

Improving the Path to Obtain Spectroscopic Parameters for the PI3K—(Platinum Complex) System: Theoretical Evidences for Using 195Pt NMR as a Probe

 ,
, 
Abstract
1. Introduction
2. Methodology
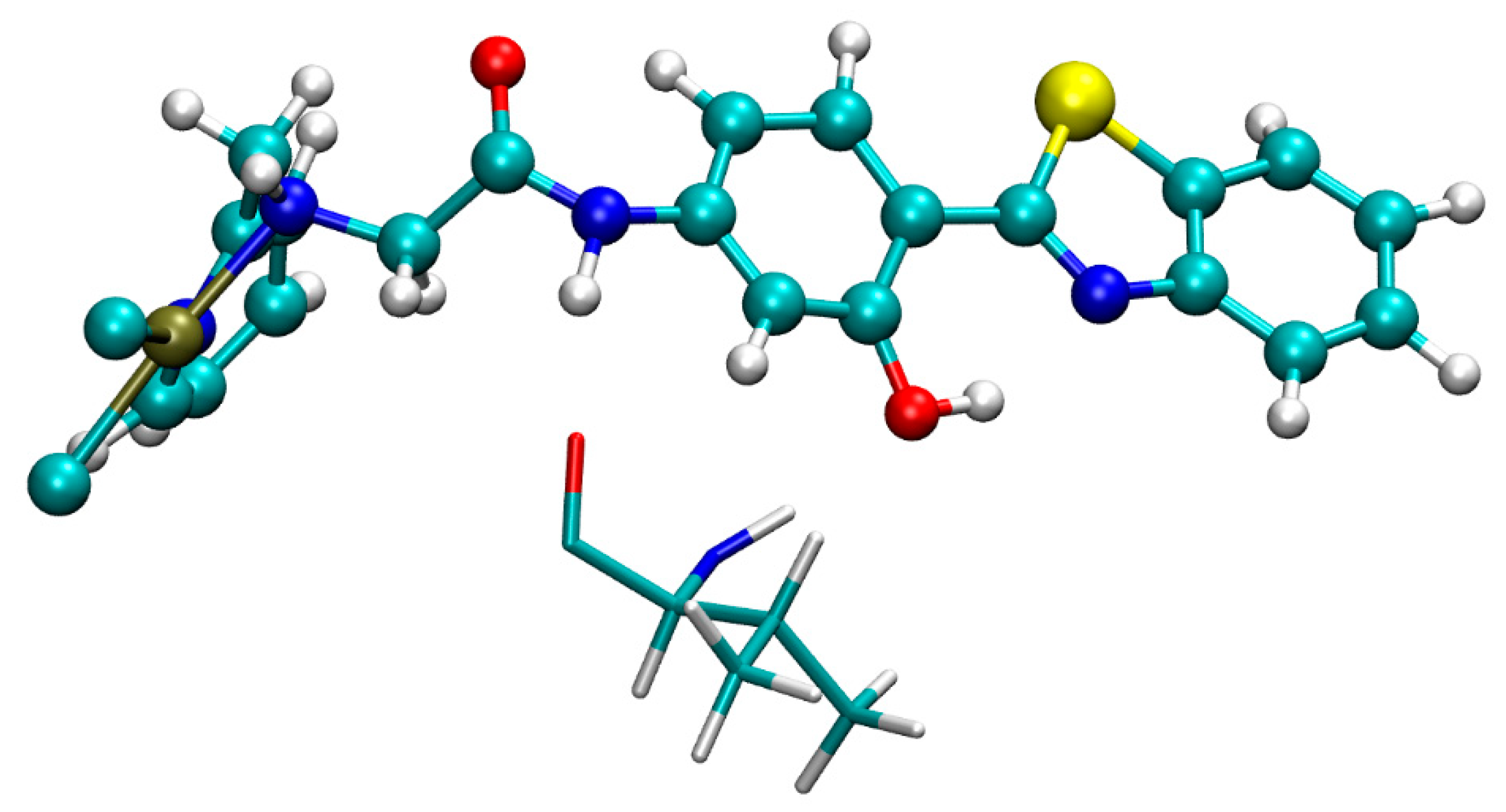
2.1. Molecular Docking
2.2. Classical Molecular Dynamics
2.2.1. Selection of the Best Snapshots and Residues
2.3. Nuclear Magnetic Resonance (NMR) Calculations
3. Results and Discussion
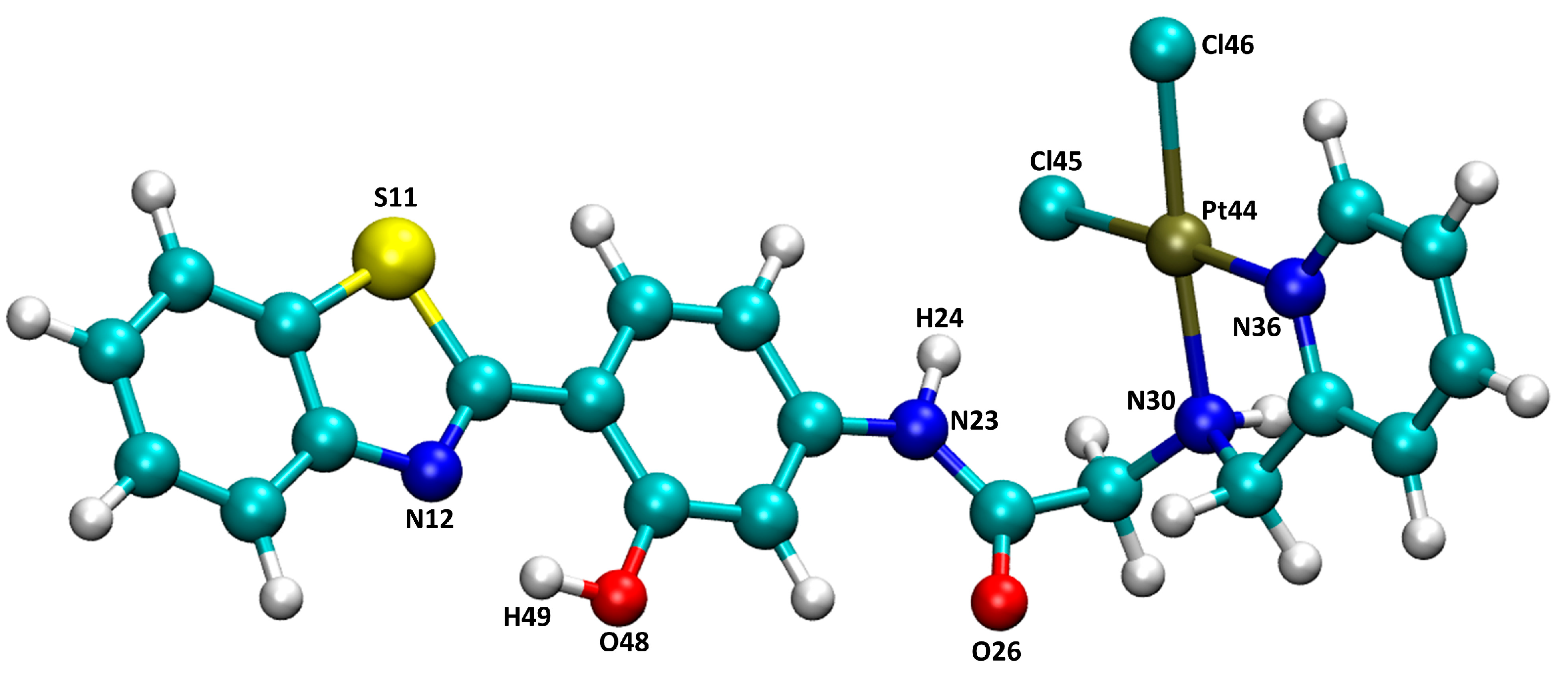
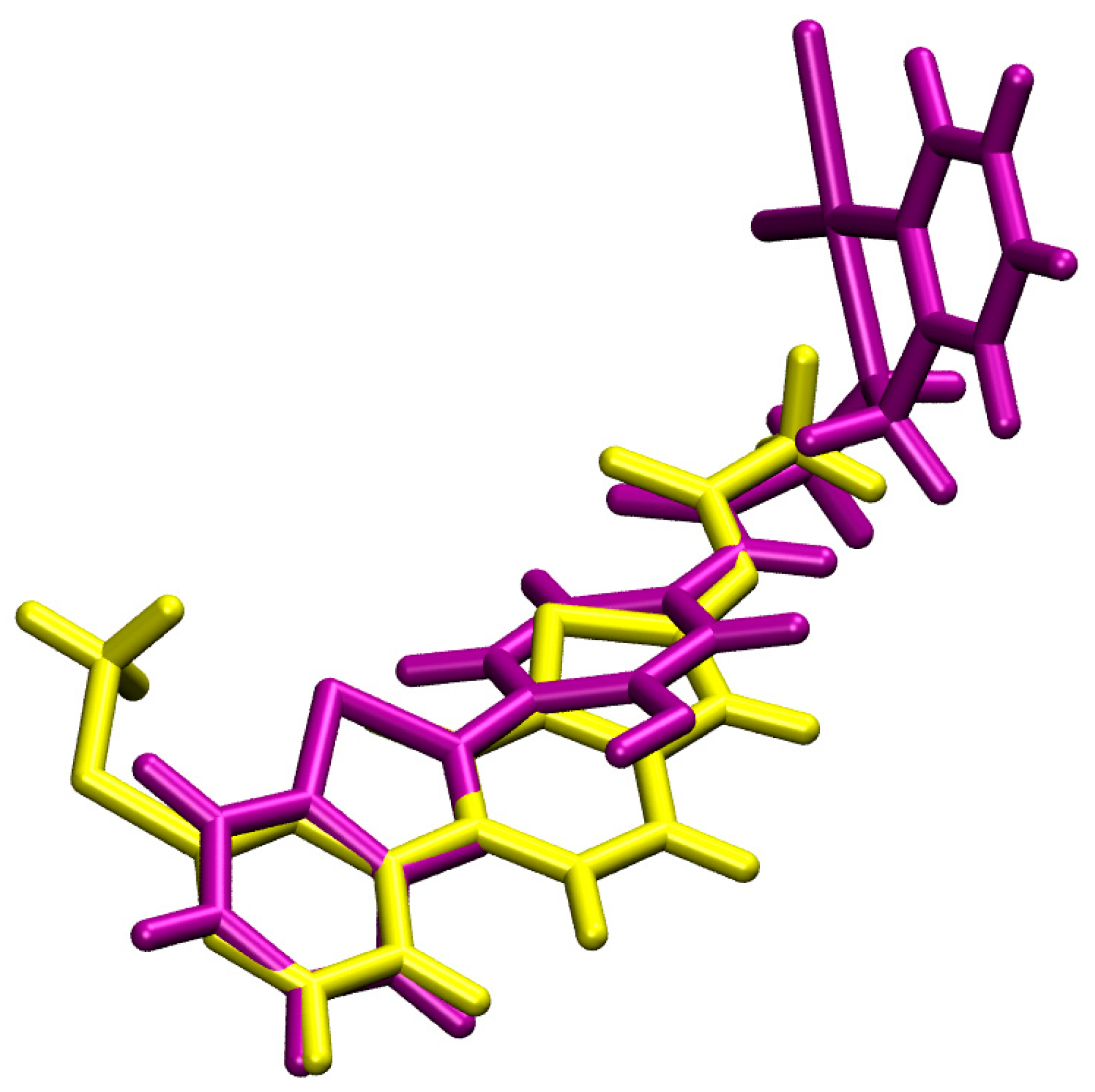
3.1. Docking
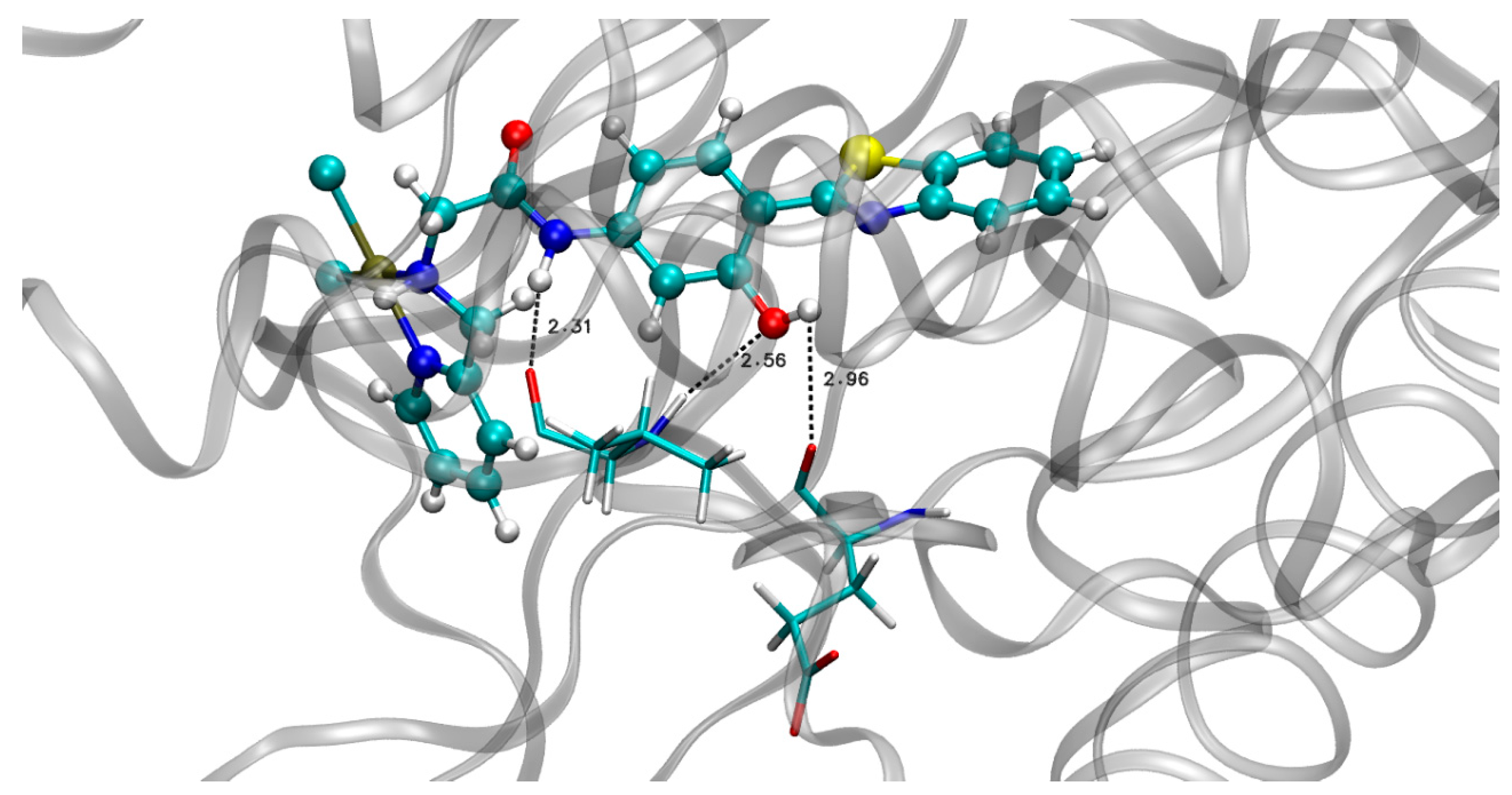
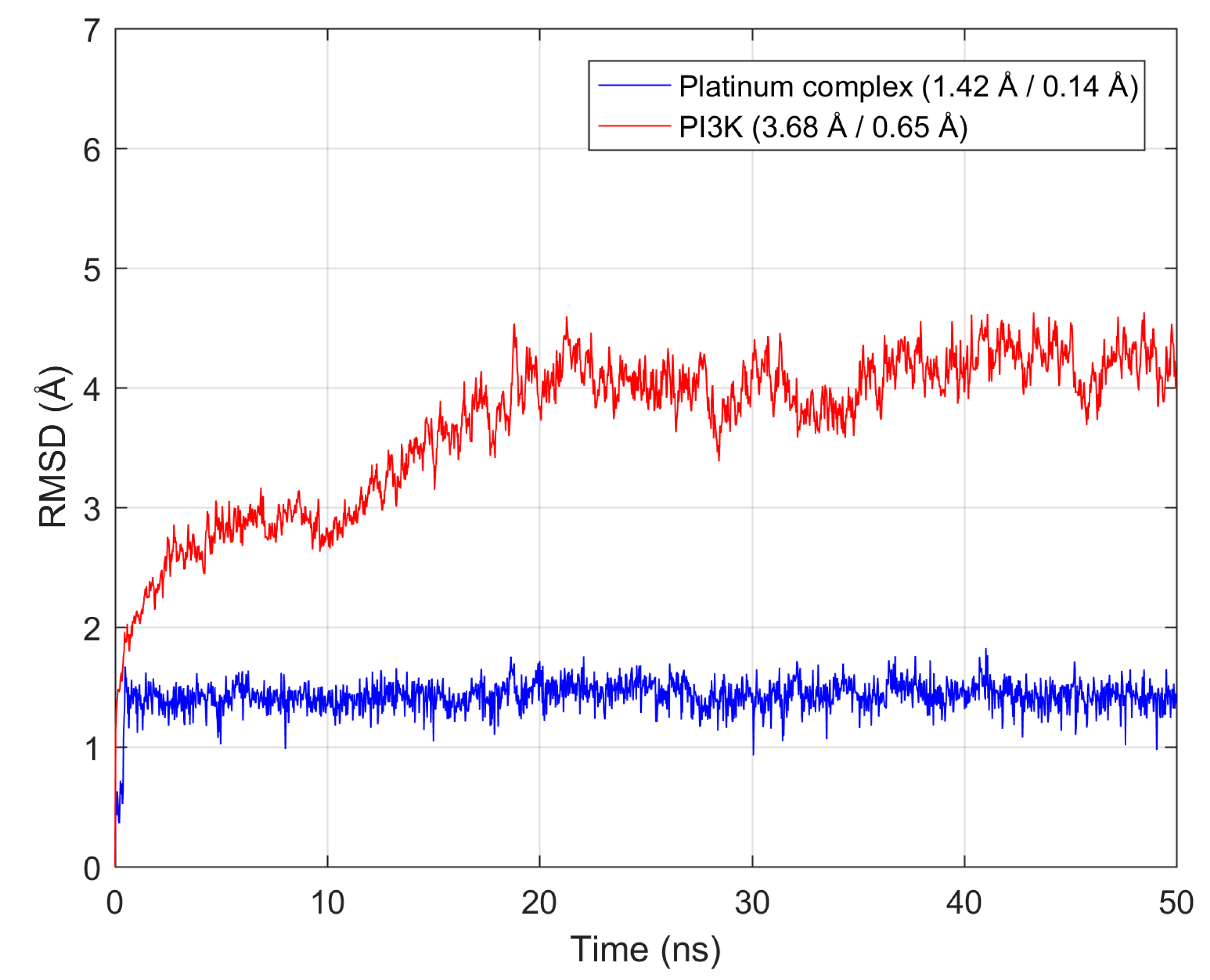
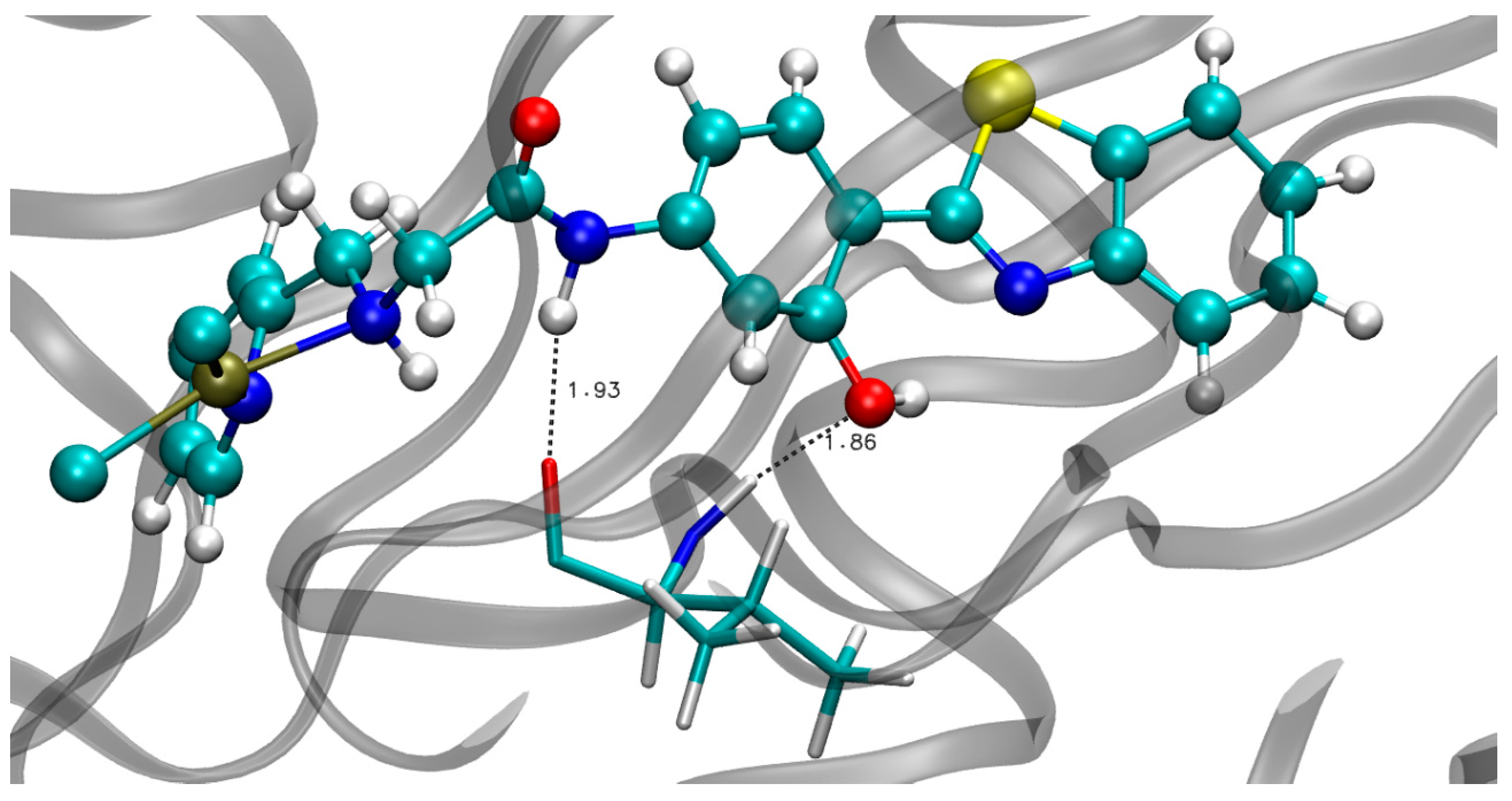
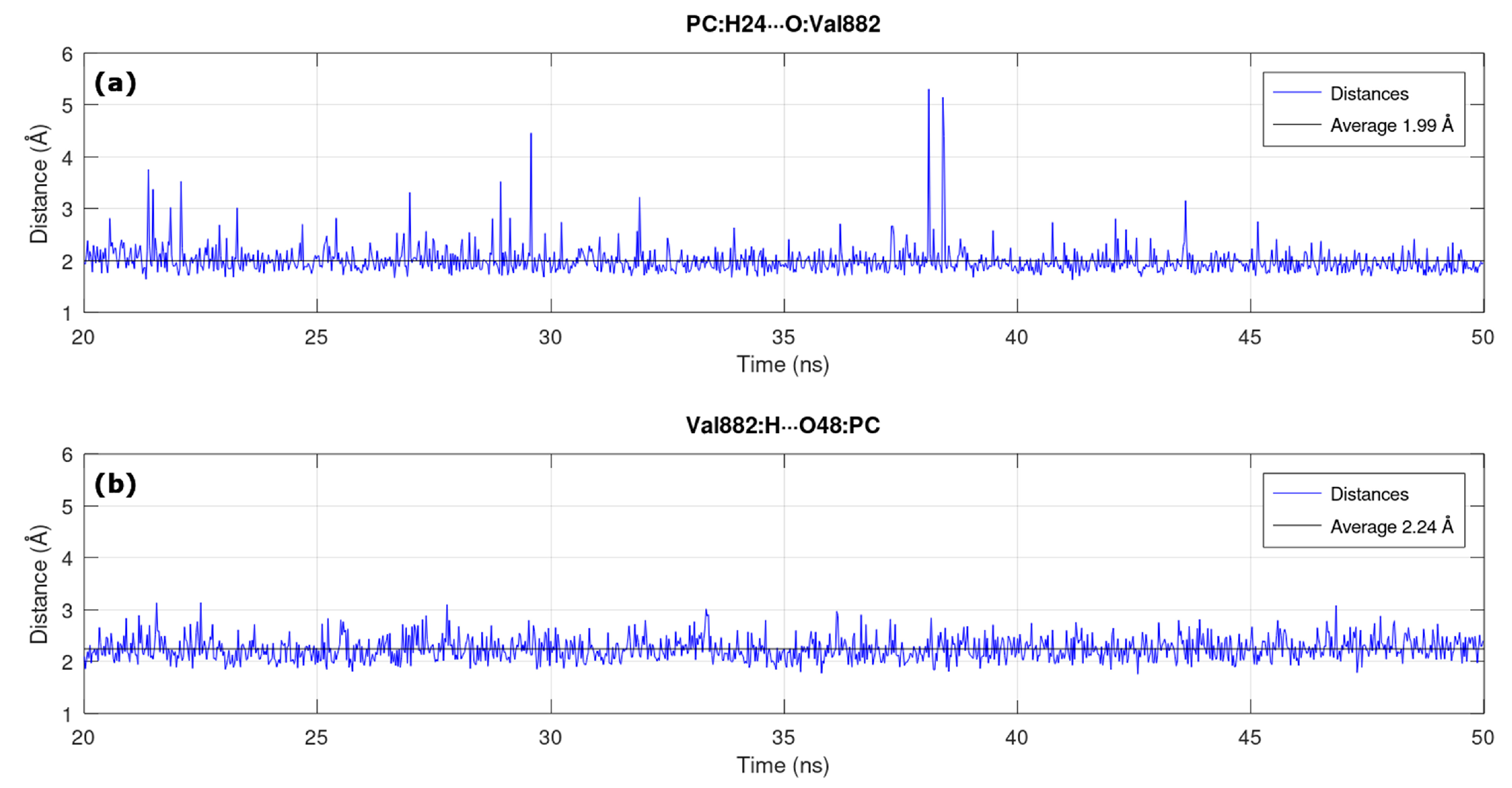
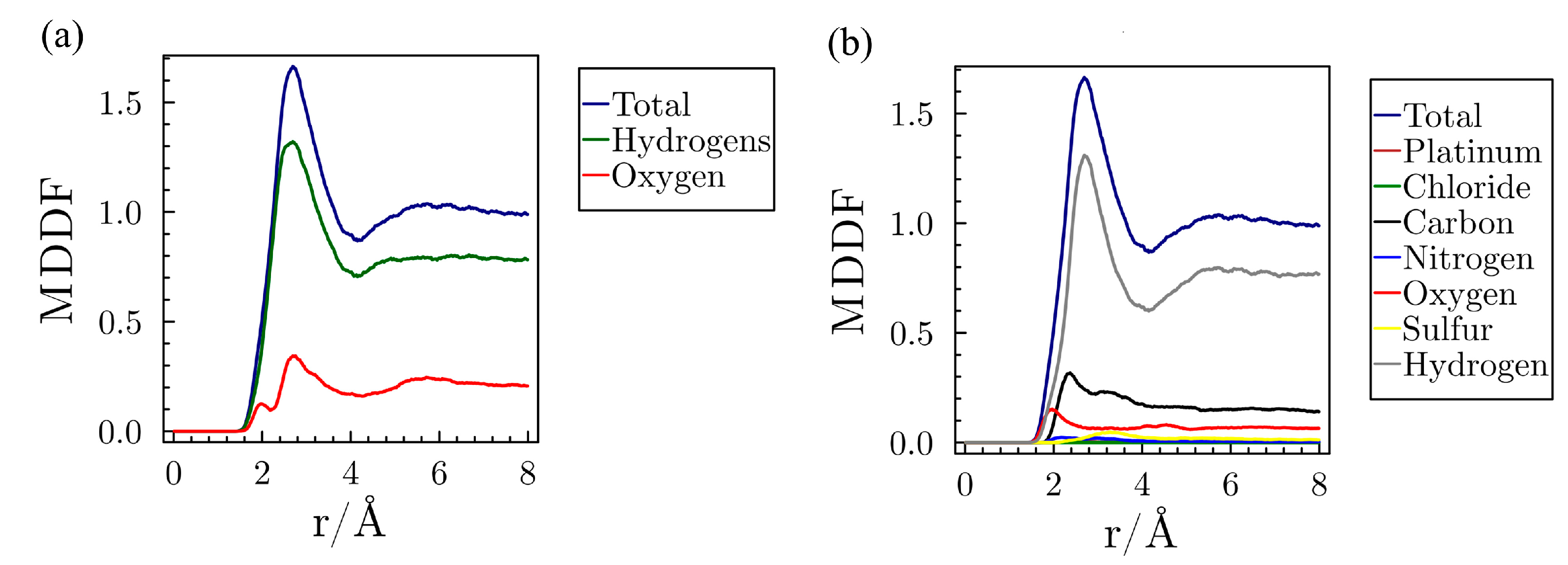
3.2. MD in Enzymatic and Aqueous Environment
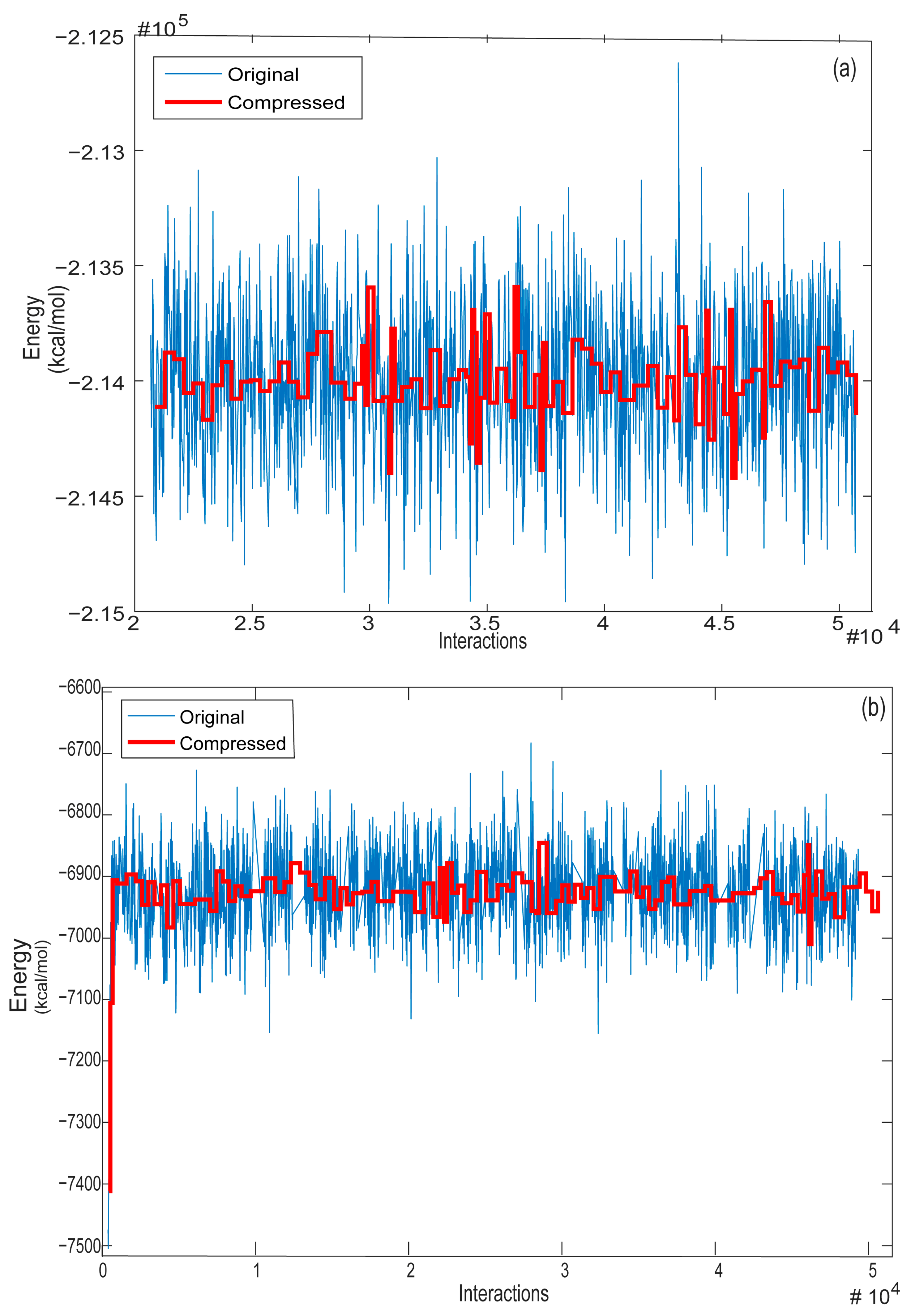
3.3. Selection of the Best MD Structures—OWSCA
