
Binder-Free Fe-N-C-O Bifunctional Electrocatalyst in Nickel Foam for Aqueous Zinc–Air Batteries



Abstract

1. Introduction
2. Materials and Methods
2.1. Sol–Gel Synthesis
2.2. Thermal Treatments
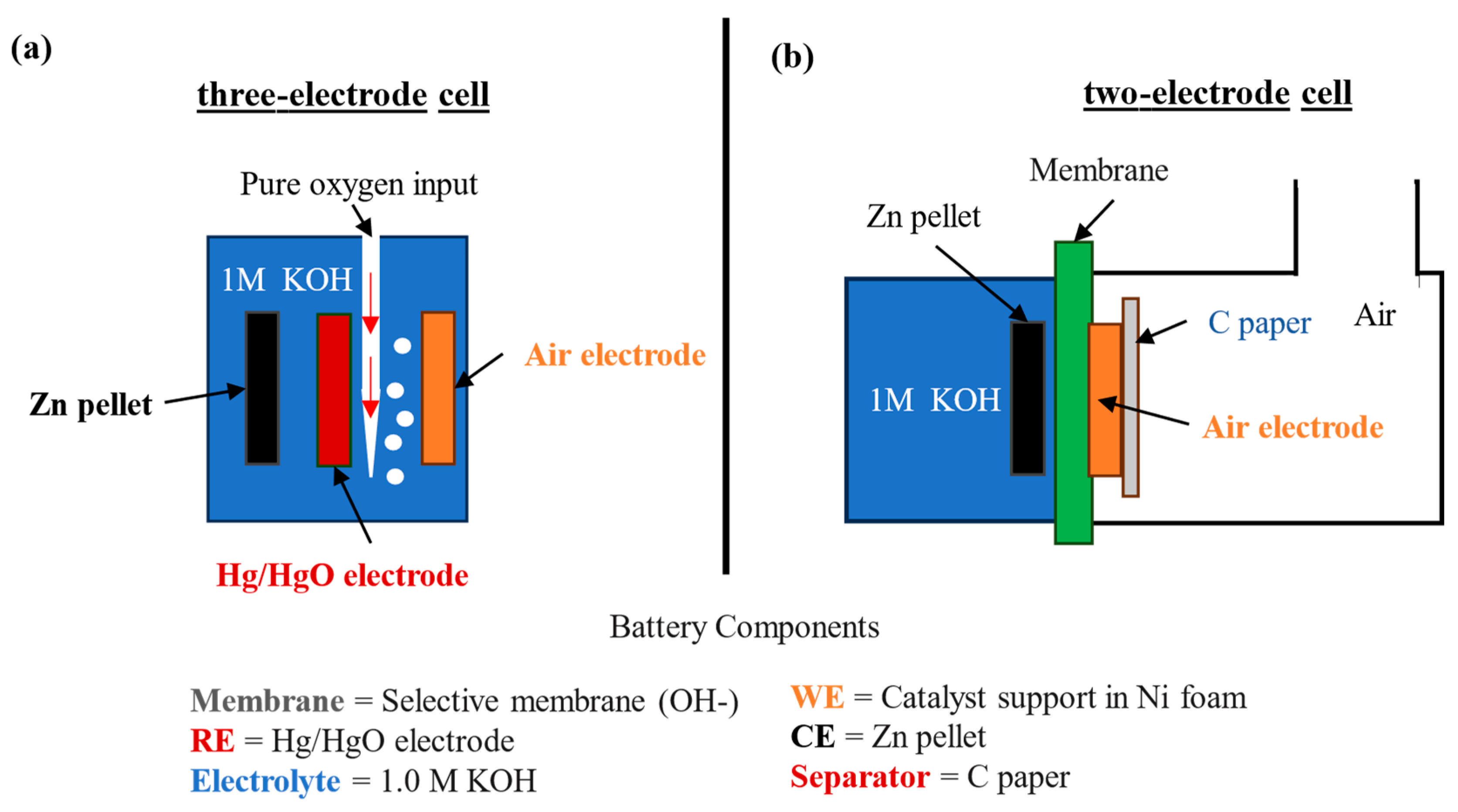
2.3. Structural and Electrochemical Characterization
3. Results and Discussion
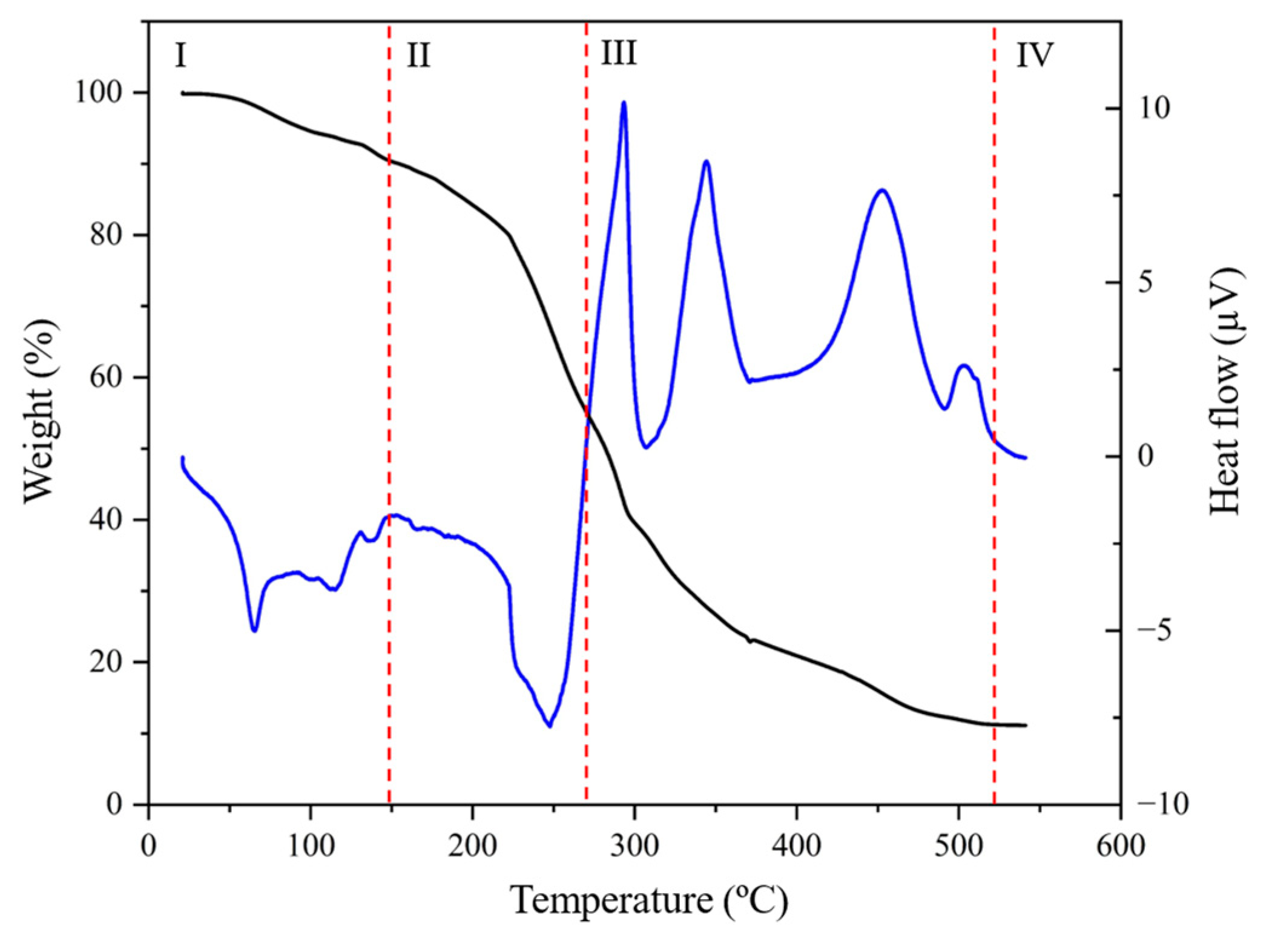
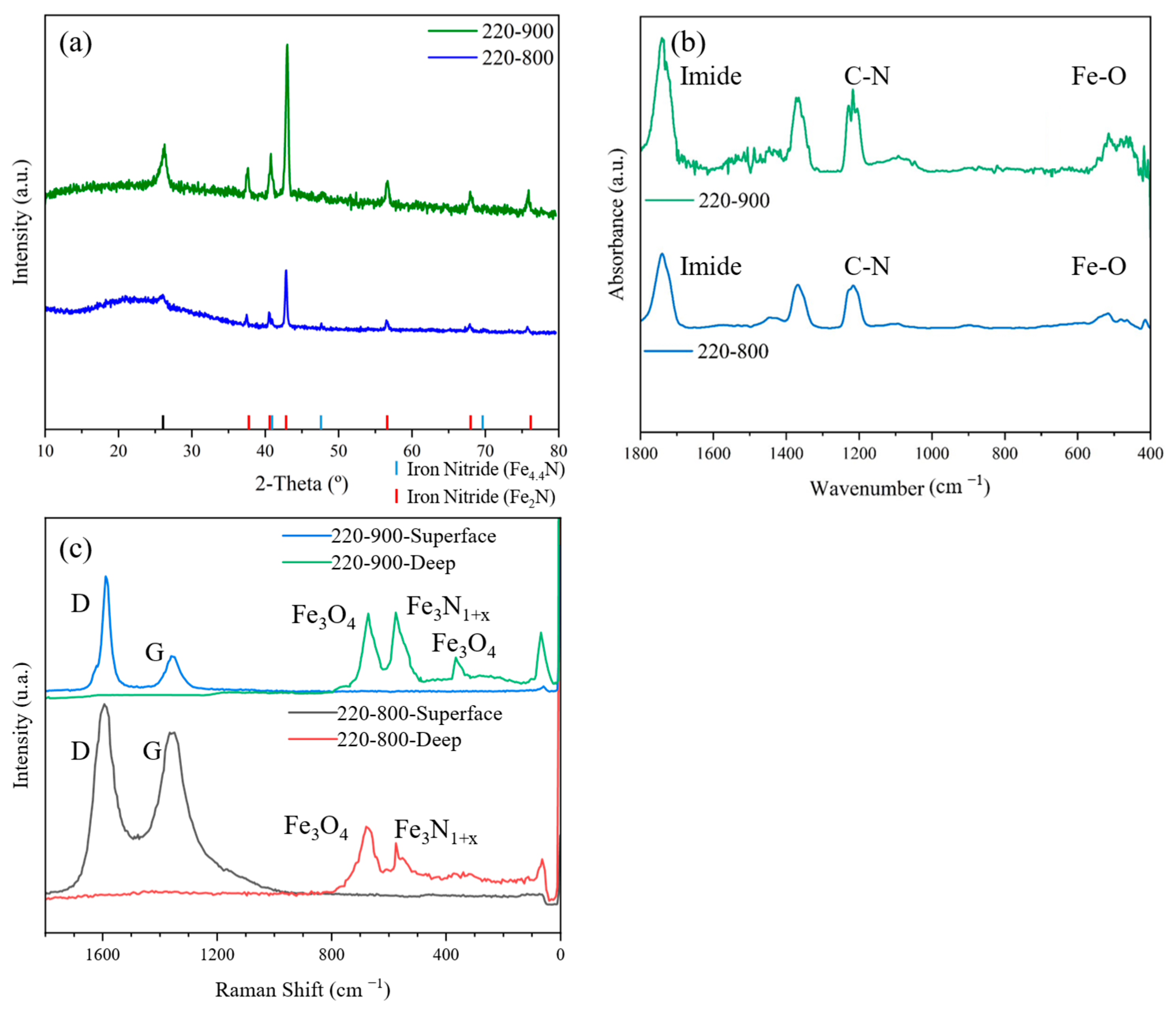
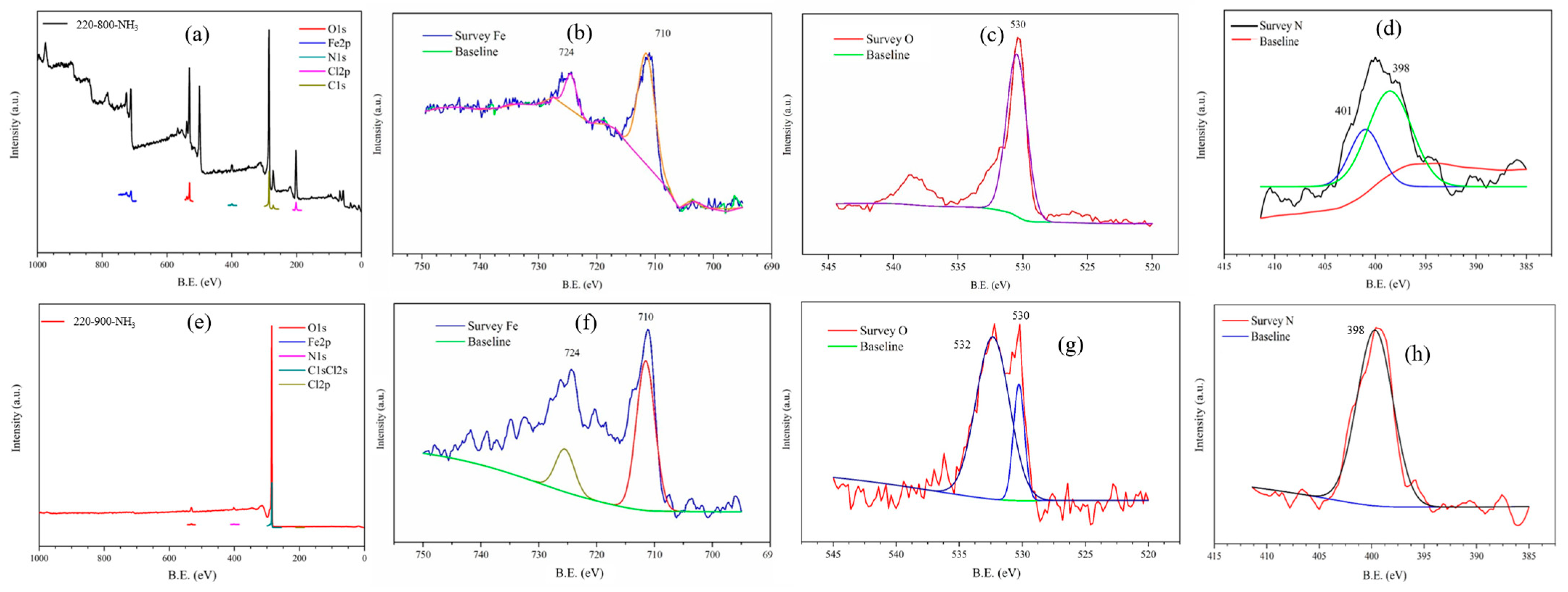
3.1. Structural and Chemical Results
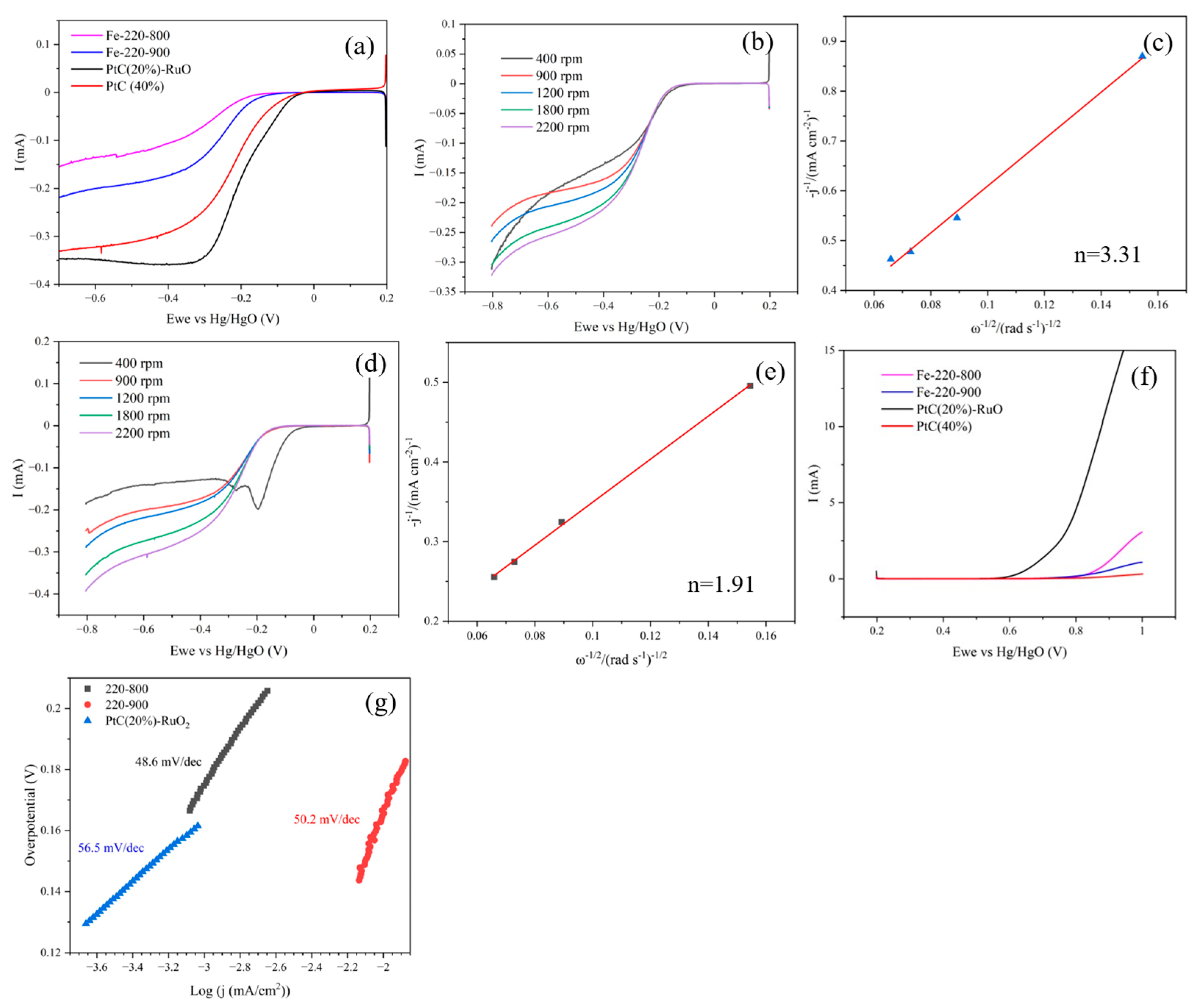
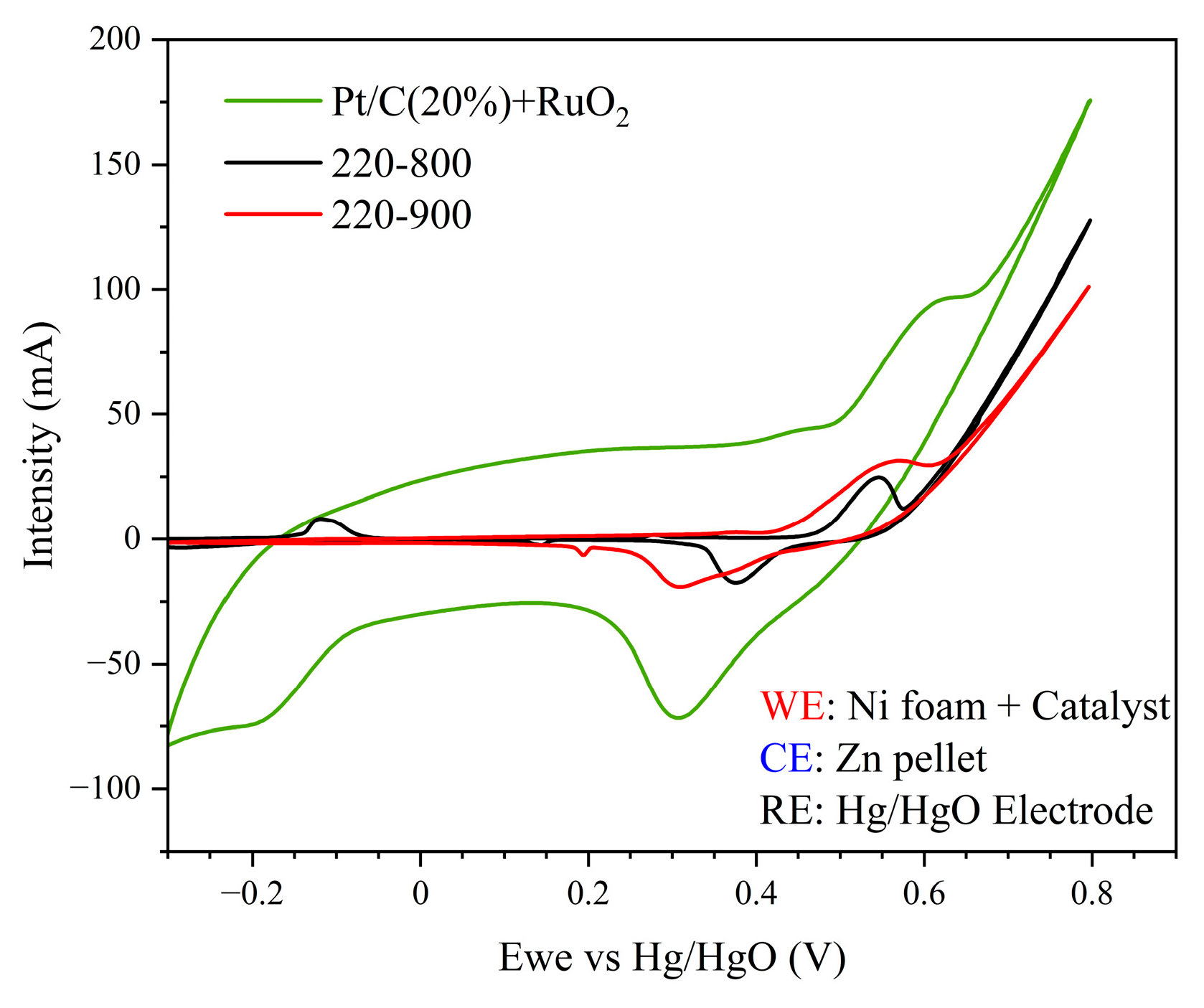
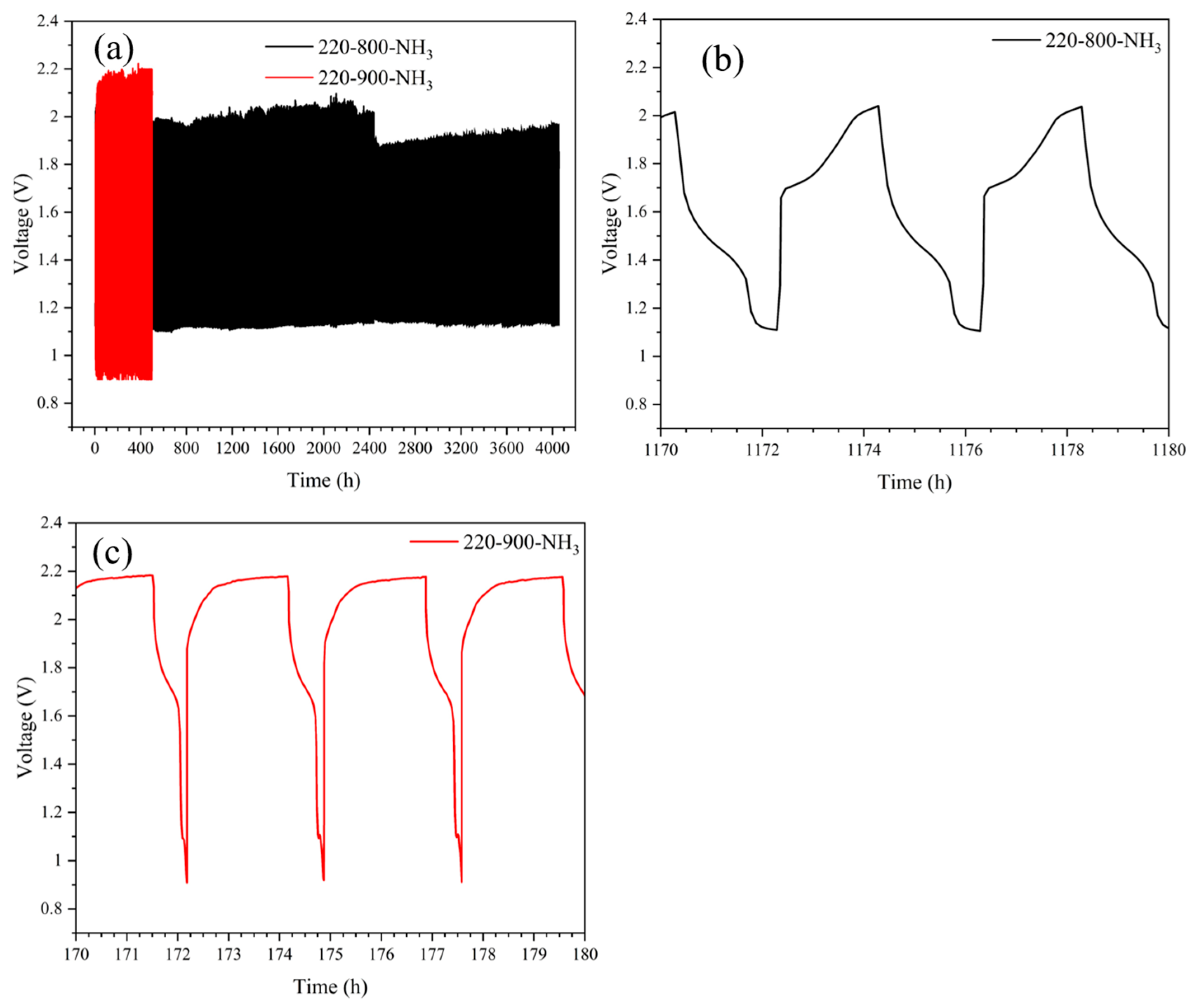
3.2. Electrochemical and “Post-Mortem” Results
3.3. Comparison with Previous Studies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Olabi, G.; Sayed, E.T.; Wilberforce, T.; Jamal, A.; Alami, A.H.; Elsaid, K.; Rahman, S.M.A.; Shah, S.K.; Abdelkareem, M.A. Metal-Air Batteries—A Review. Energies 2021, 14, 7373. [Google Scholar] [CrossRef]
- Christensen, J.; Albertus, P.; Sanchez-Carrera, R.S.; Lohmann, T.; Kozinsky, B.; Liedtke, R.; Ahmed, J.; Kojic, A. A Critical Review of Li/Air Batteries. J. Electrochem. Soc. 2012, 159, R1–R30. [Google Scholar] [CrossRef]
- Mainar, A.R.; Iruin, E.; Blázquez, J.A. High performance secondary zinc-air/silver hybrid battery. J. Energy Storage 2021, 33, 102103. [Google Scholar] [CrossRef]
- Chakkaravarthy, C.; Abdul Waheed, A.K.; Udupa, H.V.K. Zinc—Air alkaline batteries—A review. J. Power Sources 1981, 6, 203–228. [Google Scholar] [CrossRef]
- Li, Y.; Fan, X.; Liu, X.; Qu, S.; Liu, J.; Ding, J.; Han, X.; Deng, Y.; Hu, W.; Zhong, C. Long-battery-life flexible zinc–air battery with near-neutral polymer electrolyte and nanoporous integrated air electrode. J. Mater. Chem. A 2019, 7, 25449–25457. [Google Scholar] [CrossRef]
- Pan, J.; Xu, Y.Y.; Yang, H.; Dong, Z.; Liu, H.; Xia, B.Y. Advanced Architectures and Relatives of Air Electrodes in Zn–Air Batteries. Adv. Sci. 2018, 5, 1700691. [Google Scholar] [CrossRef] [PubMed]
