Abstract
Computational electron paramagnetic resonance (EPR) spectroscopy is an important field of applied quantum chemistry that contributes greatly to connecting spectroscopic observations with the fundamental description of electronic structure for open-shell molecules. However, not all EPR parameters can be predicted accurately and reliably for all chemical systems. Among transition metal ions, Cu(II) centers in inorganic chemistry and biology, and their associated EPR properties such as hyperfine coupling and g-tensors, pose exceptional difficulties for all levels of quantum chemistry. In the present work, we approach the problem of Cu(II) g-tensor calculations using double-hybrid density functional theory (DHDFT). Using a reference set of 18 structurally and spectroscopically characterized Cu(II) complexes, we evaluate a wide range of modern double-hybrid density functionals (DHDFs) that have not been applied previously to this problem. Our results suggest that the current generation of DHDFs consistently and systematically outperform other computational approaches. The B2GP-PLYP and PBE0-DH functionals are singled out as the best DHDFs on average for the prediction of Cu(II) g-tensors. The performance of the different functionals is discussed and suggestions are made for practical applications and future methodological developments.
1. Introduction
Copper is among the most abundant transition metals in bioinorganic systems [1,2,3,4,5,6,7]. Electron paramagnetic resonance (EPR) spectroscopy is a uniquely useful approach for Cu(II) systems because it provides direct insights into the electronic and geometric structure of Cu(II) centers and the nature of their ligand sphere [4,8]. Relationships between structure and EPR parameters have been previously discussed based on ligand-field theory, as well as experimental correlations [8,9,10,11,12,13]. However, such correlations are often hard to extend to complex structural cases such as those frequently encountered in enzymatic active sites. Therefore, in most cases, interpretation of EPR spectroscopy requires input from quantum chemical calculations that correlate expected EPR parameters with molecular electronic and geometric structure [14,15,16,17,18,19,20,21,22,23,24]. A primary target property for Cu(II) systems is the g-matrix (commonly referred to as g-tensor), which describes the interaction of the unpaired electron with the effective magnetic field. The limitations of classical density functional theory (DFT) methods to accurately predict g-tensors of copper systems have been recognized in multiple studies [20,21,25,26,27,28,29,30] and have been attributed to deficiencies in the description of the covalency of Cu–ligand bonds [29,31] and of the electronic excitation energies [29]. Notably, wavefunction-based methods, although systematically improvable and able to achieve arbitrary levels of accuracy, cannot at this point in time be applied to copper-based systems without the introduction of approximations that are detrimental for the accuracy of the predicted parameters [25,32,33,34,35], as well as for hyperfine coupling constants [36], thus severely restricting the utility of these methods. Hence, density functional theory (DFT) currently provides the best compromise between accuracy and cost and serves as the most promising and pragmatic platform for establishing practically useful computational protocols with predictive value.
Over the last thirty years, a plethora of density functionals (DFs) has been developed [37,38], with different levels of approximation and empirical parameters optimized for the prediction of different chemical properties. Notably, EPR parameters practically never feature in training sets of new DFs. Experience shows that local density approximation (LDA)- and generalized-gradient approximation (GGA)-type functionals systematically underestimate g-values [20,21]. Hybrid density functionals (HDFs) include a Hartree–Fock (HF) exchange component (also called exact exchange, EEX). Inclusion of this exchange term in common hybrid functionals is beneficial [20,21,28,30], but standard HDFs still cannot provide sufficient accuracy. Increasing the admixture of exact exchange (EEX) to 30–40% has been recognized as a good strategy to improve the accuracy of g-tensor calculations for 3d, 4d and 5d transition metal complexes [39,40,41,42], while the effect of changing the correlation functional is minimal in comparison. However, increasing the Hartree–Fock exchange percentage introduces spin contamination in unrestricted calculations [43], which may be detrimental for SOMOs that have metal-ligand antibonding character. Local hybrid functionals with position-dependent EEX admixture have been reported to alleviate the problem of spin polarization [44].
Double-hybrid density functionals (DHDFs) additionally introduce unoccupied Kohn–Sham orbitals via perturbation theory. Several benchmark studies have confirmed the superior performance on DHDFs on a wide range of properties, such as relative energies, thermochemistry and electronic excitation energies [45,46,47]. Based on the general idea that “pure” DFT functionals allow a better description of static correlation at the expense of dynamic correlation effects [48,49,50], and MP2 contributes a better description of dynamic correlation [45], the judicious combination of the two is expected to achieve a good balance of dynamic and non-dynamic correlation effects. DHDFs have exhibited excellent performance on response properties calculations, such as g-tensors of organic radicals [51], electric field response properties of water nanoclusters [52], hyperfine coupling constants [53,54] and NMR nuclear shielding constants [55]. However, their performance for g-tensors of transition metal systems in general and for the particularly demanding case of Cu(II) in particular remains unexplored. To determine the scope of applicability of these approximations for the title problem, in this study we investigate the performance of sixteen distinct DHDFs for the prediction of g-tensors of a reference set of eighteen Cu(II) complexes with experimentally known structures and EPR parameters. The performance of DHDFs is contrasted with the best performing hybrid functionals for g-tensor prediction for Cu(II) complexes as reported by a recent benchmark study by Sciortino et al. [26]. Based on our results, we establish that most of the recent DHDFs outperform standard global HDFs and we are able to suggest specific DHDFT-based protocols that maximize the accuracy for g-tensor calculations of copper-based enzymes.
2. Methodology
2.1. Benchmark Set of Copper Complexes
The structures of the eighteen Cu(II) complexes selected for the present study are shown in Figure 1. The set includes ligands with N, O and S coordinating atoms in different combinations. Similar sets of Cu(II) complexes have been employed in previous benchmarking studies [25,26,27,36]. The complexes are approximately square planar, except 9 and 12, which are square pyramidal, and 18, which is octahedral.
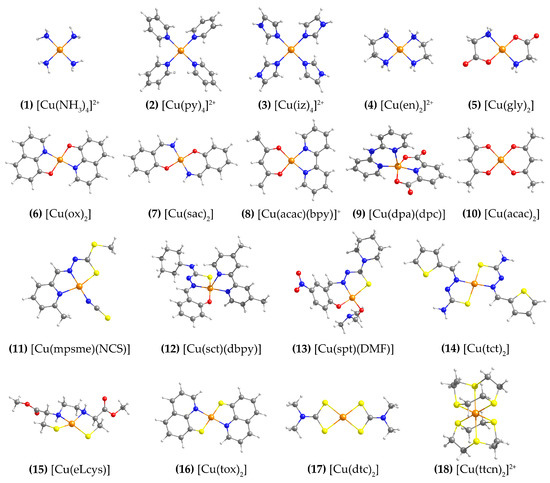
Figure 1.