3.4. NMR Spectroscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Catalan-Gomez, S.; Briones, M.; Cortijo-Campos, S.; Garcia-Mendiola, T.; de Andres, A.; Garg, S.; Kung, P.; Lorenzo, E.; Pau, J.L.; Redondo-Cubero, A. Breast cancer biomarker detection through the photoluminescence of epitaxial monolayer MoS2 flakes. Sci. Rep. 2020, 10, 16039. [Google Scholar] [CrossRef] [PubMed]
- Andolpho, G.A.; da Cunha, E.F.F.; Ramalho, T.C. Insights into the value of statistical models, solvent, and relativistic effects for investigating Re complexes of 2-(4′-aminophenyl) benzothiazole: A potential spectroscopic probe. J. Mol. Model. 2022, 28, 154. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.T.L.; Goncalves, M.A.; Mancini, D.T.; Kuca, K.; Ramalho, T.C. First Attempts of the Use of Pt-195 NMR of Phenylbenzothiazole Complexes as Spectroscopic Technique for the Cancer Diagnosis. Molecules 2019, 24, 3970. [Google Scholar] [CrossRef]
- Fuso, P.; Muratore, M.; D’Angelo, T.; Paris, I.; Carbognin, L.; Tiberi, G.; Pavese, F.; Duranti, S.; Orlandi, A.; Tortora, G.; et al. PI3K Inhibitors in Advanced Breast Cancer: The Past, The Present, New Challenges and Future Perspectives. Cancers 2022, 14, 2161. [Google Scholar] [CrossRef] [PubMed]