- Moradi, Z.; Heydarinasab, A.; Pajoum Shariati, F. First-principle study of doping effects (Ti, Cu, and Zn) on electrochemical performance of Li2MnO3 cathode materials for lithium-ion batteries. Int. J. Quantum Chem. 2020, 120, e26458. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, G.; Hu, Y.; Wang, J.; Sun, T.; Regier, T.; Qiao, J.; Sun, S. Graphitic-shell encapsulated FeNi alloy/nitride nanocrystals on biomass-derived N-doped carbon as an efficient electrocatalyst for rechargeable Zn-air battery. ChemSusChem 2021, 3, 176–187. [Google Scholar] [CrossRef]
- Vaitsis, C.; Mechili, M.; Argirusis, N.; Pandis, P.K.; Sourkouni, G.; Argirusis, C. MOF nanomaterials for battery cathodes. In Metal-Organic Framework-Based Nanomaterials for Energy Conversion and Storage; Elsevier Science: Amsterdam, The Netherlands, 2022; pp. 207–226. [Google Scholar] [CrossRef]
- Zhang, W.; Cui, L.; Liu, J. Recent advances in cobalt-based electrocatalysts for hydrogen and oxygen evolution reactions. J. Alloys Compd. 2020, 821, 153542. [Google Scholar] [CrossRef]
- Nguyen, L.N.; Thuy, U.T.; Truong, Q.D.; Honma, I.; Nguyen, Q.L.; Tran, P.D. Electrodeposited Amorphous Tungsten-doped Cobalt Oxide as an Efficient Catalyst for the Oxygen Evolution Reaction. Adv. Synth. Catal. 2018, 13, 1530–1534. [Google Scholar] [CrossRef]
- Jung, K.-N.; Hwang, S.M.; Park, M.-S.; Kim, K.J.; Kim, J.-G.; Dou, S.X.; Kim, J.H.; Lee, J.-W. One-dimensional manganese-cobalt oxide nanofibres as bi-functional cathode catalysts for rechargeable metal-air batteries. Sci. Rep. 2015, 5, 7665. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Yue, B.; Hirao, N.; Liu, Z.; Chen, B. Significant improvement in Mn2O3 transition metal oxide electrical conductivity via high pressure. Sci. Rep. 2017, 7, 44078. [Google Scholar] [CrossRef]