Structures of the eighteen Cu(II) complexes studied in this work. Ligand abbreviations: py = pyridine; iz = imidazole; en = ethylenediamine; gly = glycine; ox = 3-quinolinolato; sac = salicylaldehyde imine; acac = acetylacetone; bpy = 2,2′-bipyridine; dpa = 2,2′-dipyridylamine; dpc = dipicolinate ligand; mpsme = anionic form of the 6-methyl-2-formylpyridine Schiff base of S-methyldithiocarbazate; sct = N(4)-cyclohexyl thiosemicarbazone; dbpy = 4,4′-dimethyl-2,2′-bipyridine; spt = 5-nitrosalicylaldehyde piperidylthiosemicarbazone; tct = thiophene-2-carbaldehyde thiosemicarbazone; DMF = N,N-Dimethylformamide; eLcys = N,N′-ethylenebis(L-cysteine); tox = 3-quinolinethiolato; dtc = dimethyl-dithiocarbamate, ttcn = 1,4,7-trithiacyclononane.
It is noted that the g-tensor is expressed as:
where each component , , describes the shift of the system’s -value from the -value of the free electron () and I is the identity matrix. In this work, we express the principal components of the -tensors as -shifts in parts per thousand (ppt), computed as . The largest component is referred to as or as the parallel -tensor component, . We define the perpendicular -tensor component as the average of the two smaller components: ()/2.
The -tensor components of complexes 1–18 have been experimentally determined from EPR measurements and are given in Table 1. Since Cu(II) complexes have a d9 configuration, square planar and square pyramidal structures usually exhibit strongly axial EPR signals, i.e., < << , consistent with the high character of the singly occupied molecular orbital (SOMO). The g-shifts arise predominantly due to the interaction of the ligating atoms with the unpaired electron, so it depends on the nature of the ligands, the nature of the Cu–ligand bond, and the corresponding bond lengths. In this benchmark set we include complexes with variable coordination spheres and a wide range of values, from 83 ppt to 283 ppt. The presence of at least one S atom on the copper coordination sphere decreases the magnitude of . It should be noted that mixed ligand complexes exhibit magnetic parameters that are intermediate between the respective complexes with four of the same ligands of each type [8]. This pattern can be observed in the experimental data shown in Table 1.

Table 1.
Experimental values (ppt) of the Cu(II) complexes 1–18 studied in this work. Molecular structures of the complexes are shown in Figure 1. The complexes are arranged in groups according to the copper-coordinating atoms for each subgroup, which are also shown for convenience.
2.2. Overview of Double Hybrid Density Functionals
In double-hybrid density functional theory, a set of Kohn–Sham orbitals is first obtained by a classical HDFT calculation. Subsequently, an MP2 calculation is performed based on this set of orbitals. A general expression for the total exchange-correlation term for standard DHDFs is:
where is the amount of DFT exchange, and and are the amounts of DFT and MP2 correlation, respectively. Usually, but not always, . The parameters for all functionals studied in this work are shown in Table 2. The first DHDF was B2PLYP [47], proposed by Grimme and parametrized in order to reproduce heats of formation. The success of B2PLYP inspired further developments. mPW2PLYP [69] has a modified exchange part and higher accuracy towards the calculation of thermodynamic properties and barrier heights. B2GP-PLYP [70] has increased amounts of exact exchange and perturbative correlation, suggested for general purpose (GP) applications. B2K-PLYP and B2T-PLYP [71] were parametrized for reaction kinetics and thermochemistry. Some researchers attempted to minimize or eliminate the use of empirically defined parameters, striving instead to include parameters that have theoretical justification. Such functionals include PBE-QIDH [72], PBE0-DH [73] and the range-separated RSX-QIDH [74] and RSX-0DH [75], optimized by enforcing reproduction of the total energy of the hydrogen atom.
Table 2.
Parameters of DHDFs according to Equations (2)–(4).
As a variation of the standard MP2 approach, some functionals may treat the same-spin and opposite-spin correlation contributions separately and with unequal weights. This is referred to as a spin-component scaling (SCS). In case the same-spin contribution is entirely omitted, the approach is referred to as spin-opposite scaled (SOS). The general expression for the total exchange-correlation term for the spin-component scaled double-hybrid functionals with dispersion correction (DSD-DFT) is:
where is the amount of opposite-spin MP2, of same-spin MP2, and s6 of the dispersion correction. DSD-BLYP [76] and DSD-PBEP86 [77] have been parametrized to reproduce thermochemistry, kinetics, and dispersion forces, and were found to be more accurate than the previously most successful B2GP-PLYP functional.
In range-separated DFs [78,79,80], the interelectronic repulsion, , is separated using an error function, , into a short-range and a long-range component:
where the first term accounts for the short-range interaction and the second term accounts for the long-range interaction. ωB2PLYP and ωB2GP-PLYP [81] functionals were parametrized to reproduce electronic excitation energies. In addition, ωB88PP86 and ωPBEPP86 [82] were very recently presented, parametrized to reproduce experimental singlet-singlet as well as singlet-triplet excitations. The first range-separated DHDF that also includes spin component scaling, ωB97X-2 [83], was parametrized for thermochemistry, kinetics, and noncovalent interactions. The parameters from Equation (4) for the range separated functionals used herein are shown in Table 2.
3. Results and Discussion
3.1. Evaluation Criteria
Evaluation of the methods used for the prediction of the -shifts was based on the differences (D) of the calculated from the experimental parallel and perpendicular components:
The mean values of the above parameters for each function for the set of eighteen Cu(II) complexes under study are used to describe the performance of the functional. The mean absolute difference (MAD) of the component is defined as:
and straightforwardly describes the ability of the method to reproduce the experimental values, while the mean difference (MD) reflects the possible systematic over- or under-estimation of -shifts by the specific method.
The standard deviation of the above parameters was also considered. The standard deviation of the differences (SDD) is estimated as:
and the standard deviation of the absolute differences (SDAD):
Finally, the mean absolute percentage differences (MAPD) are examined:
3.2. Performance of Functionals
The mean values of the evaluation criteria for each functional are shown in Table 3. To begin with, we observe that the MAPDs for are very close to those for , therefore we focus our analysis on the parallel components for simplicity and the conclusions largely reflect also the performance for the perpendicular components of the -tensor. Besides, since the parallel component is the largest, it also has the largest D values. We start the evaluation of the relative performances by examining the MADs and MDs. The MAD and MD values for the 19 functionals are plotted in Figure 2, top. Complete results are provided in the Supporting Information, Tables S1–S19.
Table 3.
Mean difference (MD) and standard deviation of the differences (SDD), mean absolute difference (MAD), standard deviation of the absolute differences (SDAD) and mean absolute percent difference (MAPD), from the experimental values of the parallel and perpendicular -shifts components calculated with each functional. Values are expressed in ppt.