- Haider, K.; Rehman, S.; Pathak, A.; Najmi, A.K.; Yar, M.S. Advances in 2-substituted benzothiazole scaffold-based chemotherapeutic agents. Arch. Pharm. 2021, 354, 2100246. [Google Scholar] [CrossRef]
- Pereira, A.F.; Prandi, I.G.; Ramalho, T.C. Parameterization and validation of a new force field for Pt(II) complexes of 2-(4′-amino-2′-hydroxyphenyl)benzothiazole. Int. J. Quantum Chem. 2021, 121, e26525. [Google Scholar] [CrossRef]
- Martinez, L.; Shimizu, S. Molecular Interpretation of Preferential Interactions in Protein Solvation: A Solvent-Shell Perspective by Means of Minimum-Distance Distribution Functions. J. Chem. Theory Comput. 2017, 13, 6358–6372. [Google Scholar] [CrossRef]
- Goncalves, M.A.; Santos, L.S.; Prata, D.M.; Peixoto, F.C.; da Cunha, E.F.F.; Ramalho, T.C. Optimal wavelet signal compression as an efficient alternative to investigate molecular dynamics simulations: Application to thermal and solvent effects of MRI probes. Theor. Chem. Acc. 2016, 136, 15. [Google Scholar] [CrossRef]
- D’Angelo, N.D.; Kim, T.S.; Andrews, K.; Booker, S.K.; Caenepeel, S.; Chen, K.; D’Amico, D.; Freeman, D.; Jiang, J.A.; Liu, L.B.; et al. Discovery and Optimization of a Series of Benzothiazole Phosphoinositide 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Dual Inhibitors. J. Med. Chem. 2011, 54, 1789–1811. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Arba, M.; Sufriadin, M.; Tjahjono, D.H. Identification of Phosphatidylinositol 3-Kinase delta (PI3K delta) Inhibitor: Pharmacophore-based Virtual Screening and Molecular Dynamics Simulation. Indones. J. Chem. 2020, 20, 1070–1079. [Google Scholar] [CrossRef]
- Farrokhzadeh, A.; Akher, F.B.; Egan, T.J. Molecular Mechanism Exploration of Potent Fluorinated PI3K Inhibitors with a Triazine Scaffold: Unveiling the Unusual Synergistic Effect of Pyridine-to-Pyrimidine Ring Interconversion and CF3 Defluorination. J. Phys. Chem. B 2021, 125, 10072–10084. [Google Scholar] [CrossRef] [PubMed]