- Wang, Y.J.; Fang, B.; Zhang, D.; Li, A.; Wilkinson, D.P.; Ignaszak, A.; Zhang, L.; Zhang, J. A Review of Carbon-Composited Materials as Air-Electrode Bifunctional Electrocatalysts for Metal–Air Batteries. Electrochem. Energy Rev. 2018, 1, 1–34. [Google Scholar] [CrossRef]
- Yang, Z.K.; Lin, L.; Xu, A.W. 2D Nanoporous Fe−N/C Nanosheets as Highly Efficient Non-Platinum Electrocatalysts for Oxygen Reduction Reaction in Zn-Air Battery. Small 2016, 12, 5013–5020. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, Z.; Ren, J.; Wang, F.; Wei, C.; Xing, Q.; Yan, W.; Li, X.; Chen, Y. A facile iron-sulfur double-doping strategy to prepare high performance FeNx/S-NC electrocatalyst for oxygen reduction reaction in zinc-air battery. Appl. Surf. Sci. 2021, 573, 152255. [Google Scholar] [CrossRef]
- Ranjbar, M.; Taher, M.A.; Sam, A. Facile hydrothermal synthesis of manganese-metal organic framework nanostructures in the presence of various organic ligands for SO2 and CO2 gas adsorption. J. Porous Mater. 2016, 23, 375–380. [Google Scholar] [CrossRef]
- González-Morales, J.; Aparicio, M.; Rosero-Navarro, N.C.; Zanotto, F.M.; Franco, A.A.; Mosa, J. Electrochemical and structural properties of binder-free iron-based bifunctional catalyst for aqueous Zinc-Oxygen batteries. Open Ceramics. 2024, 100667. [Google Scholar] [CrossRef]
- Kaur Muuli, R.; Kumar, R.; Mooste, M.; Gudkova, V.; Treshchalov, A.; Piirsoo, H.-M.; Kikas, A.; Aruväli, J.; Kisand, V.; Tamm, A.; et al. Iron, cobalt, and nickel phthalocyanine tri-doped electrospun carbon nanofibre-based catalyst for rechargeable zinc–air battery air electrode. Materials 2023, 16, 4626. [Google Scholar] [CrossRef]
- Xu, J.; Brodu, N.; Mignot, M.; Youssef, B.; Taouk, B. Synthesis and characterization of phenolic resins based on pyrolysis bio-oil separated by fractional condensation and water extraction. Bioresour. Technol. Rep. 2022, 16, 106393. [Google Scholar] [CrossRef]