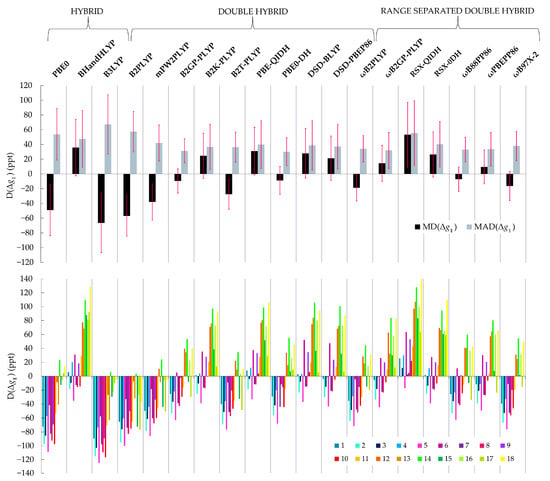
Figure 2.
(Top): Mean absolute difference (MAD), mean difference (MD) from the experimental values of the parallel -tensor component calculated with each functional. The respective standard deviations (SD) are plotted in pink. (Bottom): Differences (D) from the experimental values of the parallel -tensor component of each one of the 18 Cu(II) complexes of the set calculated with each functional. Each bar color represents a complex.
Focusing first on conventional hybrid functionals, we see that B3LYP is the worst method considered here. It shows the largest MAD value of 67 ppt, while PBE0 and BHandHLYP follow with MAD values of 54 and 48 ppt, respectively. Thus, among conventional HDFs the best performance is observed for PBE0 and BHandHLYP, which is in agreement with Sciortino et al. [26]. Only two double-hybrid functionals perform similarly to HDFs. These are B2PLYP and RSX-QIDH with MAD values of 57 and 55 ppt, respectively. Figure 2 top suggests that apart from the B2PLYP and RSX-QIDH, all remaining 14 DHDFs outperform HDFs and show similar performance in terms of the MADs, but have different MDs, which means that they show different systematic behaviour. The MAD of the other 14 functionals range between 31 and 42 ppt. The lowest MAD is 31 ppt, achieved by the B2GP-PLYP and PBE0-DH functionals. The ωB2GP-PLYP, ωB88PP86 and ωPBEPP86 follow with MADs of 32, 33 and 34 ppt, respectively. The lowest MD values among the DHDFs are observed for the PBE0-DH and ωB88PP86 functionals at −7 and −9 ppt, respectively. Notably, apart from the MAD and MD values, the respective SDs must be also considered. The differences between the individual values of the complexes 1–18, visualized in Figure 2 bottom, reflect the error of each method on reproducing the relative Δ values of the various structures. They are also described by the SDD value of each method, given in Table 3. ωB2GP-PLYP, ωPBEPP86 and ωB88PP86 have larger SD values than the B2GP-PLYP and PBE0-DH functionals, which means that there is a larger spread of D values with these functionals.
Inspection of the individual values for the complexes 1–18 obtained with each functional allows a more in-depth analysis of our results (Figure 2 bottom). This diagram shows clearly that complexes 11–18, which have at least one S coordinated ligand on copper, are described differently than complexes 1–10, which have only N and O ligating atoms. Specifically, most functionals underestimate the value of parallel -tensor component of structures 1–10, while they overestimate it for 11–18. This behavior is even more pronounced for the HDFs. Figure 2 bottom shows clearly that even though DHDFs also treat differently the Cu–S bond, they not only have smaller MADs, but also the spread of the values is smaller. This implies that DHDFs achieve a more balanced description of Cu-ligand bond covalency and this behavior is transferable among different systems, at least to a larger extent than HDFs.
In Figure 3, the D(Δ) obtained by each functional is plotted against the calculated copper Löwdin spin population of the MP2 relaxed density, ρs(Cu), for six complexes chosen as representative of different copper coordination spheres, i.e., N4, N2O2, O4, N2S2, N3OS and S4 (see Figure S1 for additional examples). Several conclusions can be extracted from these diagrams. First, it can be clearly observed that the value of D(Δ) increases following a near linear trend along with ρs(Cu). Second, each system has a different ρs(Cu) value for which the experimental Δ is reproduced. Even though no single functional predicts this most favorable ρs(Cu) value for all complexes, structures with similar coordination spheres are optimally described by the same density functionals (Figure S1). Among the DHDFs included in this study, the B2PLYP and RSX-QIDH predict the smallest and largest ρs(Cu) values, respectively, for all complexes, which directly correlates with the observed systematic negative and positive differences from the experimental values, reflected in the large MD(Δ) values shown in Figure 3. In addition, B3LYP and PBE0 in almost all cases predict a small spin population on Cu. Notably, accurate -tensor prediction does not necessarily imply prediction of the “correct” spin density, since other factors, such as higher order relativistic contributions, could also alter the D(Δ) values. We note that the possible lack of correspondence between the accuracy of DFs in the prediction of various observable properties, such as relative energies or ionization potentials, and the quality of the computed densities has been extensively debated recently [84,85,86].
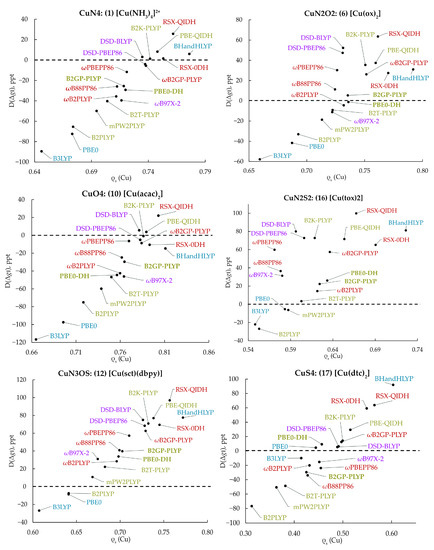
Figure 3.
Correlation of the differences, D(Δ), of the calculated -tensor from the experimental value of the parallel -shift with the Cu spin populations computed with the respective functional for complexes 1, 6, 10, 16, 12 and 17, which have different copper coordination spheres.
From the above analysis we can see that overall, the best performing functionals for this set of Cu(II) complexes are the B2GP-PLYP and PBE0-DH, because they have both the lowest MAD values, and at the same time among the lowest MD and SD values. PBE0-DH represents a definite improvement over the parent PBE0 functional, which is the best performing HDF. Interestingly, these two DHDFs represent different construction philosophies (PBE0-DH is “non empirical”) yet converge to similarly accurate behavior. Notably, even though the parameters of B2GP-PLYP (and B2PLYP) were empirically optimized, they were also suggested to be justified from a purely theoretical standpoint [87]. It is also important to draw attention to the observation that the functionals ωB2PLYP, ωB88PP86, ωB2GP-PLYP, ωPBEPP86 are among the best performing DHDFs. What these functionals have in common is that they were parametrized to reproduce experimental excitation energy reference values [81,82].
