
Synthesis of New Derivatives of BEDT-TTF: Installation of Alkyl, Ethynyl, and Metal-Binding Side Chains and Formation of Tris(BEDT-TTF) Systems †

 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
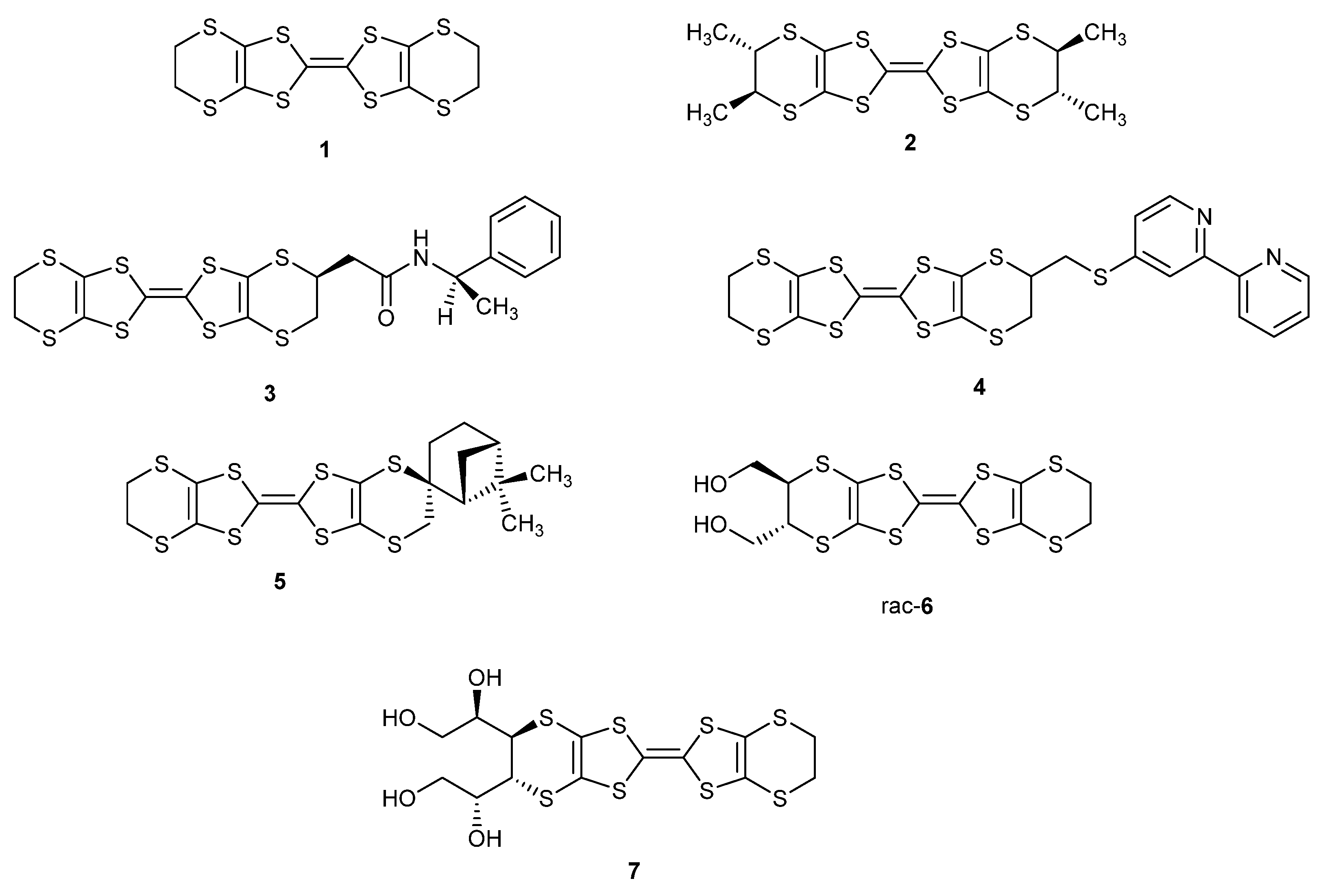
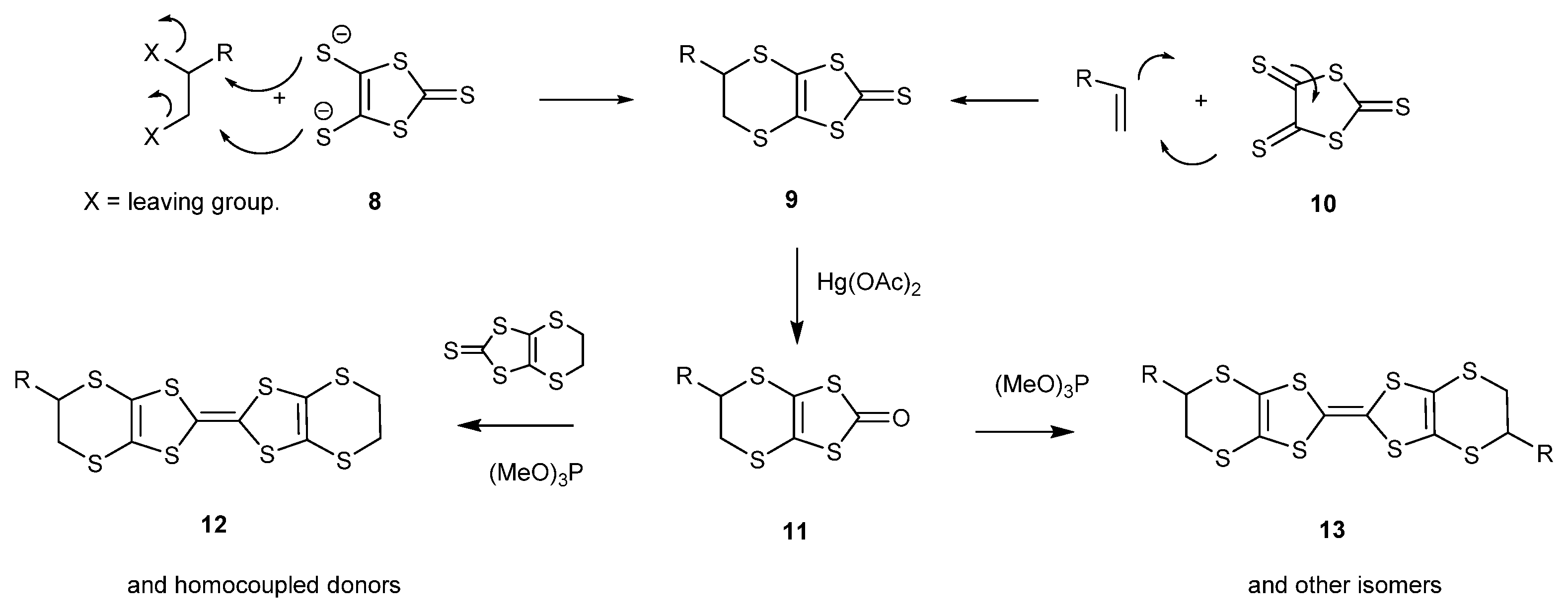
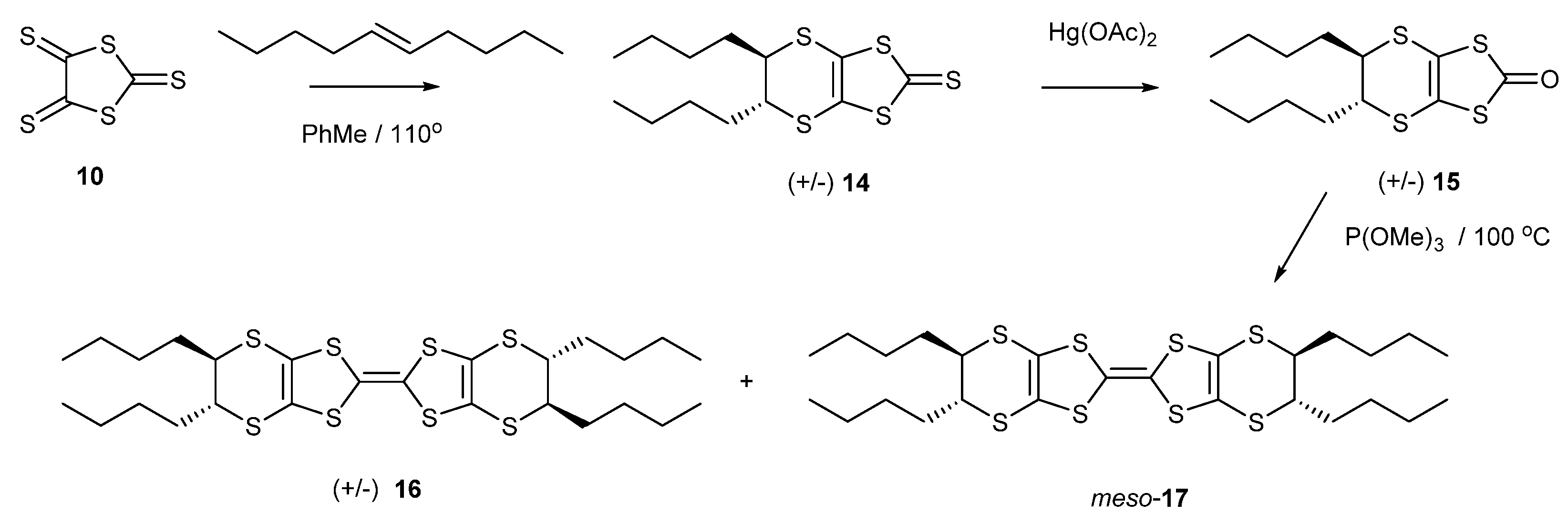
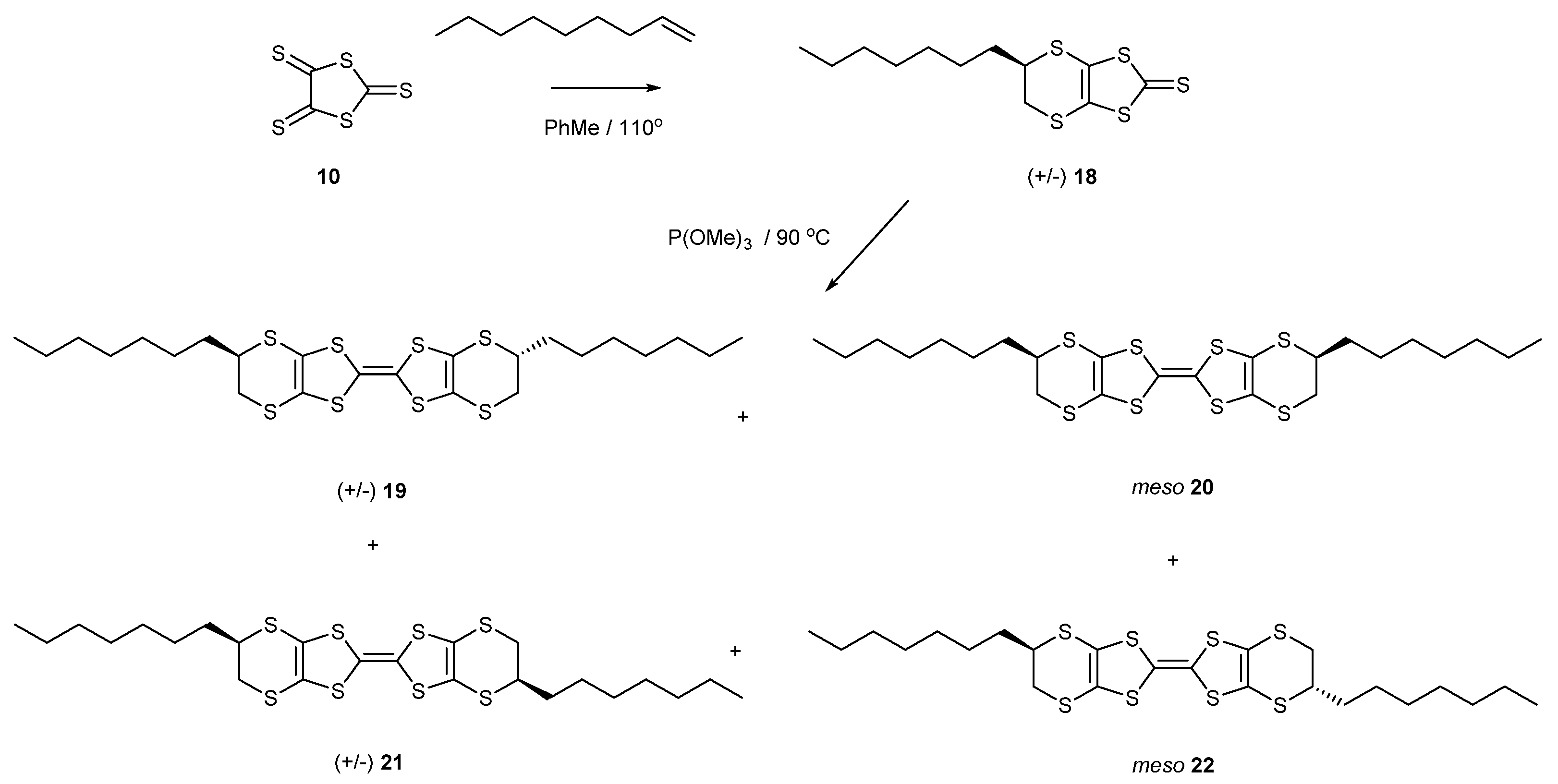
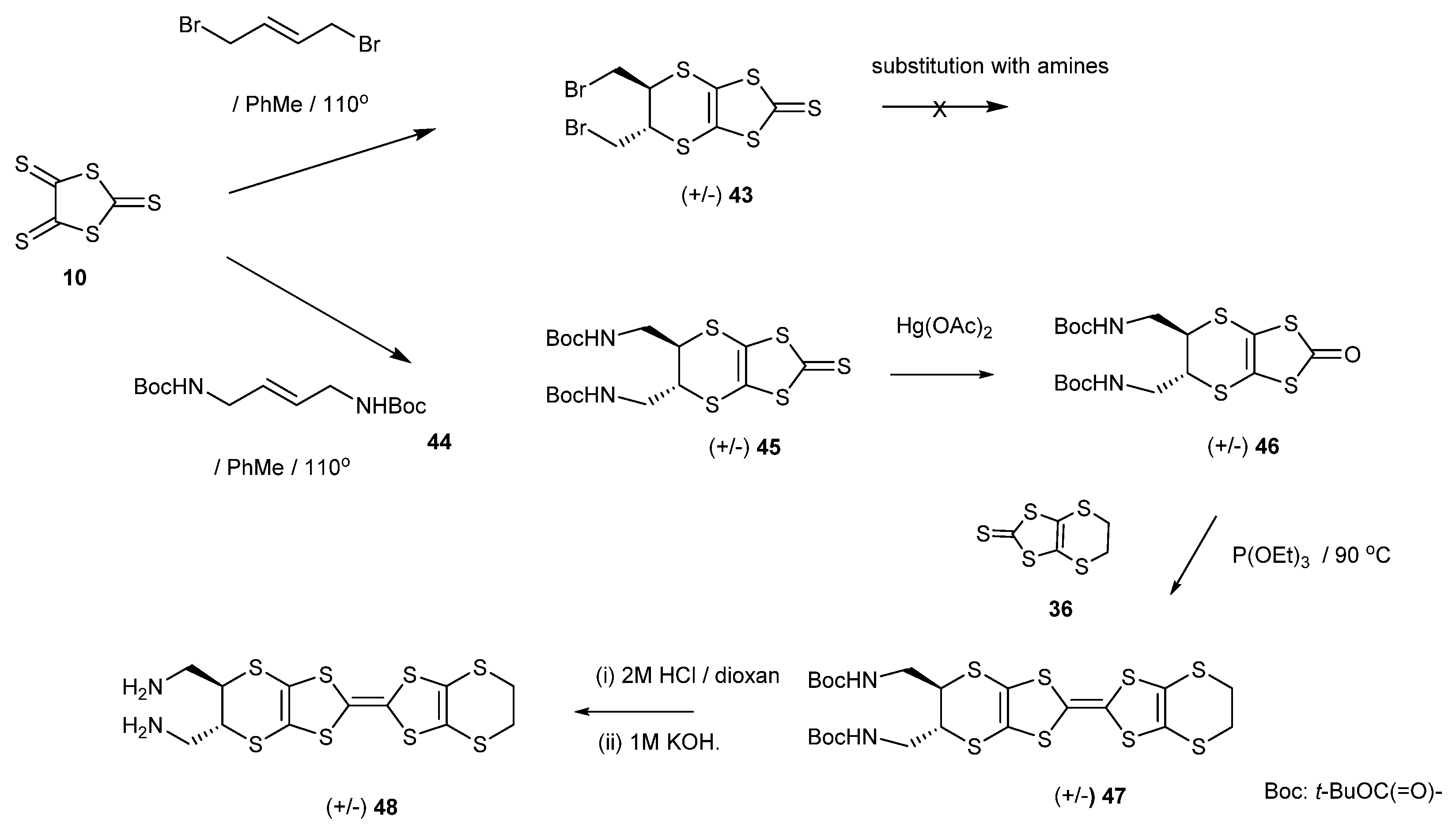
2.1. BEDT-TTF with Alkyl Chains
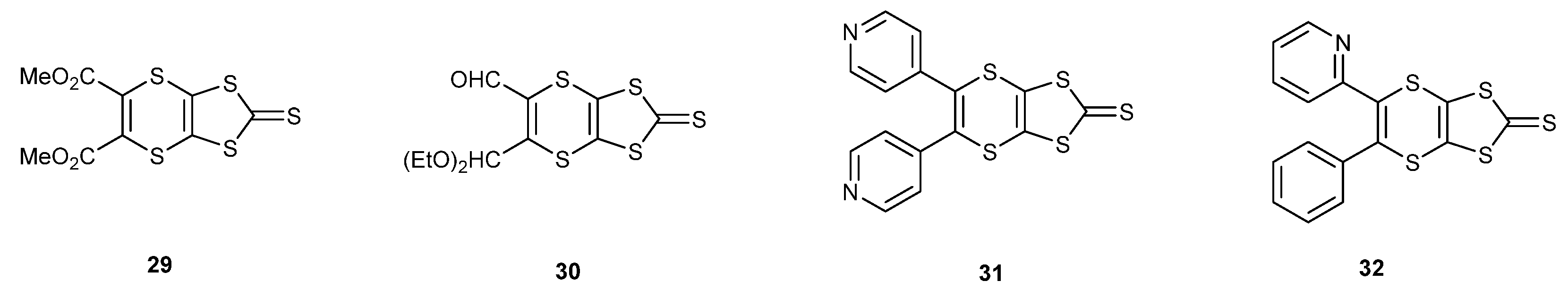
2.2. Tris-(BEDT-TTF) Donors
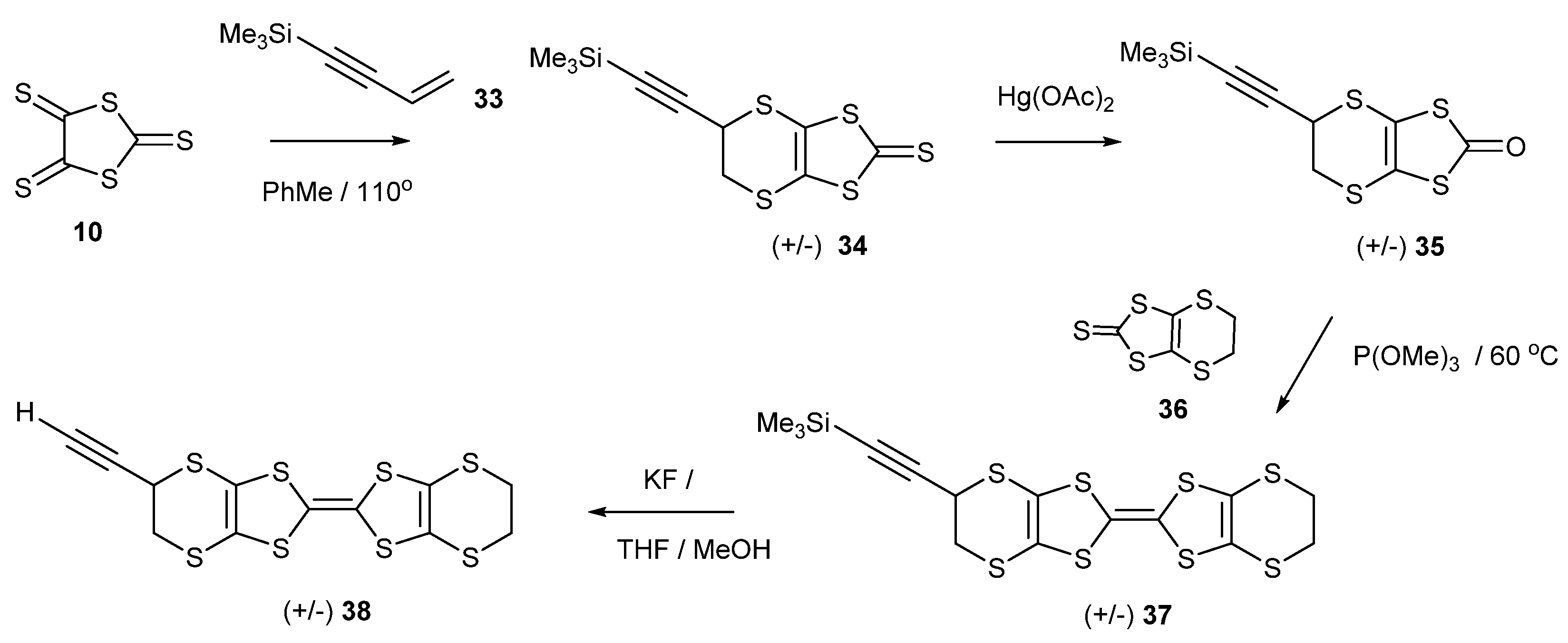
2.3. New Functionalised BEDT-TTFs: Ethynyl and Acetal Substituted Derivatives
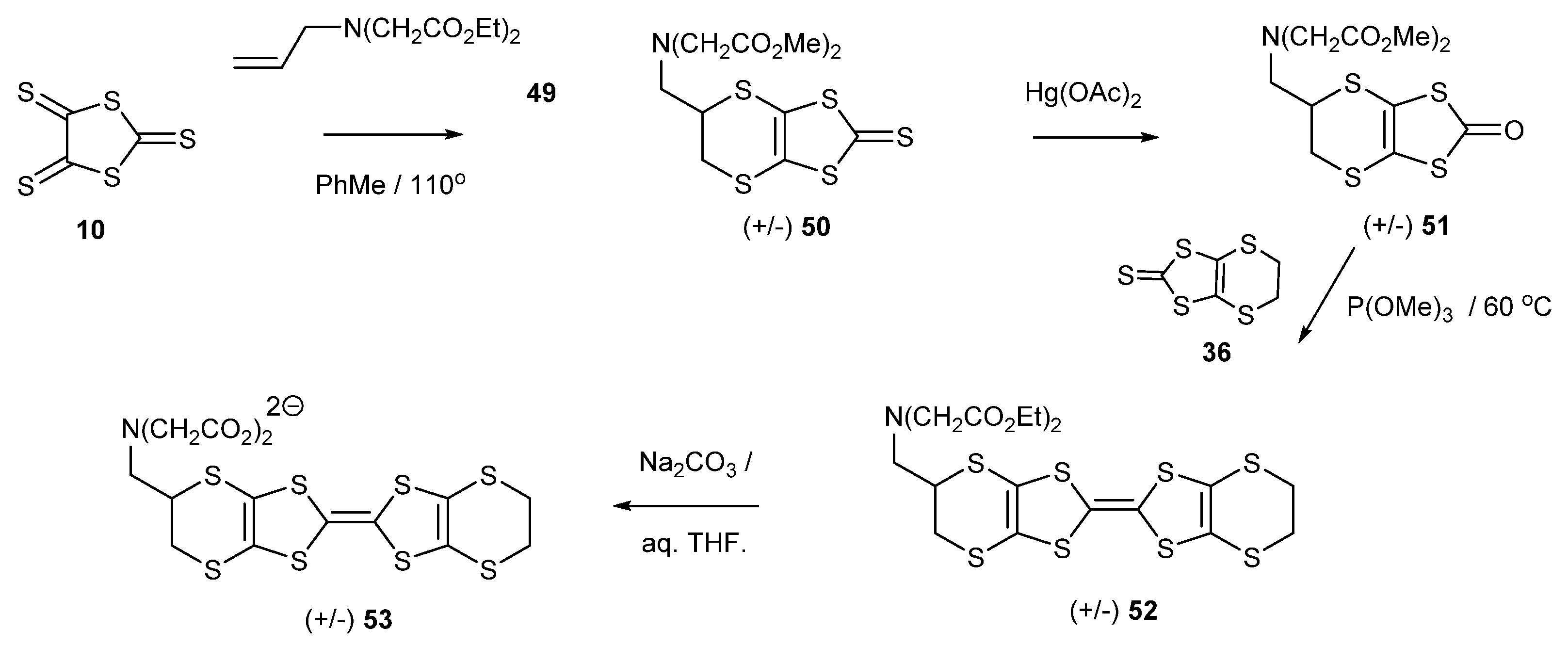
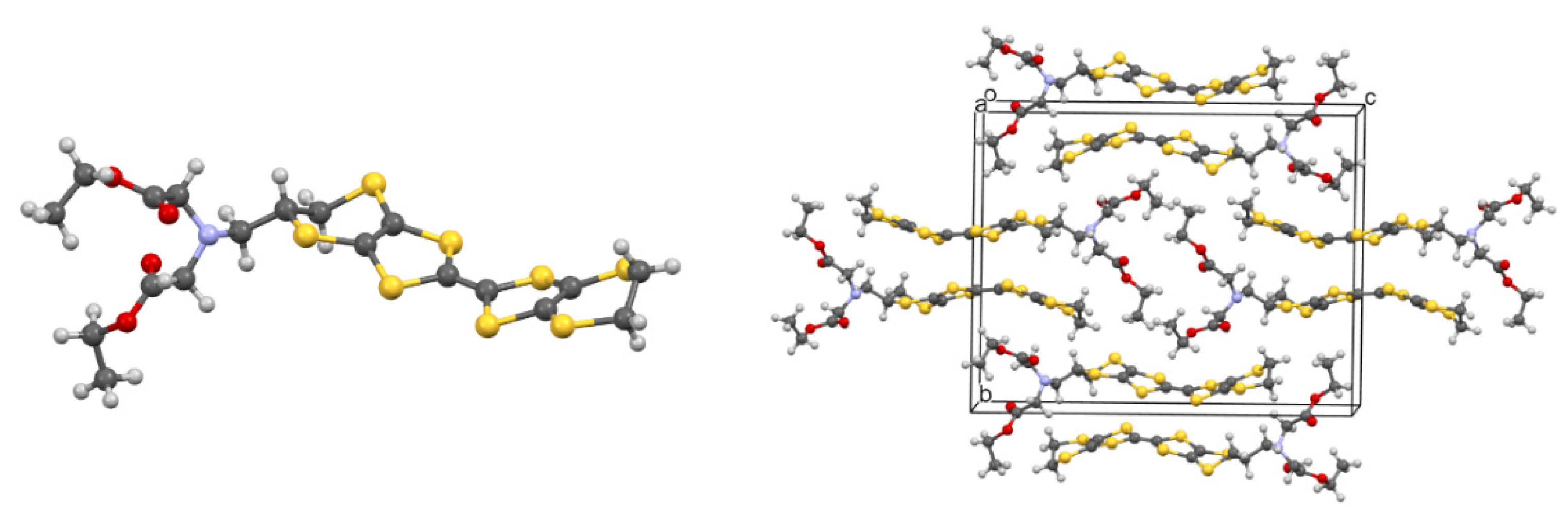
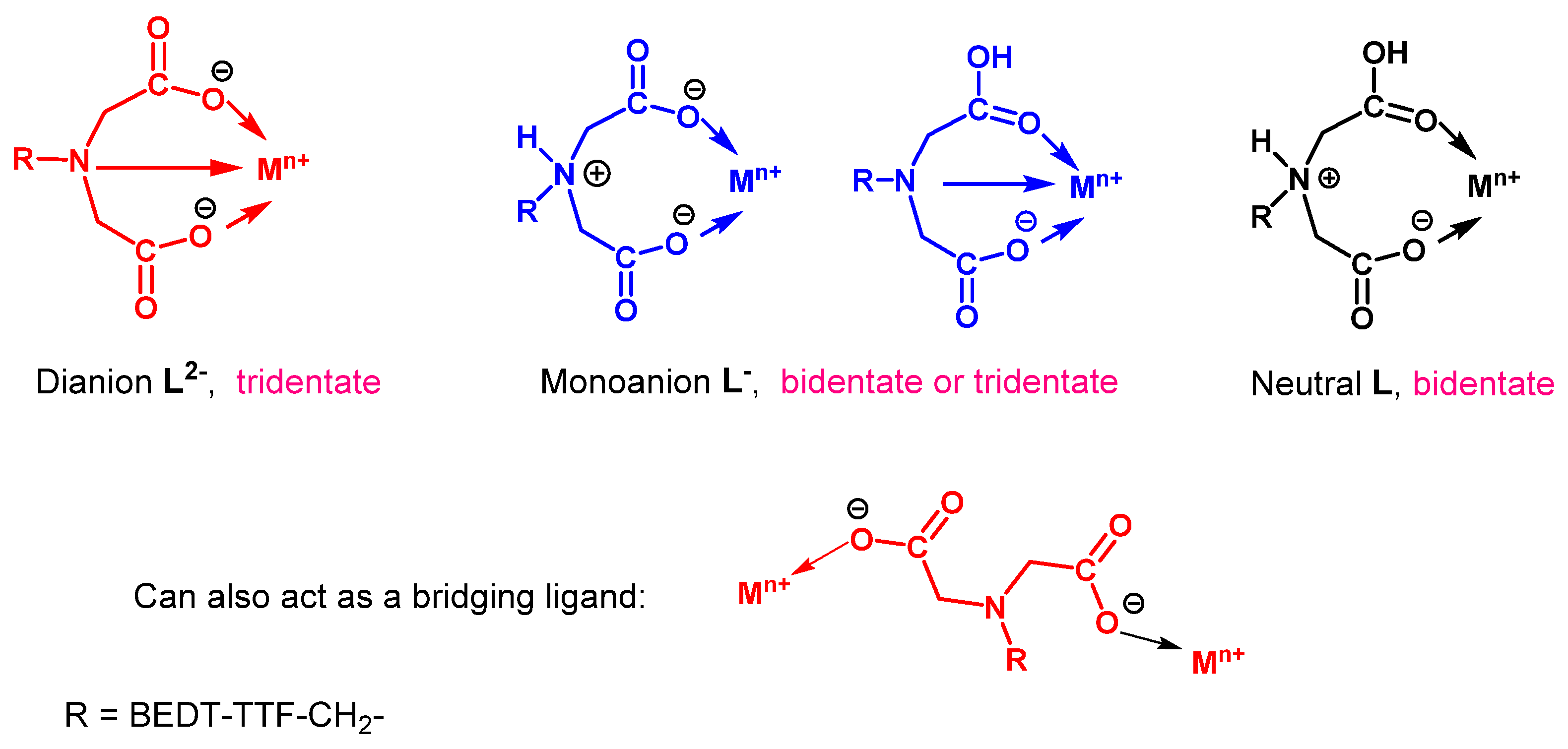
2.4. Donors with Metal Ion Binding Potential and the First Salts and Their Magnetic Properties
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Dedication
References
- Singleton, J.; Mielke, C. Quasi-Two-Dimensional Organic Superconductors. Contemp. Phys. 2002, 43, 63–96. [Google Scholar] [CrossRef] [Green Version]
- Singleton, J.; Mielke, C. Superconductors Go Organic. Phys. World 2002, 15, 35–39. [Google Scholar] [CrossRef]
- Day, P. BEDT-TTF Charge Transfer Salts: New Structures, New Functionalities. Compt. Rend. Chim. 2003, 6, 301–308. [Google Scholar] [CrossRef]
- Mori, H. Introduction to Organic Superconducting Materials. Opt. Sci. Eng. 2008, 133, 263–285. [Google Scholar]
- Mori, T. Structural Genealogy of BEDT-TTF-Based Organic Conductors. I. Parallel Molecules: β and β” Phases. Bull. Chem. Soc. Jpn. 1998, 71, 2509–2526. [Google Scholar] [CrossRef]
- Mori, T.; Mori, H.; Tanaka, S. Structural Genealogy of BEDT-TTF-Based Organic Conductors. II. Inclined Molecules: θ, α and κ Phases. Bull. Chem. Soc. Jpn. 1999, 72, 179–197. [Google Scholar] [CrossRef]