- Sun, J.F.; Wang, M.Z.; Zhao, Y.C.; Li, X.P.; Liang, B.Y. Synthesis of titanium nitride powders by reactive ball milling of titanium and urea. J. Alloys Compd. 2009, 486, 189–192. [Google Scholar] [CrossRef]
- Bashkova, S.; Bandosz, T.J. The effects of urea modification and heat treatment on the process of NO2 removal by wood-based activated carbon. J. Colloid Interface Sci. 2009, 336, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zheng, Q.; Xiang, Q.; Yu, L.; Chen, P.; Gao, D.; Liu, Q.; He, F.; Yu, D.; Liu, Y.; et al. Multielectron electrode reaction kinetics with RDE and RRDE: An advanced electrochemical laboratory experiment. J. Chem. Educ. 2021, 98, 3026–3031. [Google Scholar] [CrossRef]
- Schaber, P.M.; Colson, J.; Higgins, S.; Thielen, D.; Anspach, B.; Brauer, J. Thermal decomposition (pyrolysis) of urea in an open reaction vessel. Thermochim. Acta 2004, 419, 89–97. [Google Scholar] [CrossRef]
- Zhao, C.; Hu, Z.; Qiu, Z.; Liu, K. Urea-assisted combustion synthesis of porous Li1.2Ni0.13Co0.13Mn0.54O2 microspheres as high-performance lithium-ion battery cathodes. Micro Nano Lett. 2015, 10, 662–665. [Google Scholar] [CrossRef]
- Yang, L.; May, P.W.; Yin, L.; Smith, J.A.; Rosser, K.N. Ultra fine carbon nitride nanocrystals synthesized by laser ablation in liquid solution. J. Nanopart. Res. 2007, 9, 1181–1185. [Google Scholar] [CrossRef]
- Hsiao, S.H.; Yang, C.P.; Chen, C.W.; Liou, G.S. Synthesis and properties of novel poly(amide-imide)s derived from 2,4-diaminotriphenylamine and imide ring-preformed dicarboxylic acids. J. Polym. Res. 2005, 12, 289–294. [Google Scholar] [CrossRef]
- Dong, H.T. Synthesis and Reactivity of Non-Heme Iron-Nitrosyl Complexes That Model the Active Sites of NO Reductases. Ph.D. Thesis, University of Michigan, Ann Arbor, MI, USA, 2021. [Google Scholar] [CrossRef]