Another interesting point concerns the calculated SDD values in Table 3. These show that the B2PLYP functional has the smallest SDD value of 28 ppt. This is visually represented in Figure 2b, where it can be observed that even though B2PLYP performs poorly, systematically underestimating the g-shifts, its behavior is distinct in that the D values of complexes 11–18 with S coordinating ligands are not very different from complexes 1–10 which have N and O ligands only. By contrast, this is not observed for RSX-QIDH, the other DHDF of this set that performs poorly, systematically overestimating g-shifts. Therefore, we can conclude that B2PLYP achieves a more balanced description of the Cu-ligand bond covalency between different ligand donors.
Having compared the relative accuracies of the examined DHDFs on predicting the experimental g-tensor components, one may consider whether correlations exist between their construction parameters (Table 2) and their performance. However, at this point it is not possible to discern any obvious correlation. Perhaps a way to approach this problem is to investigate how reference spin densities, computed with high-level ab initio methods, are reproduced by density functionals with respect to the parameters included in their definition [86]. Although clear choices emerge from the present study for practical applications, further systematic studies are required for the development of improved functionals optimized for spin-density dependent properties.
4. Conclusions
We investigated the performance of sixteen DHDF approximations for the prediction of experimental g-shifts of a set of eighteen Cu(II) complexes. The DHDFs results were compared with the best performing HDFs for Cu(II) complexes [26]. Our results show that most DHDFs perform significantly better than the best performing standard HDFs, establishing a new standard for the prediction of g-tensors of copper systems. With the exception of B2PLYP and RSX-QIDH, all other DHDFs perform significantly better than HDFs. Based on the criteria defined in Section 3.1, we found that the ranking of the DHDFs with respect to g-tensor prediction is: B2GP-PLYP ~ PBE0-DH > ωB2PLYP ~ ωB88PP86 > B2T-PLYP ~ ωB2GP-PLYP ~ ωPBEPP86 > B2K-PLYP ~ PBE-QIDH ~ DSD-BLYP ~ DSD-PBEP86 ~ RSX-0DH ~ ωB97X-2 > mPW2PLYP > B2PLYP > RSX-QIDH.
The best-performing functionals represent different approaches toward construction of DHDFs. B2GP-PLYP is a general purpose empirically fitted functional based on B2PLYP, while PBE0-DH is based on PBE0 and obtained by purely theoretical arguments. Nevertheless, they converge to similarly good performance. Moreover, the equally good performance of the range-separated ωB2PLYP, ωB88PP86, ωB2GP-PLYP and ωPBEPP86 directs our attention to functionals optimized for excitation energies. We suggest that such functionals can be used as a starting point for further refinement, taking into account a more extensive set of transition metal reference compounds for g-tensors, and perhaps also for other spin-dependent properties.
Overall, the double-hybrid methodologies significantly outperform conventional hybrid approaches, offering a good balance between accuracy and computational cost. We note that due to the steep scaling of MP2 (O(N5) where N is a measure of the system size), the computational cost of DHDFs on large (>100 atoms) systems increases more steeply than HDFs in terms of both time and required memory. Hence, for large systems we propose the use of multilevel approaches, where the metal and first coordination sphere ligands are treated with a DHDFT method and the surrounding protein matrix can be treated with a cheaper DFT method. Based on our results, we recommend the use of B2GP-PLYP and PBE0-DH functionals for g-tensor calculations on Cu(II) complexes bearing N, O and S ligands, which are usually encountered in bioinorganic systems. Evidently, the uncertainty of even the best functionals may exceed the uncertainty of experimental values. This suggests that the successful use of these approaches will depend on the type of chemical problem under investigation and in the combination with complementary data. Nevertheless, the present study clearly defines the current state-of-the-art in quantum chemical calculations of g-tensors for Cu(II) systems and encourages further developments in refining double-hybrid DFT for spin density dependent properties.
5. Computational Details
All calculations were performed with Orca 5 [88]. Geometry optimizations of the eighteen Cu(II) complexes of the benchmark set was performed with the B3LYP [89,90,91] functional, starting from the crystallographic structures obtained from the Cambridge Structural database [92]. Scalar relativistic effects were treated using the zeroth-order regular approximation (ZORA) [93,94]. The resolution of identity (RI) approximation [95] and the chain-of-spheres approximation to exact exchange (COSX) [96] were used throughout to reduce computational time. Tight convergence criteria and high-quality grids (DefGrid3 in Orca convention) were used throughout. The ZORA-recontracted [97] version of the def2-TZVPP basis set [98] was used for Cu and the ZORA-def2-TZVP for all other atoms, along with fully decontracted def2/J auxiliary basis sets [99]. Since MP2 convergence with basis set size can be slower than DFT, the basis set dependence of DHDFs may increase with increasing fraction of the PT2 component [46,47]. Το investigate the dependence of calculated g-shifts on the basis set size, we carried out systematic studies on complexes 3, 10 and 17, which have N4, O4 and S4 coordination spheres, respectively, using the B2PLYP and DSD-PBEP86 functionals. In line with previous studies on Cu(II) -tensor calculations [25], the dependence on the basis set is very weak past the polarized triple-zeta level and the use of the largest available ZORA-def2-QZVPP basis set leads to negligible differences (of the order of 1–2 ppt) in the results compared to the triple-zeta basis sets. The maximum difference in computed g-shifts upon use of the quadruple-zeta basis set (5 ppt) was observed for 17 with the DSD-PBEP86 functional. Since this difference is of the same order of magnitude as the experimental uncertainty, the basis sets used here are judged to be essentially converged for all practical purposes. For the calculation of g-tensors, the DFT functionals tested in this work are: the hybrid functionals PBE0 [100,101,102], BHandHLYP [91], B3LYP [89,90,91], the double-hybrid functionals: B2PLYP [47], mPW2PLYP [69], B2GP-PLYP [70], B2K-PLYP [71], B2T-PLYP [71], PBE-QIDH [72], PBE0-DH [73], DSD-BLYP [76], DSD-PBEP86 [77], and the range-separated double-hybrid functionals: ωB2PLYP [81], ωB2GP-PLYP [81], RSX-QIDH [74], RSX-0DH [75], ωB88PP86 [82], ωPBEPP86 [82], ωB97X-2 [83]. Spin contamination values for HDFs were smaller than 0.01 for all complexes, which suggests that the ground state of all complexes can be adequately described by a single determinant. For the calculations with double-hybrid functionals the NoFrozenCore option was used. In terms of computational costs, we mention as an example that for complexes 1 (17 atoms), 18 (43 atoms) and 12 (63 atoms) the PBE0 calculation needs 1, 16 and 48 min, respectively, using 10 processing cores, while the PBE0-DH calculation needs 3, 184 and 927 min, respectively. The part of the calculation that is responsible for most of the additional computational effort is the calculation of the relaxed MP2 response density, which incorporates orbital relaxation and is consistent with first order properties as analytic derivatives. The spin-orbit coupling was treated using the spin-orbit mean-field (SOMF) operator [102] with the 1X-approximation [103,104] (SOCType 3 in ORCA convention). For the construction of the potential one-electron terms were included, the Coulomb term was computed using the RI approximation, exchange terms were incorporated via one-center exact integrals including the spin-other orbit interaction, and without local DFT correlation terms (SOCFlags 1,3,3,0 in ORCA). Picture change effects were also taken into account.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/magnetochemistry8040036/s1, Figure S1: Correlation of the differences, D(Δ), of the calculated -tensor from the experimental value of the parallel -shift with the Cu spin populations computed with the respective functional for pairs of complexes with the same copper-coordinating atoms 1 and 3, 14 and 16, and with similar coordination spheres 12 and 13. Tables S1–S19: Detailed results for each functional.
