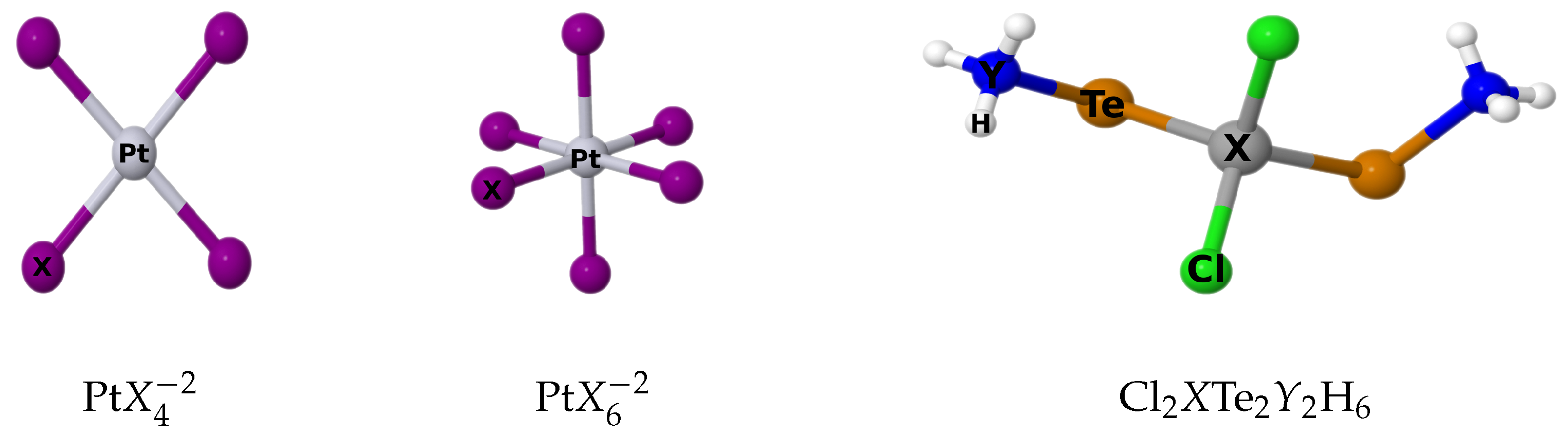
4.1. Platinum Complexes
In
Table 3, we present the results of calculations of the isotropic nuclear magnetic shielding of the Pt atom,
(Pt), obtained using the LRESC and 4C-RPA methods. Both the paramagnetic and diamagnetic components are shown for comparison between the two calculation schemes. Additionally, we include the chemical shifts,
(Pt), with respect to the PtCl
molecule, which serves as a reference for both theoretical and experimental results.
From
Table 3, it can be observed that the LRESC values are not in close agreement with the 4C-RPA values. However, the overall sign of the total shielding value is consistent for the entire set of molecules, except for PtF
and PtBr
. It is known that the LRESC scheme encounters some difficulties in accurately reproducing magnetic shielding for nuclei in the 6th row of the periodic table, although it does capture the correct trends [
21,
27,
37].
A more insightful analysis can be performed by considering the chemical shifts instead of the absolute values and comparing them with the experimental results. In this regard, the LRESC chemical shifts exhibit closer agreement with the experimental values compared to the 4C-RPA results.
Figure 2 demonstrates that
follows the same trend as
across the entire set of molecules studied. However, it is important to note that these calculations were conducted under static conditions, and several effects were not taken into account.
The primary differences between LRESC and 4C-RPA total values originate from the paramagnetic component, as the diamagnetic component exhibits a nearly constant difference. Specifically, the LRESC diamagnetic component is consistently approximately 1400 ppm lower than the corresponding 4c diamagnetic contributions. The variation of the paramagnetic component is not constant and is influenced by the ligand-dependent contributions as illustrated in
Table 4 and
Table 5.
The primary electronic mechanisms responsible for ligand-dependent relativistic effects are the well-known contribution and the contribution. Both of these mechanisms exhibit significant variations with the atomic number of the halogen atom, particularly in planar systems. The contribution is widely recognized as being important in numerous atomic and molecular systems. However, the contribution is also of comparable magnitude but with an opposite sign. As a result, the overall relativistic effects depend on the difference between these two contributions. In the case of the Pt molecule, the contribution is greater than the contribution, resulting in a negative value. Moreover, this negative value becomes more pronounced (in absolute value) as the influence of the halogen substituent increases. This analysis tell us that one must include the non-SO contributions to the shieldings of heavy atoms when they are bonded to heavy atoms.