- Mori, T. Structural Genealogy of BEDT-TTF-Based Organic Conductors. III. Twisted Molecules: δ and α’ Phases. Bull. Chem. Soc. Jpn. 1999, 72, 2011–2027. [Google Scholar] [CrossRef]
- Williams, J.M.; Wang, H.H.; Kini, A.M.; Carlson, K.D.; Beno, M.A.; Geiser, U.; Whangbo, M.-H.; Jung., D.; Evain, M.; Novoa, J.J. Recent Progress in the Development of Structure-Property Correlations for κ-Phase Organic Superconductors. Mol. Crys. Liq. Crys. 1999, 181, 59–64. [Google Scholar] [CrossRef]
- Ishiguro, T.; Yamaji, K.; Saito, G. Organic Superconductors; Springer: Berlin, Germany, 1998. [Google Scholar]
- Mori, H. Materials Viewpoint of Organic Superconductors. J. Phys. Soc. Jpn. 2006, 75, 051003-15. [Google Scholar] [CrossRef]
- Kurmoo, M.; Graham, A.W.; Day, P.; Coles, S.J.; Hursthouse, M.B.; Caulfield, J.L.; Singleton, J.; Pratt, F.L.; Hayes, W.; Ducasse, L.; et al. Superconducting and Semiconducting Magnetic Charge Transfer Salts: (BEDT-TTF)4AFe(C2O4)3·C6H5CN (A = H2O, K, NH4). J. Am. Chem. Soc. 1995, 117, 12209–12217. [Google Scholar] [CrossRef]
- Coronado, E.; Day, P. Magnetic molecular conductors. Chem. Rev. 2004, 104, 5419–5448. [Google Scholar] [CrossRef]
- Martin, L. Molecular Conductors of BEDT-TTF with Tris(oxalato)metallate Anions. Coord. Chem. Rev. 2018, 376, 277–291. [Google Scholar] [CrossRef] [Green Version]
- Sahadevan, S.A.; Abherve, A.; Monni, N.; Auban-Senzier, P.; Cano, J.; Lloret, F.; Julve, M.; Cui, H.; Kato, R.; Canadell, E.; et al. Magnetic Molecular Conductors Based on Bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF) with the Tris(chlorocyananilato)ferrate(III) Complex. Inorg. Chem. 2019, 58, 15359–15370. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.-P.; Wallis, J.D. Substituted BEDT-TTF Derivatives: Synthesis, Chirality, Properties and Potential Applications. J. Mater. Chem. 2005, 15, 347–365. [Google Scholar]
- Wallis, J.D.; Karrer, A.; Dunitz, J.D. Chiral Metals? A Chiral Substrate for Organic Conductors and Superconductors. Helv. Chim. Acta 1986, 69, 69–70. [Google Scholar] [CrossRef]