- Hwang, S.W.; Umar, A.; Dar, G.N.; Kim, S.H.; Badran, R.I. Synthesis and characterization of iron oxide nanoparticles for phenyl hydrazine sensor applications. Sens. Lett. 2014, 12, 97–101. [Google Scholar] [CrossRef]
- Hanesch, M. Raman spectroscopy of iron oxides and (oxy)hydroxides at low laser power and possible applications in environmental magnetic studies. Geophys. J. Int. 2009, 177, 941–948. [Google Scholar] [CrossRef]
- Cambier, S.; Verreault, D.; Frankel, G.S. Raman investigation of anodic undermining of coated steel during environmental exposure. Corrosion 2014, 70, 1219–1229. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 255, 4747–4752. [Google Scholar] [CrossRef]
- Nohira, H.; Tsai, W.; Besling, W.; Tuominen, M. Characterization of ALCVD-Al2O3 and ZrO2 layer using X-ray photoelectron spectroscopy. J. Non-Cryst. Solids 2002, 303, 83–87. [Google Scholar] [CrossRef]
- Jansen, R.J.J.; van Bekkum, H. XPS of nitrogen-containing functional groups on activated carbon. Carbon 1995, 33, 1105–1110. [Google Scholar] [CrossRef]
- Hellgren, N.; Haasch, R.T.; Schmidt, S.; Hultman, L.; Petrov, I. Interpretation of X-ray photoelectron spectra of carbon-nitride thin films: New insights from in situ XPS. Carbon 2016, 108, 91–97. [Google Scholar] [CrossRef]
- Han, G.-Q.; Liu, Y.-R.; Hu, W.-H.; Dong, B.; Li, X.; Shang, X.; Chai, Y.-M.; Liu, Y.-Q. Three-dimensional nickel oxides/nickel structure by in situ electro-oxidation of nickel foam as robust electrocatalyst for oxygen evolution reaction. Appl. Surf. Sci. 2016, 363, 93–101. [Google Scholar] [CrossRef]
- Wang, X.; Sunarso, J.; Lu, Q.; Zhou, Z.; Dai, J.; Guan, D.; Zhou, W.; Shao, Z. High-performance platinum-perovskite composite bifunctional oxygen electrocatalyst for rechargeable Zn–air battery. Adv. Energy Mater. 2019, 10, 1903271. [Google Scholar] [CrossRef]
