorczyk, K.; Lee, S.B.; Rubloff, G.W. Nanoscale Protection Layers To Mitigate Degradation in High-Energy Electrochemical Energy Storage Systems. Acc. Chem. Res. 2018, 51, 97–106. [Google Scholar] [CrossRef]
- Manandhar, K.; Ren, Y.; Stasak, D.; Hou, H.; Kirsch, D.; Sarker, S.; Mehta, A.; Sardar, S.; Xiao, M.; Weaver, J.L.; et al. High-Throughput Exploration of Lithium-Alloy Protection Layers for High-Performance Lithium-Metal Batteries. ACS Appl. Energy Mater. 2020, 3, 2547–2555. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, S.; Zhou, G.; Chen, W.; Zhou, F.; Rong, Z.; Wu, Y.; Li, J.; Guo, J.; Zhang, Y. Solubility-Dependent Protective Effects of Binary Alloys for Lithium Anode. ACS Appl. Energy Mater. 2020, 3, 2278–2284. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, J.; Zhao, K.; Ma, Y.; Zhang, H.; Song, D.; Shi, X.; Zhang, L.; Ding, Y. A novel dual-protection interface based on gallium-lithium alloy enables dendrite-free lithium metal anodes. Energy Storage Mater. 2021, 39, 403–411. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, W.; Zheng, W.; Cui, X.; Rojo, T.; Zhang, Q. Towards High-Safe Lithium Metal Anodes: Suppressing Lithium Dendrites via Tuning Surface Energy. Adv. Sci. 2017, 4, 1600168. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Lu, Z.; Lee, H.-W.; Xiong, F.; Hsu, P.-C.; Li, Y.; Zhao, J.; Chu, S.; Cui, Y. Selective deposition and stable encapsulation of lithium through heterogeneous seeded growth. Nat. Energy 2016, 1, 16010. [Google Scholar] [CrossRef]
- Qiu, H.; Tang, T.; Asif, M.; Li, W.; Zhang, T.; Hou, Y. Stable lithium metal anode enabled by lithium metal partial alloying. Nano Energy 2019, 65, 103989. [Google Scholar] [CrossRef]
- Choi, S.H.; Lee, S.J.; Yoo, D.-J.; Park, J.H.; Park, J.-H.; Ko, Y.N.; Park, J.; Sung, Y.-E.; Chung, S.-Y.; Kim, H.; et al. Marginal Magnesium Doping for High-Performance Lithium Metal Batteries. Adv. Energy Mater. 2019, 9, 1902278. [Google Scholar] [CrossRef]
- Xu, T.; Gao, P.; Li, P.; Xia, K.; Han, N.; Deng, J.; Li, Y.; Lu, J. Fast-Charging and Ultrahigh-Capacity Lithium Metal Anode Enabled by Surface Alloying. Adv. Energy Mater. 2020, 10, 1902343. [Google Scholar] [CrossRef]
- Peng, Z.; Song, J.; Huai, L.; Jia, H.; Xiao, B.; Zou, L.; Zhu, G.; Martinez, A.; Roy, S.; Murugesan, V.; et al. Enhanced Stability of Li Metal Anodes by Synergetic Control of Nucleation and the Solid Electrolyte Interphase. Adv. Energy Mater. 2019, 9, 1901764. [Google Scholar] [CrossRef]
- Chen, Z.; Luo, Y.; Yang, D.; Hu, Y.; Hou, H.; Koratkar, N.; Zhou, G.; Liu, W. Intraparticle alloying-plating reaction for high-performing lithium metal batteries with low volume expansion. Mater. Today, 2025; in press. [Google Scholar] [CrossRef]
- Zhang, X.; Gong, Y.; Xu, C.; Qu, P.; Wang, G. Thickness dependence of high volumetric energy density lithium ion battery based on Sn–Zn eutectic alloy foil anode. Ionics 2022, 28, 2685–2692. [Google Scholar] [CrossRef]