Table 4 and
Table 5 provide insights into the ligand-dependent and non-ligand-dependent contributions to the nuclear magnetic shieldings of the platinum atom, focusing on the localized molecular orbitals. These contributions can be categorized into core orbitals, lone pairs, and bond orbitals. It is evident that the non-ligand-dependent corrections remain unaffected by the substituent atom, with the primary contribution originating from the platinum core orbitals. On the other hand, for the ligand-dependent contribution, the core orbitals of platinum and Pt-X bond orbitals play crucial roles in the
correction, while
arises from the platinum core orbitals. However, when the weight of the halogen atom increases, such a contribution is reduced and the contribution of the lone pairs becomes important, reaching the 50% of the platinum core contributions. As shown in that Tables, the main contribution to
arises from the core(Pt) and the bonding Pt-
X being
X = F, Cl, Br, I. There is also a contribution from the LP
of the atom
X. When
X = I,
is around 3 times the contribution of
. This becomes 1.5 times when
X = F. The importance of HAVHA effects are clearly shown in this case.
At the NR level, the diamagnetic component of platinum (Pt) exhibits a similar trend for the whole set of molecules and experiences an increase of less than 600 ppm from F to I atoms. The core orbital contributions of platinum remain independent of the substituent halogen atom, constituting approximately 92% to 98% of the total . However, the core orbitals of the substituent atom play a crucial role in determining the variation in this component, ranging from 40 ppm for F to 662 ppm for I. The combination of these contributions determines the overall behavior of .
On the other hand, the NR paramagnetic component of platinum shielding (Pt) displays a significant variation depending on the substituent halogen atom, ranging from −15,838 ppm for F to −8055 ppm for I. Analysis of the molecular orbitals reveals that the core orbitals of Pt are consistently negative and dominant, ranging from −15,772 ppm for F to −8754 ppm for I. The core orbitals of the halogen atoms also contribute negatively, with a negligible contribution for F but progressively increasing with the atomic weight of the halogen, accounting for up to 4% of the platinum core orbitals contribution in the case of I.
Table 4 and
Table 5 further illustrate the distinct behavior of the remaining MOs. The Pt-
X bond orbitals consistently contribute positively but at one or two orders of magnitude lower than the platinum core orbitals. In contrast, the
-type and
-type lone pair contributions exhibit similar behavior, with negative values for F substituents that transition to positive values for the other halogens. These lone-pair contributions can collectively account for up to 10% of the platinum core orbitals contribution in the case of the PtCl
molecule.
In
Figure 3, the contributions of localized molecular orbitals to the
and
relativistic corrections are presented for the entire set of molecules under study. Additionally, for Pt
, the contribution of
is also shown. The
mechanism originates from the core electrons of platinum, but as the weight of the halogen increases, the lone-pair orbitals become significant and change sign from negative to positive. The primary contribution to the
mechanism also arises from the core orbitals of platinum. Furthermore, the
mechanism exhibits a comparable magnitude to
in PtF
and PtCl
molecules, with the core orbitals of platinum and Pt-X bonds playing a significant role.
We will now analyze the
(Pt) in Pt
X (
X = F, Cl, Br, I) molecules, as shown in
Table 3. This allows us to observe the influence of the number of substituent halogen atoms as well as the influence of the change in geometry.
The core contributions to the paramagnetic and diamagnetic components are the same as those in Pt molecules. These contributions arise from the core orbitals of Pt and are responsible for these effects.
The diamagnetic component, (Pt), in Pt molecules is approximately 1% larger than in Pt molecules. However, the contribution of platinum core orbitals slightly decreases with an increasing number of bonds, resulting in a slightly higher contribution from the other molecular orbitals. These orbitals contribute a larger percentage to the total values compared to Pt molecules. Additionally, the core value of the halogen atoms increases by 31% when Pt is bonded to six halogen atoms. This analysis highlights the effect of the increased number of halogen substituents on the electronic mechanisms.
Regarding the paramagnetic component,
Table 6 and
Table 7 reveal that the
relativistic correction is less significant than the
corrections and has an opposite sign. As a result, the ligand-dependent corrections to the paramagnetic component have negative values with the exceptions of PtF
and PtCl
.
In Pt
molecules with D
symmetry, both
and
corrections decrease as the weight of the substituent halogen atom increases. However, in Pt
molecules with O
symmetry, the behavior is opposite as illustrated in
Figure 4.