- York, D.M.; Kollman, P.A. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for nmr chemical-shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Paschoal, D.; Guerra, C.F.; de Oliveira, M.A.L.; Ramalho, T.C.; Dos Santos, H.F. Predicting Pt-195 NMR Chemical Shift Using New Relativistic All-Electron Basis Set. J. Comput. Chem. 2016, 37, 2360–2373. [Google Scholar] [CrossRef] [PubMed]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.J.; Plastaras, J.P.; Marzilli, L.G. A molecular mechanics amber-type force-field for modeling platinum complexes of guanine derivatives. Inorg. Chem. 1994, 33, 6061–6077. [Google Scholar] [CrossRef]
- Scheeff, E.D.; Briggs, J.M.; Howell, S.B. Molecular modeling of the intrastrand guanine-guanine DNA adducts produced by cisplatin and oxaliplatin. Mol. Pharmacol. 1999, 56, 633–643. [Google Scholar] [CrossRef]
- Yesylevskyy, S.; Cardey, B.; Kraszewski, S.; Foley, S.; Enescu, M.; da Silva, A.M.; Dos Santos, H.F.; Ramseyer, C. Empirical force field for cisplatin based on quantum dynamics data: Case study of new parameterization scheme for coordination compounds. J. Mol. Model. 2015, 21, 268. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Ponomarev, S.Y.; Kaminski, G.A. Polarizable Simulations with Second-Order Interaction Model (POSSIM) Force Field: Developing Parameters for Alanine Peptides and Protein Backbone. J. Chem. Theory Comput. 2011, 7, 1415–1427. [Google Scholar] [CrossRef]
- Cvitkovic, J.P.; Kaminski, G.A. Developing Multisite Empirical Force Field Models for Pt(II) and Cisplatin. J. Comput. Chem. 2017, 38, 161–168. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.A.; Goncalves, A.S.; Franca, T.C.C.; Santana, M.S.; Da Cunha, E.F.F.; Ramalho, T.C. Improved Protocol for the Selection of Structures from Molecular Dynamics of Organic Systems in Solution: The Value of Investigating Different Wavelet Families. J. Chem. Theory Comput. 2022, 18, 5810–5818. [Google Scholar] [CrossRef] [PubMed]