- Jin, S.; Ye, Y.; Niu, Y.; Xu, Y.; Jin, H.; Wang, J.; Sun, Z.; Cao, A.; Wu, X.; Luo, Y.; et al. Solid-Solution-Based Metal Alloy Phase for Highly Reversible Lithium Metal Anode. J. Am. Chem. Soc. 2020, 142, 8818–8826. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Liu, M.; Naik, D.; Gole, J.L. Electrochemical properties of Li-Mg alloy electrodes for lithium batteries. J. Power Sources 2001, 92, 70–80. [Google Scholar] [CrossRef]
- Seo, J.; Lim, J.; Chang, H.; Lee, J.; Woo, J.; Jung, I.; Kim, Y.; Kim, B.; Moon, J.; Lee, H. Sustaining Surface Lithiophilicity of Ultrathin Li-Alloy Coating Layers on Current Collector for Zero-Excess Li-Metal Batteries. Small 2024, 20, 2402988. [Google Scholar] [CrossRef] [PubMed]
- Bach, P.; Stratmann, M.; Valencia-Jaime, I.; Romero, A.H.; Renner, F.U. Lithiation and Delithiation Mechanisms of Gold Thin Film Model Anodes for Lithium Ion Batteries: Electrochemical Characterization. Electrochim. Acta 2015, 164, 81–89. [Google Scholar] [CrossRef]
- Kulova, T.L.; Skundin, A.M.; Kozhevin, V.M.; Yavsin, D.A.; Gurevich, S.A. A Study of Lithium Insertion into Electrodes with Thin Gold Films. Russ. J. Electrochem. 2010, 46, 877–881. [Google Scholar] [CrossRef]
- Hertle, J.; Walther, F.; Mogwitz, B.; Schroder, S.; Richter, H.; Janek, J. Miniaturization of Reference Electrodes for Solid-State Lithium-Ion Batteries. J. Electrochem. Soc. 2023, 170, 040519. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, S.; Yang, Y.; Sun, Z.-T.; Yu, X.; Guan, C.; Ouyang, R.; Guo, Y.; Bo, S.-H.; Zhu, H. Unlocking the Potential of Li-Ag Alloys: Phase Selection and Practical Application. Energy Mater. Adv. 2024, 5, 0108. [Google Scholar] [CrossRef]
- Jungjohann, K.L.; Gannon, R.N.; Goriparti, S.; Randolph, S.J.; Merrill, L.C.; Johnson, D.C.; Zavadil, K.R.; Harris, S.J.; Harrison, K.L. Cryogenic laser ablation reveals short-circuit mechanism in lithium metal batteries. ACS Energy Lett. 2021, 6, 2138–2144. [Google Scholar] [CrossRef]
- Merrill, L.C.; Rosenberg, S.G.; Jungjohann, K.L.; Harrison, K.L. Uncovering the Relationship between Aging and Cycling on Lithium Metal Battery Self-Discharge. ACS Appl. Energy Mater. 2021, 4, 7589–7598. [Google Scholar] [CrossRef]
- Kim, M.; Lee, S.; Park, D.; Kang, H.; Kam, D.; Park, J.-H.; Oh, S.H.; Jung, H.-G.; Choi, W. Tuning Lithiophilic Sites of Ag-Embedded N-Doped Carbon Hollow Spheres via Intentional Blocking Strategy for Ultrastable Li Metal Anode in Rechargeable Batteries. ACS Sustain. Chem. Eng. 2023, 11, 1785–1796. [Google Scholar] [CrossRef]
- Kim, H.; Choi, M.; Kang, H.; Choi, W. Accelerating Lithium Deposition Kinetics Via Lithiophilic Ag-Decorated Graphitic Carbon Nitride Spheres for Stable Lithium Metal Anode. Energy Environ. Sci. 2024, 8, e12830. [Google Scholar] [CrossRef]
- Taillades, G.; Sarradin, J. Silver: High perfromance anode for thin film lithium ion batteries. J. Power Sources 2004, 125, 199–205. [Google Scholar] [CrossRef]