4.2. Cadmium and Mercury Complexes
Table 8 presents the paramagnetic and diamagnetic contributions to
(
X) and
(Te) in the Cl
XTe
YH
(
X = Cd, Hg;
Y = N, P) molecular systems, calculated using both the LRESC and 4C-RPA methods. For the Cd atom, which belongs to the 5th row of the periodic table, the LRESC values are very close to the 4C-RPA values, with differences ranging from 0.2% to 1%. This indicates a good agreement between the two methods for Cd atom. On the other hand, for the Hg atom belonging to the 6th row of the periodic table, larger differences are observed between the LRESC and 4C-RPA values. The differences range from 16% to 18%, which is consistent with the expected behavior for an atom in the 6th row. In addition, the nuclear magnetic shielding of the Te atom is included in the table to analyze the HAVHA effect. When the total relativistic contribution cannot be explained by only including the SO mechanism, that effect is then in action. In other words, the HAVHA effects become important when the SO mechanism does not explain the trends found for the shielding of HAs that are bonded to another HA.
The diamagnetic component of Cd and Hg in the ClXTeYH (X = Cd, Hg; Y = N, P) molecular systems shows quite close values when the nitrogen (N) atom is replaced by the phosphorus (P) atom, both at the LRESC and 4C-RPA levels. This similarity indicates the core nature of this component. In contrast, the paramagnetic component of Cd and Hg exhibits a small variation when the N atom is replaced by the P atom due to the two-bond coupling between X and Y. However, the influence of the surrounding environment on the total value is not significant, with values of approximately 163 ppm for Cd and 380 ppm for Hg at the 4C-RPA level. This results in a relative increase of approximately 3.7% for Cd and 2.8% for Hg when the N atom is replaced by the P atom.
As mentioned above, the influence of replacing Cd with Hg on (Te) provides a measure of the HAVHA effect on this parameter. Additionally, it is interesting to analyze the replacement of the atom N with the atom P. At the 4C-RPA level, the substitution of Cd by Hg in ClXTeNH molecules leads to a 15% decrease in (Te), while such a substitution in ClXTePH molecules results in only a 0.5% decrease. These values demonstrate a significant HAVHA effect on (Te) when nitrogen is present in the molecule but a negligible effect when phosphorus is present. However, the most substantial change occurs when N is replaced by P, resulting in an increase of approximately 2900 ppm for Cd-centered molecules and 3230 ppm for Hg-centered molecules. This represents a variation of over 100% in (Te) from ClXTeNH to ClXTePH molecules.
Upon analyzing the orbitals responsible for the nonrelativistic diamagnetic component () of transition metals Cd and Hg, it becomes evident that this component is primarily attributed to the contribution of core molecular orbitals. However, the core orbitals of Te contribute approximately 6% of the total value for Cd and 3% for Hg. On the other hand, the non-ligand-dependent contributions to the nonrelativistic paramagnetic component () solely originate from the core orbitals of Cd or Hg. The behavior of and the ligand-dependent contributions to this component warrant further analysis.