- Coronado, E.; Galán-Mascarós, J.R.; Coldea, A.I.; Goddard, P.; Singleton, J.; Wallis, J.D.; Coles, S.J.; Alberola, A. A Chiral Ferromagnetic Molecular Metal. J. Am. Chem. Soc. 2010, 132, 9271–9273. [Google Scholar]
- Pop, F.; Laroussi, S.; Cauchy, T.; Gomez-Garcia, C.J.; Wallis, J.D.; Avarvari, N. Tetramethyl-Bis(ethylenedithio)-Tetrathiafulvalene (TM-BEDT-TTF) Revisited: Crystal Structures, Chiroptical Properties, Theoretical Calculations and a Complete Series of Conducting Radical Cattion Salts. Chirality 2013, 25, 466–474. [Google Scholar] [CrossRef]
- Pop, F.; Meziere, C.; Allain, M.; Auban-Senzier, P.; Tajima, N.; Hirobe, D.; Yamamoto, H.; Canadell, E.; Avarvari, N. Unusual Stoichiometry, Band Structure and Band Filling in Conducting Enantiopure Radical Cation Salts of TM-BEDT-TTF Showing Helical Packing of the Donors. J. Mater. Chem. C. 2021. online. [Google Scholar] [CrossRef]
- Short, J.; Blundell, T.J.; Krivickas, S.J.; Yang, S.; Wallis, J.D.; Akutsu, H.; Nakazawa, Y.; Martin, L. Chiral Molecular Conductor with an Insulator–Metal Transition Close to Room Temperature. Chem. Commun. 2020, 56, 9497–9500. [Google Scholar] [CrossRef]
- Wang, Q.; Martin, L.; Blake, A.J.; Day, P.; Akutsu, H.; Wallis, J.D. Coordination Chemistry of 2,2′-Bipyridyl- and 2,2′: 6′,2″-Terpyridyl-substituted BEDT-TTFs: Formation of a Supramolecular Capsule Motif by the Iron (II) Tris Complex of 2,2′-Bipyridine-4-thiomethyl-BEDT-TTF. Inorg. Chem. 2016, 55, 8543–8551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, J.-P.; Nie, H.; Brown, R.J.; Day, P.; Wallis, J.D. Synthetic Strategies to Chiral Organosulfur Donors related to Bis(ethylenedithio)tetrathiafulvalene. Org. Biomolec. Chem. 2005, 3, 2155–2166. [Google Scholar] [CrossRef]