- González-Morales, J.; Aparicio, M.; Rosero-Navarro, N.C.; Mosa, J. A Stable Rechargeable Aqueous Zn–Oxygen Battery with New Mn-Based Bifunctional Electrocatalyst. ACS Appl. Energy Mater. 2024, 7, 16. [Google Scholar] [CrossRef]
- González-Morales, J.; Mosa, J.; Ishiyama, S.; Rosero-Navarro, N.C.; Miura, A.; Tadanaga, K.; Aparicio, M. Carbon-Free Cathode Materials Based on Titanium Compounds for Zn-Oxygen Aqueous Batteries. Batteries 2024, 10, 94. [Google Scholar] [CrossRef]
- Ran, L.; Xu, Y.; Zhu, X.; Chen, S.; Qiu, X. Mn single-atom tuning Fe-N-C catalyst enables highly efficient and durable oxygen electrocatalysis and zinc–air batteries. ACS Nano 2024, 18, 750–760. [Google Scholar] [CrossRef]
- Li, C.; Wu, M.; Liu, R. High-performance bifunctional oxygen electrocatalysts for zinc-air batteries over mesoporous Fe/Co-N-C nanofibers with embedding FeCo alloy nanoparticles. Appl. Catal. B Environ. 2019, 244, 926–934. [Google Scholar] [CrossRef]
- Hang, L.; Liu, J.; Mo, F.; Men, D.; Wen, H.; Jiang, G. Developing flexible solid-state zinc air batteries based on NiFe@NC catalyst and dual network hydrogel electrolyte. Mater. Today Phys. 2023, 42, 101288. [Google Scholar] [CrossRef]
- Tsai, J.-E.; Hong, W.-X.; Pourzolfaghar, H.; Wang, W.-H.; Li, Y.-Y. A Fe-Ni-Zn triple single-atom catalyst for efficient oxygen reduction and oxygen evolution reaction in rechargeable Zn-air batteries. Chem. Eng. J. 2023, 462, 141868. [Google Scholar] [CrossRef]
- Chen, Y.; He, T.; Liu, Q.; Hu, Y.; Gu, H.; Deng, L.; Liu, H.; Liu, Y.; Liu, Y.-N.; Zhang, Y.; et al. Highly durable iron single-atom catalysts for low-temperature zinc-air batteries by electronic regulation of adjacent iron nanoclusters. Appl. Catal. B Environ. 2022, 316, 122163. [Google Scholar] [CrossRef]