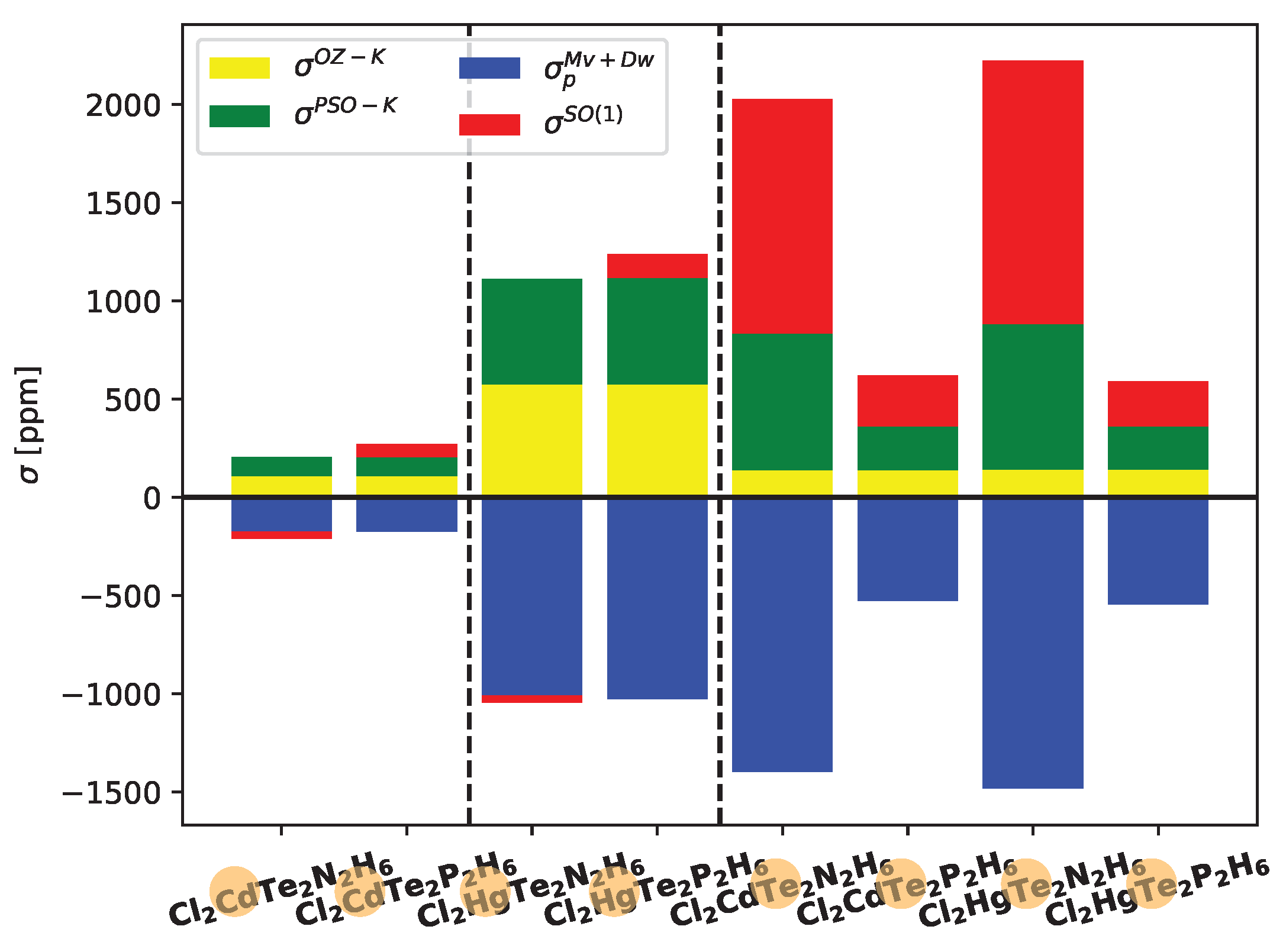
In
Figure 5, the contributions of the most significant ligand-dependent electronic mechanisms, namely
,
,
, and
, are shown for Cd, Hg, and Te in Cl
XTe
Y
H
systems at the LRESC level. The ligand-dependent contributions on Cd are relatively small compared to those on Hg in both analyzed molecules, but the behavior of each electronic mechanism is similar. Due to the comparable magnitudes of the main electronic mechanisms with opposite signs, the total ligand-dependent value is very small. Specifically,
(Hg) amounts to only 66 ppm in the Cl
XTe
N
H
molecule and 209 ppm in the Cl
XTe
P
H
molecule. The difference arises from the change in sign of
, which is negative in the first molecule and positive in the second.
Figure 5 also provides a clear demonstration of the HAVHA effect on
(Te). As mentioned earlier, the HAVHA effect is approximately 15% in Cl
XTe
N
H
molecules, and the contributions of each electronic mechanism to
(Te) are very similar when Cd or Hg atoms are present in the molecule. However, a significant difference arises when N is replaced by P, particularly in the reduction of
,
, and
(in absolute value) contributions. The molecular orbitals responsible for these changes are the
X-Te bond orbitals, and in particular, the lone pairs of tellurium (LP
Te) as shown in Tables provided in the electronic supporting information (ESI).
Table 9 and
Table 10 present the contributions of localized molecular orbitals to
,
,
, and
on Cd in Cl
CdTe
YH
(
Y = N, P) molecules. Similarly,
Table 11 and
Table 12 display the corresponding contributions of the Hg atom in Cl
HgTe
YH
molecules. These tables provide detailed information about the specific molecular orbitals involved in each mechanism and their respective contributions to the overall electronic behavior of the system.
For the Cd atom, the , , and relativistic electronic mechanisms show similar values in both molecules when the N atom is replaced by P. The contribution of each molecular orbital follows the same trend. However, the mechanism changes its sign between the two molecules, and the Cd–Te bond orbitals are responsible for the change in the final value. A similar behavior is observed for the Hg atom, where the major change occurs when N is replaced by P in the mechanism, attributed to the Hg–Te bond orbitals. The contributions change from −186 ppm in ClHgTeNH to 47 ppm in ClHgTePH, even including the negative increase due to the Hg–Cl bondings.
As mentioned earlier, the replacement of the p-orbital element produces significant changes in some relativistic electronic mechanisms and, consequently, in the total magnetic shielding value. This effect is more significant than the variation in the weight of the transition metal atom in this kind of molecule.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}