- Griffiths, J.; Arnal, A.A.; Appleby, G.; Wallis, J.D. Synthesis and Reactivity of Amino-substituted BEDT-TTF Donors as Building Blocks for Bifunctional Materials. Tetrahedron Lett. 2004, 45, 2813–2816. [Google Scholar] [CrossRef]
- Saygili, N.; Brown, R.J.; Day, P.; Hoelzl, R.; Kathirgamanathan, P.; Mageean, E.R.; Ozturk, T.; Pilkington, M.; Qayyum, M.M.B.; Turner, S.S.; et al. Functionalised Organosulfur Donor Molecules: Synthesis of Racemic Hydroxymethyl-, Alkoxymethyl- and Dialkoxymethyl-bis(ethylenedithio)tetrathiafulvalenes. Tetrahedron 2001, 57, 5015–5026. [Google Scholar] [CrossRef]
- Wang, Q.; Zecchini, M.; Wallis, J.D.; Wu, Y.; Rawson, J.M.; Pilkington, M. A Family of Unsymmetrical Hydroxyl-substituted BEDT-TTF Donors: Syntheses, Structures and Preliminary Thin Film Studies. RSC Adv. 2015, 5, 40205–40218. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.J.; Brooks, A.C.; Griffiths, J.-P.; Vital, B.; Day, P.; Wallis, J.D. Synthesis of Bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF) Derivatives with Two, Four or Eight Hydroxyl Groups. Org. Biomolec. Chem. 2007, 5, 3172–3182. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, D.; Zhang, B.; Yao, Y.; Xu, W.; Zhu, D.; Wang, Z. Synthesis of New Electron Donors with Hydroxymethyl Groups and Studies on Their Cation-Radical Salts. J. Mater. Chem. 2000, 10, 2063–2067. [Google Scholar] [CrossRef]
- Li, H.; Zhang, D.; Xu, W.; Fan, L.; Zhu, D. New Electron Donor: Bis(ethylenedithio)tetrathiafulvalene Derivative with Four Hydroxyl Groups. Syn. Met. 1999, 106, 111–114. [Google Scholar] [CrossRef]
- Berridge, R.; Serebryakov, I.M.; Skabara, P.J.; Orti, E.; Viruela, R.; Pou-Amerigo, R.; Coles, S.J.; Hursthouse, M.B. A New Series of π-Extended Tetrathiafulvalene Derivatives Incorporating Fused Furanodithiino and Thienodithiino Units: A Joint Experimental and Theoretical Study. J. Mater. Chem. 2004, 14, 2822–2830. [Google Scholar] [CrossRef]