- Murugavel, S.; Ravikumar, C.; Jaabil, G.; Alagusundaram, P. Synthesis, crystal structure analysis, spectral investigations (NMR, FT-IR, UV), DFT calculations, ADMET studies, molecular docking and anticancer activity of 2-(1-benzyl-5-methyl-1H-1,2,3-triazol-4-yl)-4-(2-chlorophenyl)-6-methoxypyridine—A novel potent human topoisomerase IIα inhibitor. J. Mol. Struct. 2019, 1176, 729–742. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Environment | δ(195Pt) (ppm) |
|---|---|
| Enzymatic | −1306.58 ± 25.33 |
| Aqueous | −2911.25 ± 9.66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, T.M.R.; Andolpho, G.A.; Tavares, C.A.; Gonçalves, M.A.; Ramalho, T.C. Improving the Path to Obtain Spectroscopic Parameters for the PI3K—(Platinum Complex) System: Theoretical Evidences for Using 195Pt NMR as a Probe. Magnetochemistry 2023, 9, 89. https://doi.org/10.3390/magnetochemistry9040089
Santos TMR, Andolpho GA, Tavares CA, Gonçalves MA, Ramalho TC. Improving the Path to Obtain Spectroscopic Parameters for the PI3K—(Platinum Complex) System: Theoretical Evidences for Using 195Pt NMR as a Probe. Magnetochemistry. 2023; 9(4):89. https://doi.org/10.3390/magnetochemistry9040089
Chicago/Turabian StyleSantos, Taináh M. R., Gustavo A. Andolpho, Camila A. Tavares, Mateus A. Gonçalves, and Teodorico C. Ramalho. 2023. "Improving the Path to Obtain Spectroscopic Parameters for the PI3K—(Platinum Complex) System: Theoretical Evidences for Using 195Pt NMR as a Probe" Magnetochemistry 9, no. 4: 89. https://doi.org/10.3390/magnetochemistry9040089
APA StyleSantos, T. M. R., Andolpho, G. A., Tavares, C. A., Gonçalves, M. A., & Ramalho, T. C. (2023). Improving the Path to Obtain Spectroscopic Parameters for the PI3K—(Platinum Complex) System: Theoretical Evidences for Using 195Pt NMR as a Probe. Magnetochemistry, 9(4), 89. https://doi.org/10.3390/magnetochemistry9040089