- Sim, W.J.; Nguyen, M.T.; Huang, Z.; Kheawhom, S.; Wattanakit, C.; Yonezawa, T. Efficient iron–cobalt oxide bifunctional electrode catalysts in rechargeable high current density zinc–air batteries. Nanoscale 2022, 14, 8012–8022. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, Y.; Wang, W.; Zhou, T.; Guo, Z. A highly durable zinc-air battery from a directly integrated FexNC@NiFe(OH)x bifunctional catalyst. Inorg. Chem. Front. 2023, 10, 1758–1768. [Google Scholar] [CrossRef]
- Jin, T.; Chen, W.; Zhuo, H.; Li, W.; Fu, Z.; Feng, L.; Chen, Y. Fe single atoms encased in nanoscale ZIF-8 dodecahedra for oxygen reduction and flexible zinc–air batteries. ACS Appl. Nano Mater. 2024, 7, 13547–13556. [Google Scholar] [CrossRef]
- Adhikari, A.; Chhetri, K.; Rai, R.; Acharya, D.; Kunwar, J.; Bhattarai, R.M.; Jha, R.K.; Kandel, D.; Kim, H.Y.; Kandel, M.R. (Fe-Co-Ni-Zn)-Based Metal–Organic Framework-Derived Electrocatalyst for Zinc–Air Batteries. Nanomaterials 2023, 13, 2612. [Google Scholar] [CrossRef]
- Li, Y.-W.; Zhang, W.-J.; Li, J.; Ma, H.-Y.; Du, H.-M.; Li, D.-C.; Wang, S.-N.; Zhao, J.-S.; Dou, J.-M.; Xu, L. Fe-MOF-Derived Efficient ORR/OER Bifunctional Electrocatalyst for Rechargeable Zinc–Air Batteries. ACS Appl. Mater. Interfaces 2020, 12, 44710–44719. [Google Scholar] [CrossRef]
- Wei, Y.-S.; Zhang, M.; Kitta, M.; Liu, Z.; Horike, S.; Xu, Q. A Single-Crystal Open-Capsule Metal–Organic Framework. J. Am. Chem. Soc. 2019, 141, 7906–7916. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | S.A. (m2/g) | %N | %C |
|---|---|---|---|
| 220–800 | 90.7 | 6.7 | 25.2 |
| 220–900 | 92.4 | 6.1 | 36.5 |
| Catalyst | Electrolyte | Capacity | n | ORR Half-Wave/ Onset | OER Overpot. | CV ORR/ OER | CD | Ref. |
|---|---|---|---|---|---|---|---|---|
| 220–800 | KOH | 80 mAh/g (1 mA) | 3.3 | 0.75/0.85 V | 1.5 V | Yes/Yes | 4100 h | This paper |
| 220–900 | KOH | 80 mAh/g (1 mA) | 1.9 | 0.8/0.85 V | 1.5 V | Yes/Yes | 400 h | This paper |
| (Fe-N-C)-Mn | KOH + Zn(Ac)2 | 788 mAh/g | 4.0 | 0.8/0.84 V | 1.5 V | Yes/No | 1500 CV cycles | [40] |