- Aoyagi, I.; Katsuhara, M.; Mori, T. Synthesis and Structure of Highly Soluble Bis(ethylenedithio)tetrathiafulvalene Molecules with Alkyl Chains. Sci. Tech. Adv. Mater. 2004, 5, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Kini, A.M.; Parakka, J.P.; Geiser, U.; Wang, H.-H.; Rivas, F.; DiNiro, E.; Thomas, S.; Dudek, J.D.; Williams, J.M. Tetra-alkyl and Di-alkyl Substituted BEDT-TTF Derivatives and Their Cation-Radical Salts: Synthesis, Structure and Properties. J. Mater. Chem. 1999, 9, 883–892. [Google Scholar] [CrossRef]
- Troitsky, V.I.; Berzina, T.S.; Katsen, Y.Y.; Neilands, O.Y.; Nicolini, C. Conducting Langmuir-Blodgett Films of Heptadecylcarboxymethyl-BEDT-TTF. Syn. Metals 1995, 74, 1–6. [Google Scholar] [CrossRef]
- Goldenberg, L.M.; Khodorkovsky, V.Y.; Vladimir, Y.; Becker, J.Y.; Lukes, P.J.; Bryce, M.R.; Petty, M.C.; Yarwood, J. Highly Conducting Langmuir-Blodgett Films of an Amphiphilic Bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF) Derivative: BEDT-TTF-C18H37. Chem. Mater. 1994, 6, 1426–1431. [Google Scholar] [CrossRef]
- Khodorkovskii, B.Y.; Pukitis, G.; Puplovskii, A.Y.; Edzina, A.; Neilands, O.Y. Synthesis and Properties of the Hexadecyl Derivative of Bis(ethylenedithio)tetrathiafulvalene. Khim. Geterotsikl. Soedin. 1990, 131–132. [Google Scholar]
- Yang, X.; Rauchfuss, T.B.; Wilson, S. The Chemistry of C6S10: A Channel Structure for C6S10.(CS2)0.5 and Access to the Versatile DMAD-C3S4O. Chem. Commun. 1990, 34–36. [Google Scholar] [CrossRef]
- Leriche, P.; Gorgues, A.; Jubault, M.; Becher, J.; Orduna, J.; Garin, J. Cycloaddition of Acetylenedicarbaldehyde Monoacetal and 2,4,5-Trithioxo-1,3-dithiole: Ready Access to Novel Highly Extended and Sulfur-rich Analogues of Tetrathiafulvalene (TTF). Tetrahedron Lett. 1995, 36, 1275–1278. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Miyamoto, T.; Yoshida, A.; Kawada, Y.; Nakazaki, J.; Izuoka, A.; Sugawara, T. New Synthesis of 2-(1,3-Dithiol-2-ylidene)-5,6-dihydro-1,3-dithiolo[4,5-b][1,4]dithiiins with Formyl Group on Fused Benzene, [1,4]dithiin or Thiophene Ring. Tetrahedron Lett. 1999, 40, 8819–8822. [Google Scholar] [CrossRef]