Author Contributions
Conceptualization, D.A.P. and M.D.; methodology, D.A.P. and M.D.; formal analysis, M.D., D.A.P. and M.O.; investigation, M.D.; data curation, M.D.; writing—original draft preparation, M.D.; writing—review and editing, D.A.P. and M.D.; supervision, C.A.M.; project administration, D.A.P., C.A.M. and M.O.; All authors have read and agreed to the published version of the manuscript.
Funding
M.D. acknowledges support by the Hellenic Foundation for Research and Innovation (HFRI) under the HFRI PhD Fellowship grant (Fellowship number: 16199). D.A.P. acknowledges support by the Max Planck Society. M.O. and D.A.P. gratefully acknowledge financial support of this work by the French National Research Agency and the Deutsche Forschungsgemeinschaft (CUBISM, grant No. ANR-18 CE092_0040_01/DFG project No. 406697875), and from the France-Germany Hubert Curien Program—German Academic Exchange Service (DAAD) (Procope 2019–2020 project 42525PB/DAAD project 57445526). The authors acknowledge support from COST Action 18234 supported by European Cooperation in Science and Technology.
Data Availability Statement
Data is contained within the article or supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Karlin, K.D.; Tyeklár, Z. Bioinorganic Chemistry of Copper; Springer: Dordrecht, The Netherlands, 2013; ISBN 978-94-011-6875-5. [Google Scholar]
- Kaim, W.; Schwederski, B.; Klein, A. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life: An Introduction and Guide, 2nd ed.; Inorganic Chemistry: A Wiley Series of Advanced Textbooks; Wiley: Chichester, UK, 2013; ISBN 978-0-470-97524-4. [Google Scholar]
- Festa, R.A.; Thiele, D.J. Copper: An Essential Metal in Biology. Curr. Biol. 2011, 21, R877–R883. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef]
- Belle, C.; Rammal, W.; Pierre, J. Sulfur Ligation in Copper Enzymes and Models. J. Inorg. Biochem. 2005, 99, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, I.S.; Murphy, M.E.P. Type-2 Copper-Containing Enzymes. Cell. Mol. Life Sci. 2007, 64, 2887–2899. [Google Scholar] [CrossRef]
- Pretzler, M.; Rompel, A. What Causes the Different Functionality in Type-III-Copper Enzymes? A State of the Art Perspective. Inorg. Chim. Acta 2018, 481, 25–31. [Google Scholar] [CrossRef]
- Peisach, J.; Blumberg, W.E. Structural Implications Derived from the Analysis of Electron Paramagnetic Resonance Spectra of Natural and Artificial Copper Proteins. Arch. Biochem. Biophys. 1974, 165, 691–708. [Google Scholar] [CrossRef]
- Solomon, E.I.; Hare, J.W.; Dooley, D.M.; Dawson, J.H.; Stephens, P.J.; Gray, H.B. Spectroscopic Studies of Stellacyanin, Plastocyanin, and Azurin. Electronic Structure of the Blue Copper Sites. J. Am. Chem. Soc. 1980, 102, 168–178. [Google Scholar] [CrossRef]
- Gewirth, A.A.; Cohen, S.L.; Schugar, H.J.; Solomon, E.I. Spectroscopic and Theoretical Studies of the Unusual EPR Parameters of Distorted Tetrahedral Cupric Sites: Correlations to x-Ray Spectral Features of Core Levels. Inorg. Chem. 1987, 26, 1133–1146. [Google Scholar] [CrossRef]
- Shadle, S.E.; Penner-Hahn, J.E.; Schugar, H.J.; Hedman, B.; Hodgson, K.O.; Solomon, E.I. X-Ray Absorption Spectroscopic Studies of the Blue Copper Site: Metal and Ligand K-Edge Studies to Probe the Origin of the EPR Hyperfine Splitting in Plastocyanin. J. Am. Chem. Soc. 1993, 115, 767–776. [Google Scholar] [CrossRef]
- Andersson, K.K.; Schmidt, P.P.; Katterle, B.; Strand, K.R.; Palmer, A.E.; Lee, S.-K.; Solomon, E.I.; Gräslund, A.; Barra, A.-L. Examples of High-Frequency EPR Studies in Bioinorganic Chemistry. J. Biol. Inorg. Chem. 2003, 8, 235–247. [Google Scholar] [CrossRef]
- de Almeida, K.J.; Rinkevicius, Z.; Hugosson, H.W.; Ferreira, A.C.; Ågren, H. Modeling of EPR Parameters of Copper(II) Aqua Complexes. Chem. Phys. 2007, 332, 176–187. [Google Scholar] [CrossRef]
- Kaupp, M.; Bühl, M.; Malkin, V.G. (Eds.) Calculation of NMR and EPR Parameters: Theory and Applications, 1st ed.; Wiley: Hoboken, NJ, USA, 2004; ISBN 978-3-527-30779-1. [Google Scholar]
- Sinnecker, S.; Neese, F. Theoretical Bioinorganic Spectroscopy. In Atomistic Approaches in Modern Biology; Reiher, M., Ed.; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2007; Volume 268, pp. 47–83. ISBN 978-3-540-38082-5. [Google Scholar]
- Neese, F. Prediction of Molecular Properties and Molecular Spectroscopy with Density Functional Theory: From Fundamental Theory to Exchange-Coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Orio, M.; Pantazis, D.A. Successes, Challenges, and Opportunities for Quantum Chemistry in Understanding Metalloenzymes for Solar Fuels Research. Chem. Commun. 2021, 57, 3952–3974. [Google Scholar] [CrossRef] [PubMed]
- Remenyi, C.; Reviakine, R.; Kaupp, M. Density Functional Study of EPR Parameters and Spin-Density Distribution of Azurin and Other Blue Copper Proteins. J. Phys. Chem. B 2007, 111, 8290–8304. [Google Scholar] [CrossRef] [PubMed]
- Sinnecker, S.; Neese, F. QM/MM Calculations with DFT for Taking into Account Protein Effects on the EPR and Optical Spectra of Metalloproteins. Plastocyanin as a Case Study. J. Comput. Chem. 2006, 27, 1463–1475. [Google Scholar] [CrossRef]
- Ames, W.M.; Larsen, S.C. DFT Calculations of the EPR Parameters for Cu(Ii) DETA Imidazole Complexes. Phys. Chem. Chem. Phys. 2009, 11, 8266. [Google Scholar] [CrossRef]
- Ames, W.M.; Larsen, S.C. Density Functional Theory Investigation of EPR Parameters for Tetragonal Cu(II) Model Complexes with Oxygen Ligands. J. Phys. Chem. A 2009, 113, 4305–4312. [Google Scholar] [CrossRef]