- Thomas, J.; Behara, S.S.; Van der Ven, A. Thermodynamic and Kinetic Properties of the Lithium-Silver System. Chem. Mater. 2024, 36, 8936–8948. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-J.; Qu, J.; Chang, Y.; Yang, C.-Y.; Zhai, X.-Z.; Yu, Z.-Z.; Li, X. Engineering Lithiophilic Silver Sponge Integrated with Ion-Conductive PVDF/LiF Protective Layer for Dendrite-Free and High Performance Lithium Metal Batteries. ACS Appl. Energy Mater. 2023, 6, 519–529. [Google Scholar] [CrossRef]
- Fatahine, M.; Guay, D.; Roue, L. Study of lithium metal-based electrodes by electrochemical dilatometry. J. Appl. Electrochem. 2021, 52, 149–157. [Google Scholar] [CrossRef]
- Lohrberg, O.; Maletti, S.; Heubner, C.; Schneider, M.; Michaelis, A. Understanding Li Plating and Stripping Behavior in Zero-Excess Li Metal Batteries Using Operando Dilatometry. J. Electrochem. Soc. 2022, 169, 030543. [Google Scholar] [CrossRef]
- Gopinadh, S.V.; Phanendra, P.V.R.L.; V, A.; John, B.; TD, M. Progress, Challenges, and Perspectives on Alloy-Based Anode Materials for Lithium Ion Battery: A Mini-Review. Energy Fuels 2024, 38, 17253–17277. [Google Scholar] [CrossRef]
- Gireaud, L.; Grugeon, S.; Laruelle, S.; Yrieix, B.; Tarascon, J.M. Lithium metal stripping/plating mechanisms studies: A metallurgical approach. Electrochem. Commun. 2006, 8, 1639–1649. [Google Scholar] [CrossRef]
- Bassett, K.L.; Small, K.A.; Long, D.M.; Merrill, L.C.; Warren, B.; Harrison, K.L. Interfacial pressure improves calendar aging of lithium metal anodes. Front. Batter. Electrochem. 2023, 2, 1292639. [Google Scholar] [CrossRef]
- Lu, B.; Bao, W.; Yao, W.; Doux, J.-M.; Fang, C.; Meng, Y.S. Editors’ Choice—Methods—Pressure Control Apparatus for Lithium Metal Batteries. J. Electrochem. Soc. 2022, 169, 070537. [Google Scholar] [CrossRef]
- Cai, M.; Dai, F. Best practices in lithium battery cell preparation and evaluation. Commun. Mater. 2022, 3, 64. [Google Scholar] [CrossRef]
- Schommer, A.; Corzo, M.O.; Henshell, P.; Morrey, D.; Collier, G. Stack pressure on lithium-ion pouch cells: A comparative study of constant pressure and fixed displacement devices. J. Power Sources 2025, 629, 236019. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merrill, L.C.; Craig, R.L.; Cummings, D.P.; Deitz, J.I. Lithium Growth on Alloying Substrates and Effect on Volumetric Expansion. Batteries 2025, 11, 249. https://doi.org/10.3390/batteries11070249
Merrill LC, Craig RL, Cummings DP, Deitz JI. Lithium Growth on Alloying Substrates and Effect on Volumetric Expansion. Batteries. 2025; 11(7):249. https://doi.org/10.3390/batteries11070249
Chicago/Turabian StyleMerrill, Laura C., Robert L. Craig, Damion P. Cummings, and Julia I. Deitz. 2025. "Lithium Growth on Alloying Substrates and Effect on Volumetric Expansion" Batteries 11, no. 7: 249. https://doi.org/10.3390/batteries11070249
APA StyleMerrill, L. C., Craig, R. L., Cummings, D. P., & Deitz, J. I. (2025). Lithium Growth on Alloying Substrates and Effect on Volumetric Expansion. Batteries, 11(7), 249. https://doi.org/10.3390/batteries11070249