| Fe/Co-N-C | KOH + Zn(Ac)2 | NA | 3.8 | 0.8/0.85 V | 1.1 V | Yes/No | 24 h | [41] |
| NiFe@NCx | PANa + PVA(s) | 708 mAh/g | 3.9 | 0.68/0.82 V | 1.4 V | Yes/No | 500 h | [42] |
| Fe-Ni-Zn | KOH | 1086 Wh/kg | NA | 0.7/0.86 V | 1.5 V | Yes/No | 1800 cycles | [43] |
| NCA/FeSA + NC | PAA + PAM | NA | 3.9 | 0.78/0.85 V | 1.5 V | NA | 2400 cycles | [44] |
| (Fe-Co)O | KOH + ZnCl2 | NA | 2.8 | 0.75/0.86 V | 1.5 V | NA | 70 cycles | [45] |
| FeNC@NiFe(OH)x | KOH | 785 mAh/g Zn | NA | 0.8/0.98 V | 1.45 V | NA | 375 h | [46] |
| Fe-C-N | PVA-KOH | NA | ≈4 | 0.85/0.9 V | NA | No | 10 h | [47] |
| (Fe-Co-Ni-Zn)-Based MOFs | KOH | 732 mAh/g | 2 | 0.65/0.7 V | 1.3 V | No | NA | [48] |
| FeS/Fe3C@NS-C-900 | M) + Zn(OAc)2 2H2O | 750 mAh/g | 4.2 | 0.85/0.9 V | 1.4 V | NA | 865 h | [49] |
| FeNiP/NCH | KOH + M ZnCl2 | NA | NA | 0.75/0.8 V | 1.5 V | NA | 500 h | [50] |
| FeNx/S-NC | KOH + H2SO4 | 740 mAh/g | NA | 0.8/0.9 V | NA | NA | NA | [16] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Morales, J.; Mosa, J.; Aparicio, M. Binder-Free Fe-N-C-O Bifunctional Electrocatalyst in Nickel Foam for Aqueous Zinc–Air Batteries. Batteries 2025, 11, 159. https://doi.org/10.3390/batteries11040159
González-Morales J, Mosa J, Aparicio M. Binder-Free Fe-N-C-O Bifunctional Electrocatalyst in Nickel Foam for Aqueous Zinc–Air Batteries. Batteries. 2025; 11(4):159. https://doi.org/10.3390/batteries11040159
Chicago/Turabian StyleGonzález-Morales, Jorge, Jadra Mosa, and Mario Aparicio. 2025. "Binder-Free Fe-N-C-O Bifunctional Electrocatalyst in Nickel Foam for Aqueous Zinc–Air Batteries" Batteries 11, no. 4: 159. https://doi.org/10.3390/batteries11040159
APA StyleGonzález-Morales, J., Mosa, J., & Aparicio, M. (2025). Binder-Free Fe-N-C-O Bifunctional Electrocatalyst in Nickel Foam for Aqueous Zinc–Air Batteries. Batteries, 11(4), 159. https://doi.org/10.3390/batteries11040159