- Courtade, G.; Ciano, L.; Paradisi, A.; Lindley, P.J.; Forsberg, Z.; Sørlie, M.; Wimmer, R.; Davies, G.J.; Eijsink, V.G.H.; Walton, P.H.; et al. Mechanistic Basis of Substrate–O2 Coupling within a Chitin-Active Lytic Polysaccharide Monooxygenase: An Integrated NMR/EPR Study. Proc. Natl. Acad. Sci. USA 2020, 117, 19178–19189. [Google Scholar] [CrossRef]
- Bissaro, B.; Streit, B.; Isaksen, I.; Eijsink, V.G.H.; Beckham, G.T.; DuBois, J.L.; Røhr, Å.K. Molecular Mechanism of the Chitinolytic Peroxygenase Reaction. Proc. Natl. Acad. Sci. USA 2020, 117, 1504–1513. [Google Scholar] [CrossRef]
- Theibich, Y.A.; Sauer, S.P.A.; Leggio, L.L.; Hedegård, E.D. Estimating the Accuracy of Calculated Electron Paramagnetic Resonance Hyperfine Couplings for a Lytic Polysaccharide Monooxygenase. Comput. Struct. Biotechnol. J. 2021, 19, 555–567. [Google Scholar] [CrossRef]
- Singh, S.K.; Atanasov, M.; Neese, F. Challenges in Multireference Perturbation Theory for the Calculations of the g -Tensor of First-Row Transition-Metal Complexes. J. Chem. Theory Comput. 2018, 14, 4662–4677. [Google Scholar] [CrossRef] [PubMed]
- Sciortino, G.; Lubinu, G.; Maréchal, J.-D.; Garribba, E. DFT Protocol for EPR Prediction of Paramagnetic Cu(II) Complexes and Application to Protein Binding Sites. Magnetochemistry 2018, 4, 55. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, D.; Gagliardi, L.; Truhlar, D.G. Calculation of the Zeeman Effect for Transition-Metal Complexes by Multiconfiguration Pair-Density Functional Theory. J. Chem. Theory Comput. 2021, 17, 5050–5063. [Google Scholar] [CrossRef] [PubMed]
- Ames, W.M.; Larsen, S.C. Insight into the Copper Coordination Environment in the Prion Protein through Density Functional Theory Calculations of EPR Parameters. J. Biol. Inorg. Chem. 2009, 14, 547–557. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, R.P.; Venugopalan, P.; Witwicki, M.; Ferretti, V. Synthesis, Characterization, Single Crystal X-Ray Structure, EPR and Theoretical Studies of a New Hybrid Inorganic-Organic Compound [Cu(Hdien)2(H2O)2](Pnb)4·4H2O and Its Structural Comparison with Related [Cu(En)2(H2O)2](Pnb)2. J. Mol. Struct. 2016, 1123, 124–132. [Google Scholar] [CrossRef]
- Atanasov, M.; Daul, C.A.; Rohmer, M.-M.; Venkatachalam, T. A DFT Based Ligand Field Study of the EPR Spectra of Co(II) and Cu(II) Porphyrins. Chem. Phys. Lett. 2006, 427, 449–454. [Google Scholar] [CrossRef][Green Version]
- Ames, W.M.; Larsen, S.C. DFT Calculations of EPR Parameters for Copper(II)-Exchanged Zeolites Using Cluster Models. J. Phys. Chem. A 2010, 114, 589–594. [Google Scholar] [CrossRef]
- Vancoillie, S.; Malmqvist, P.-Å.; Pierloot, K. Calculation of EPR g Tensors for Transition-Metal Complexes Based on Multiconfigurational Perturbation Theory (CASPT2). Chem. Phys. Chem. 2007, 8, 1803–1815. [Google Scholar] [CrossRef]
- Bolvin, H. An Alternative Approach to the G-Matrix: Theory and Applications. Chem. Eur. J. Chem. Phys. 2006, 7, 1575–1589. [Google Scholar] [CrossRef]
- Sayfutyarova, E.R.; Chan, G.K.-L. Electron Paramagnetic Resonance G-Tensors from State Interaction Spin-Orbit Coupling Density Matrix Renormalization Group. J. Chem. Phys. 2018, 148, 184103. [Google Scholar] [CrossRef]
- Sayfutyarova, E.R.; Chan, G.K.-L. A State Interaction Spin-Orbit Coupling Density Matrix Renormalization Group Method. J. Chem. Phys. 2016, 144, 234301. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Piñeiro, R.J.; Pantazis, D.A.; Orio, M. Comparison of Density Functional and Correlated Wave Function Methods for the Prediction of Cu(II) Hyperfine Coupling Constants. ChemPhysChem 2020, 21, 2667–2679. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, N.; Head-Gordon, M. Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Perdew, J.P. Jacob’s Ladder of Density Functional Approximations for the Exchange-Correlation Energy. In AIP Conference Proceedings; AIP: Antwerp, Belgium, 2001; Volume 577, pp. 1–20. [Google Scholar] [CrossRef]
- Kaupp, M.; Reviakine, R.; Malkina, O.L.; Arbuznikov, A.; Schimmelpfennig, B.; Malkin, V.G. Calculation of Electronic G-Tensors for Transition Metal Complexes Using Hybrid Density Functionals and Atomic Meanfield Spin-Orbit Operators. J. Comput. Chem. 2002, 23, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Fritscher, J.; Hrobárik, P.; Kaupp, M. Computational Studies of Electron Paramagnetic Resonance Parameters for Paramagnetic Molybdenum Complexes. 1. Method Validation on Small and Medium-Sized Systems. J. Phys. Chem. B 2007, 111, 4616–4629. [Google Scholar] [CrossRef]
- Fritscher, J.; Hrobárik, P.; Kaupp, M. Computational Studies of EPR Parameters for Paramagnetic Molybdenum Complexes. II. Larger Mo V Systems Relevant to Molybdenum Enzymes. Inorg. Chem. 2007, 46, 8146–8161. [Google Scholar] [CrossRef]
- Gohr, S.; Hrobárik, P.; Repiský, M.; Komorovský, S.; Ruud, K.; Kaupp, M. Four-Component Relativistic Density Functional Theory Calculations of EPR g—and Hyperfine-Coupling Tensors Using Hybrid Functionals: Validation on Transition-Metal Complexes with Large Tensor Anisotropies and Higher-Order Spin–Orbit Effects. J. Phys. Chem. A 2015, 119, 12892–12905. [Google Scholar] [CrossRef]
- Munzarová, M.; Kaupp, M. A Critical Validation of Density Functional and Coupled-Cluster Approaches for the Calculation of EPR Hyperfine Coupling Constants in Transition Metal Complexes. J. Phys. Chem. A 1999, 103, 9966–9983. [Google Scholar] [CrossRef]