- Brooks, A.C.; Day, P.; Dias, S.I.G.; Rabaca, S.; Santos, I.C.; Henriques, R.T.; Wallis, J.D.; Almeida, M. Pyridine-functionalised (Vinylenedithio)tetrathiafulvalene (VDT-TTF) Derivatives and Their Dithiolene Analogues. Eur. J. Inorg. Chem. 2009, 3084–3093. [Google Scholar] [CrossRef]
- Niu, Z.-G.; He, L.-R.; Li, L.; Cheng, W.-F.; Li, X.-Y.; Chen, H.-H.; Li, G.-N. Synthesis, Characterisation and DFT Studies of Two New π-Conjugated Pyridine-based Tetrathiafulvalene Derivatives. Acta Chim. Sloven. 2014, 61, 786–791. [Google Scholar]
- Marshallsay, G.J.; Bryce, M.R.; Cooke, G.; Joergensen, T.; Becher, J.; Reynolds, C.D.; Wood, S. Functionalised Tetrathiafulvalene (TTF) Systems Derived from 4, 5-(Propylenedithio)-1, 3-dithiole Units. Tetrahedron 1993, 49, 6849–6862. [Google Scholar] [CrossRef]
- Arenzano, J.A.; Virues, J.O.; Colorado-Peralta, R.; Ramirez-Montes, P.I.; Santillan, R.; Sanchez, M.; Rivera, J.M. Heterometallic Coordination Framework by Sodium Carboxylate Subunits and Cobalt(III) Centres Obtained from a Highly Hydrogen Bonding Stabilized Cobalt(II) Monomeric Complex. Inorg. Chem. Commun. 2015, 51, 55–60. [Google Scholar] [CrossRef]
- Puentes, R.; Torres, J.; Kremer, C.; Cano, J.; Lloret, F.; Capucci, D.; Bacchi, A. Mononuclear and Polynuclear Complexes Ligated by an Iminodiacetic Acid Derivative: Synthesis, Structure, Solution Studies and Magnetic Properties. Dalton Trans. 2016, 45, 5356–5373. [Google Scholar] [CrossRef]
- Yousuf, I.; Zeeshan, M.; Arjmand, F.; Rizvi, M.A.; Tabassum, S. Synthesis, Structural Investigations and DNA Cleavage Properties of a New Water Soluble Cu(II)-iminodiacetate Complex. Inorg. Chem. Commun. 2019, 106, 48–53. [Google Scholar] [CrossRef]