- Schattenberg, C.J.; Maier, T.M.; Kaupp, M. Lessons from the Spin-Polarization/Spin-Contamination Dilemma of Transition-Metal Hyperfine Couplings for the Construction of Exchange-Correlation Functionals. J. Chem. Theory Comput. 2018, 14, 5653–5672. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Double-Hybrid Density Functionals: Double-Hybrid Density Functionals. WIREs Comput. Mol. Sci. 2014, 4, 576–600. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Santra, G. Empirical Double-Hybrid Density Functional Theory: A ‘Third Way’ in Between WFT and DFT. Isr. J. Chem. 2020, 60, 787–804. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical Hybrid Density Functional with Perturbative Second-Order Correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [PubMed]
- Radoń, M. Revisiting the Role of Exact Exchange in DFT Spin-State Energetics of Transition Metal Complexes. Phys. Chem. Chem. Phys. 2014, 16, 14479–14488. [Google Scholar] [CrossRef] [PubMed]
- Pinter, B.; Chankisjijev, A.; Geerlings, P.; Harvey, J.N.; De Proft, F. Conceptual Insights into DFT Spin-State Energetics of Octahedral Transition-Metal Complexes through a Density Difference Analysis. Chem. Eur. J. 2018, 24, 5281–5292. [Google Scholar] [CrossRef]
- Cremer, D. Density Functional Theory: Coverage of Dynamic and Non-Dynamic Electron Correlation Effects. Mol. Phys. 2001, 99, 1899–1940. [Google Scholar] [CrossRef]
- Tran, V.A.; Neese, F. Double-Hybrid Density Functional Theory for g-Tensor Calculations Using Gauge Including Atomic Orbitals. J. Chem. Phys. 2020, 153, 054105. [Google Scholar] [CrossRef]
- Alipour, M. How Well Can Parametrized and Parameter-Free Double-Hybrid Approximations Predict Response Properties of Hydrogen-Bonded Systems? Dipole Polarizabilities of Water Nanoclusters as a Working Model. J. Phys. Chem. A 2013, 117, 4506–4513. [Google Scholar] [CrossRef]
- Kossmann, S.; Kirchner, B.; Neese, F. Performance of Modern Density Functional Theory for the Prediction of Hyperfine Structure: Meta-GGA and Double Hybrid Functionals. Mol. Phys. 2007, 105, 2049–2071. [Google Scholar] [CrossRef]
- Witwicki, M.; Walencik, P.K.; Jezierska, J. How Accurate Is Density Functional Theory in Predicting Spin Density? An Insight from the Prediction of Hyperfine Coupling Constants. J. Mol. Model. 2020, 26, 10. [Google Scholar] [CrossRef]
- Dittmer, A.; Stoychev, G.L.; Maganas, D.; Auer, A.A.; Neese, F. Computation of NMR Shielding Constants for Solids Using an Embedded Cluster Approach with DFT, Double-Hybrid DFT, and MP2. J. Chem. Theory Comput. 2020, 16, 6950–6967. [Google Scholar] [CrossRef]
- Scholl, H.J.; Huettermann, J. ESR and ENDOR of Copper(II) Complexes with Nitrogen Donors: Probing Parameters for Prosthetic Group Modeling of Superoxide Dismutase. J. Phys. Chem. 1992, 96, 9684–9691. [Google Scholar] [CrossRef]
- Yordanov, N.D.; Stankova, M.; Shopov, D. EPR Study of Bis(8-Quinolinethiolato) Copper(II) and Bis(8-Quinolinolato) Copper(II) Complexes. Chem. Phys. Lett. 1976, 39, 174–176. [Google Scholar] [CrossRef]
- Kuźniarska-Biernacka, I.; Kurzak, K.; Kurzak, B.; Jezierska, J. Spectrochemical Properties of Noncubical Transition Metal Complexes in Solutions. XV. Solution Properties of Bis (Salicylideneaniline)Copper(II). J. Solut. Chem. 2003, 32, 719–741. [Google Scholar] [CrossRef]
- Folli, A.; Ritterskamp, N.; Richards, E.; Platts, J.A.; Murphy, D.M. Probing the Structure of Copper(II)-Casiopeina Type Coordination Complexes [Cu(O-O)(N-N)]+ by EPR and ENDOR Spectroscopy. J. Catal. 2021, 394, 220–227. [Google Scholar] [CrossRef]
- Uçar, İ.; Bulut, A.; Büyükgüngör, O. Synthesis, Crystal Structure, EPR and Electrochemical Studies of Copper(II) Dipicolinate Complex with 2,2′-Dipyridylamine Ligand. J. Phys. Chem. Solids 2007, 68, 2271–2277. [Google Scholar] [CrossRef]
- Ritterskamp, N.; Sharples, K.; Richards, E.; Folli, A.; Chiesa, M.; Platts, J.A.; Murphy, D.M. Understanding the Coordination Modes of [Cu(Acac)2(Imidazole)n=1,2] Adducts by EPR, ENDOR, HYSCORE, and DFT Analysis. Inorg. Chem. 2017, 56, 11862–11875. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Mirza, A.H.; Fereday, R.J.; Butcher, R.J.; Fuller, J.M.; Drew, S.C.; Gahan, L.R.; Hanson, G.R.; Moubaraki, B.; Murray, K.S. Synthetic, EPR Spectroscopic, Magnetic and X-Ray Crystallographic Structural Studies on Copper(II) Complexes of the Tridentate N2S Donor Ligand Formed from 6-Methyl-2-Formylpyridine and S-Methyldithiocarbazate (Hmpsme). Inorg. Chim. Acta 2005, 358, 3937–3948. [Google Scholar] [CrossRef]
- Seena, E.B.; Kurup, M.R.P. Spectral and Structural Studies of Mono- and Binuclear Copper(II) Complexes of Salicylaldehyde N(4)-Substituted Thiosemicarbazones. Polyhedron 2007, 26, 829–836. [Google Scholar] [CrossRef]
- West, D.X.; Salberg, M.M.; Bain, G.A.; Liberta, A.E.; Valdés-Martínez, J.; Hernández-Ortega, S. Binuclear Copper(II) Complexes of 5-Nitrosalicylaldehyde N(3)-Substituted Thiosemicarbazones. Transit. Met. Chem. 1996, 21, 206–212. [Google Scholar] [CrossRef]
- García-Tojal, J.; García-Orad, A.; Serra, J.L.; Pizarro, J.L.; Lezama, L.; Arriortua, M.I.; Rojo, T. Synthesis and Spectroscopic Properties of Copper(II) Complexes Derived from Thiophene-2-Carbaldehyde Thiosemicarbazone. Structure and Biological Activity of [Cu(C6H6N3S2)2]. J. Inorg. Biochem. 1999, 75, 45–54. [Google Scholar] [CrossRef]
- Bharadwaj, P.K.; Potenza, J.A.; Schugar, H.J. Characterization of [Dimethyl N,N′-Ethylenebis(L-Cysteinato)(2-)-S,S’]Copper(II), a Stable Copper(II) Aliphatic Dithiolate. J. Am. Chem. Soc. 1986, 108, 1351–1352. [Google Scholar] [CrossRef]
- Suzuki, Y.; Fujii, S.; Tominaga, T.; Yoshimoto, T.; Yoshimura, T.; Kamada, H. The Origin of an EPR Signal Observed in Dithiocarbamate-Loaded Tissues. Biochim. Biophys. Acta Gen. Subj. 1997, 1335, 242–245. [Google Scholar] [CrossRef]
- Glass, R.S.; Steffen, L.K.; Swanson, D.D.; Wilson, G.S.; de Gelder, R.; de Graaff, R.A.G.; Reedijk, J. Bis(Trithiacyclononane)Metal(II) Compounds and Jahn-Teller Distortions from Octahedral Geometry, Electrochemistry, Spectroscopy, and Crystal Structures of the Copper Bis(Tetrafluoroborate) Bis(Acetonitrile) Complex at 177 K and the Cadmium Bis(Tetrafluoroborate) and Copper Bis(Tetrafluoroborate) Bis(Nitromethane) Complexes at 300 K. Inorg. Chim. Acta 1993, 207, 241–252. [Google Scholar] [CrossRef]
- Schwabe, T.; Grimme, S. Towards Chemical Accuracy for the Thermodynamics of Large Molecules: New Hybrid Density Functionals Including Non-Local Correlation Effects. Phys. Chem. Chem. Phys. 2006, 8, 4398. [Google Scholar] [CrossRef]
- Karton, A.; Tarnopolsky, A.; Lamère, J.-F.; Schatz, G.C.; Martin, J.M.L. Highly Accurate First-Principles Benchmark Data Sets for the Parametrization and Validation of Density Functional and Other Approximate Methods. Derivation of a Robust, Generally Applicable, Double-Hybrid Functional for Thermochemistry and Thermochemical Kinetics. J. Phys. Chem. A 2008, 112, 12868–12886. [Google Scholar] [CrossRef]
- Tarnopolsky, A.; Karton, A.; Sertchook, R.; Vuzman, D.; Martin, J.M.L. Double-Hybrid Functionals for Thermochemical Kinetics. J. Phys. Chem. A 2008, 112, 3–8. [Google Scholar] [CrossRef]
- Brémond, É.; Sancho-García, J.C.; Pérez-Jiménez, Á.J.; Adamo, C. Communication: Double-Hybrid Functionals from Adiabatic-Connection: The QIDH Model. J. Chem. Phys. 2014, 141, 031101. [Google Scholar] [CrossRef]
- Brémond, E.; Adamo, C. Seeking for Parameter-Free Double-Hybrid Functionals: The PBE0-DH Model. J. Chem. Phys. 2011, 135, 024106. [Google Scholar] [CrossRef]
- Brémond, É.; Savarese, M.; Pérez-Jiménez, Á.J.; Sancho-García, J.C.; Adamo, C. Range-Separated Double-Hybrid Functional from Nonempirical Constraints. J. Chem. Theory Comput. 2018, 14, 4052–4062. [Google Scholar] [CrossRef]
- Brémond, É.; Pérez-Jiménez, Á.J.; Sancho-García, J.C.; Adamo, C. Range-Separated Hybrid Density Functionals Made Simple. J. Chem. Phys. 2019, 150, 201102. [Google Scholar] [CrossRef]
- Kozuch, S.; Gruzman, D.; Martin, J.M.L. DSD-BLYP: A General Purpose Double Hybrid Density Functional Including Spin Component Scaling and Dispersion Correction. J. Phys. Chem. C 2010, 114, 20801–20808. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. DSD-PBEP86: In Search of the Best Double-Hybrid DFT with Spin-Component Scaled MP2 and Dispersion Corrections. Phys. Chem. Chem. Phys. 2011, 13, 20104. [Google Scholar] [CrossRef] [PubMed]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A Long-Range Correction Scheme for Generalized-Gradient-Approximation Exchange Functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange–Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef]
- Casanova-Páez, M.; Dardis, M.B.; Goerigk, L. ΩB2PLYP and ΩB2GPPLYP: The First Two Double-Hybrid Density Functionals with Long-Range Correction Optimized for Excitation Energies. J. Chem. Theory Comput. 2019, 15, 4735–4744. [Google Scholar] [CrossRef]
- Casanova-Páez, M.; Goerigk, L. Time-Dependent Long-Range-Corrected Double-Hybrid Density Functionals with Spin-Component and Spin-Opposite Scaling: A Comprehensive Analysis of Singlet–Singlet and Singlet–Triplet Excitation Energies. J. Chem. Theory Comput. 2021, 17, 5165–5186. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Double-Hybrid Density Functionals. J. Chem. Phys. 2009, 131, 174105. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density Functional Theory Is Straying from the Path toward the Exact Functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Kepp, K.P. Comment on “Density Functional Theory Is Straying from the Path toward the Exact Functional”. Science 2017, 356, 496. [Google Scholar] [CrossRef]
- Savarese, M.; Brémond, É.; Ciofini, I.; Adamo, C. Electron Spin Densities and Density Functional Approximations: Open-Shell Polycyclic Aromatic Hydrocarbons as Case Study. J. Chem. Theory Comput. 2020, 16, 3567–3577. [Google Scholar] [CrossRef] [PubMed]
- Sharkas, K.; Toulouse, J.; Savin, A. Double-Hybrid Density-Functional Theory Made Rigorous. J. Chem. Phys. 2011, 134, 064113. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Regular Two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Total Energy Using Regular Approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Neese, F. An Improvement of the Resolution of the Identity Approximation for the Formation of the Coulomb Matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, Approximate and Parallel Hartree–Fock and Hybrid DFT Calculations. A ‘Chain-of-Spheres’ Algorithm for the Hartree–Fock Exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Heß, B.A.; Marian, C.M.; Wahlgren, U.; Gropen, O. A Mean-Field Spin-Orbit Method Applicable to Correlated Wavefunctions. Chem. Phys. Lett. 1996, 251, 365–371. [Google Scholar] [CrossRef]
- Neese, F. Efficient and Accurate Approximations to the Molecular Spin-Orbit Coupling Operator and Their Use in Molecular g-Tensor Calculations. J. Chem. Phys. 2005, 122, 034107. [Google Scholar] [CrossRef]
- Neese, F. Calculation of the Zero-Field Splitting Tensor on the Basis of Hybrid Density Functional and Hartree-Fock Theory. J. Chem. Phys. 2007, 127, 164112. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).