- Puentes, R.; Torres, J.; Faccio, R.; Bacchi, A.; Kremer, C. Lanthanide Coordination Polymers Based on Flexible Ligands Derived from Iminodiacetic acid. Polyhedron 2019, 170, 683–689. [Google Scholar] [CrossRef]
- Kanehama, R.; Umemiya, M.; Iwahori, F.; Miyasaka, H.; Sugiura, K.-I.; Yamashita, M.; Yokochi, Y.; Ito, H.; Kuroda, S.-I.; Kishida, H.; et al. New ET-Coordinated Copper(I) Complexes: Synthesis, Structures and Physical Properties. Inorg. Chem. 2003, 42, 7173–7181. [Google Scholar] [CrossRef]
- Jia, C.; Zhang, D.; Liu, C.-M.; Xu, W.; Hu, H.; Zhu, D. Novel Silver(I) Complexes Derived From Tetrakis(methylthio)tetrathiafulvalene and Bis(ethylenedithio)tetrathiafulvalene with 3D and 1D Structures. New J. Chem. 2002, 26, 490–494. [Google Scholar] [CrossRef]
- Inoue, M.B.; Inoue, M.; Bruck, M.A.; Fernando, Q. Structure of Bis(ethylenedithio)tetrafulvalenium Tribromodicuprate(I), (BEDT-TTF+)Cu(I)2Br3: Coordination of the Organic Radical Cation to the Metal Ions. J. Chem. Soc. Chem. Commun. 1992, 515–516. [Google Scholar] [CrossRef]
- Pop, F.; Auban-Senzier, P.; Canadell, E.; Rikken, G.L.R.A.; Avarvari, N. Electrical Magnetochiral Anisotropy in a Bulk Chiral Molecular Conductor. Nat. Commun. 2014, 5, 3757. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Barbour, L.J. X-Seed—A Software Tool for Supramolecular Crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Wood, P.A. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | E1 (V) | E2 (V) |
|---|---|---|
| 16–17 | 0.47 | 0.89 |
| 19–22 | 0.49 | 0.89 |
| 26 | 0.48, 0.55 b | 0.88 b |
| 27 | 0.44, 0.56 b | 0.86 b |
| 28 | 0.50 | 0.88 |
| 38 | 0.52 | 0.92 |
| 42 | 0.50 | 0.93 |
| 48 | 0.72 b,c | 0.89 b,c |
| 52 | 0.53 | 0.94 |
| Metal salt, Colour. | Proposed Composition, Compound number, RMM. | Magnetic properties: a χT (cm3 K mol−1); C (cm3 K mol−1); θ (K). | Found CHN (%) | Calculated CHN (%) |
|---|---|---|---|---|
| Zn(II)triflate Red/pink. | Zn(HL)2 54 RMM 1121.4 | C: 31.95 H: 2.41 N: 2.64 | C: 32.09 H: 2.51 N: 2.49 | |
| MnCl2 Red. | Mn(H2L)2Cl2.2H2O 55 RMM 1220 | χT: 4.4 C: 4.47 θ: −6.07 | C: 29.28 H: 2.57 N: 2.58 | C: 29.50 H: 2.81 N: 2.29 |
| Mn(hfac)2 Orange /brown. | Mn(H2L)(hfac)2.H2O 56 RMM 1016.8 | χT: 4.5 C: 4.66 θ: −7.72 | C: 29.58 H: 1.88 N: 1.48 | C: 29.53 H: 1.88 N: 1.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Zecchini, M.; Brooks, A.C.; Krivickas, S.J.; Dalligos, D.M.; Matuszek, A.M.; Stares, E.L.; Pilkington, M.; Wallis, J.D. Synthesis of New Derivatives of BEDT-TTF: Installation of Alkyl, Ethynyl, and Metal-Binding Side Chains and Formation of Tris(BEDT-TTF) Systems. Magnetochemistry 2021, 7, 110. https://doi.org/10.3390/magnetochemistry7080110
Yang S, Zecchini M, Brooks AC, Krivickas SJ, Dalligos DM, Matuszek AM, Stares EL, Pilkington M, Wallis JD. Synthesis of New Derivatives of BEDT-TTF: Installation of Alkyl, Ethynyl, and Metal-Binding Side Chains and Formation of Tris(BEDT-TTF) Systems. Magnetochemistry. 2021; 7(8):110. https://doi.org/10.3390/magnetochemistry7080110
Chicago/Turabian StyleYang, Songjie, Matteo Zecchini, Andrew C. Brooks, Sara J. Krivickas, Desiree M. Dalligos, Anna M. Matuszek, Emma L. Stares, Melanie Pilkington, and John D. Wallis. 2021. "Synthesis of New Derivatives of BEDT-TTF: Installation of Alkyl, Ethynyl, and Metal-Binding Side Chains and Formation of Tris(BEDT-TTF) Systems" Magnetochemistry 7, no. 8: 110. https://doi.org/10.3390/magnetochemistry7080110
APA StyleYang, S., Zecchini, M., Brooks, A. C., Krivickas, S. J., Dalligos, D. M., Matuszek, A. M., Stares, E. L., Pilkington, M., & Wallis, J. D. (2021). Synthesis of New Derivatives of BEDT-TTF: Installation of Alkyl, Ethynyl, and Metal-Binding Side Chains and Formation of Tris(BEDT-TTF) Systems. Magnetochemistry, 7(8), 110. https://doi.org/10.3390/magnetochemistry7080110