Abstract
The syntheses of new BEDT-TTF derivatives are described. These comprise BEDT-TTF with one ethynyl group (HC≡C-), with two (n-heptyl) or four (n-butyl) alkyl side chains, with two trans acetal (-CH(OMe)2) groups, with two trans aminomethyl (-CH2NH2) groups, and with an iminodiacetate (-CH2N(CH2CO2−)2 side chain. Three transition metal salts have been prepared from the latter donor, and their magnetic properties are reported. Three tris-donor systems are reported bearing three BEDT-TTF derivatives with ester links to a core derived from benzene-1,3,5-tricarboxylic acid. The stereochemistry and molecular structure of the donors are discussed. X-ray crystal structures of two BEDT-TTF donors are reported: one with two CH(OMe)2 groups and with one a -CH2N(CH2CO2Me)2 side chain.
1. Introduction
BEDT-TTF 1 has played a significant role in the development of electroactive organic materials. A very wide range of crystalline radical cation salts, with different stoichiometries, have been prepared, some of which are semi-conductors, conductors, or low temperature superconductors [1,2,3,4]. A range of different packing modes for the donors in these salts have been identified [5,6,7], which includes a κ-phase in the superconducting salts, such as (BEDT-TTF)2Cu(NCS)2, where the donors pack in face-to-face pairs but lie roughly perpendicular to their neighbouring pairs [8,9,10]. BEDT-TTF has been used to prepare hybrid materials with conducting and magnetic properties [11,12,13,14], as pioneered initially by Day et al. who prepared salts with iron tris(oxalate) salts which also showed low temperature superconductivity [11,12].
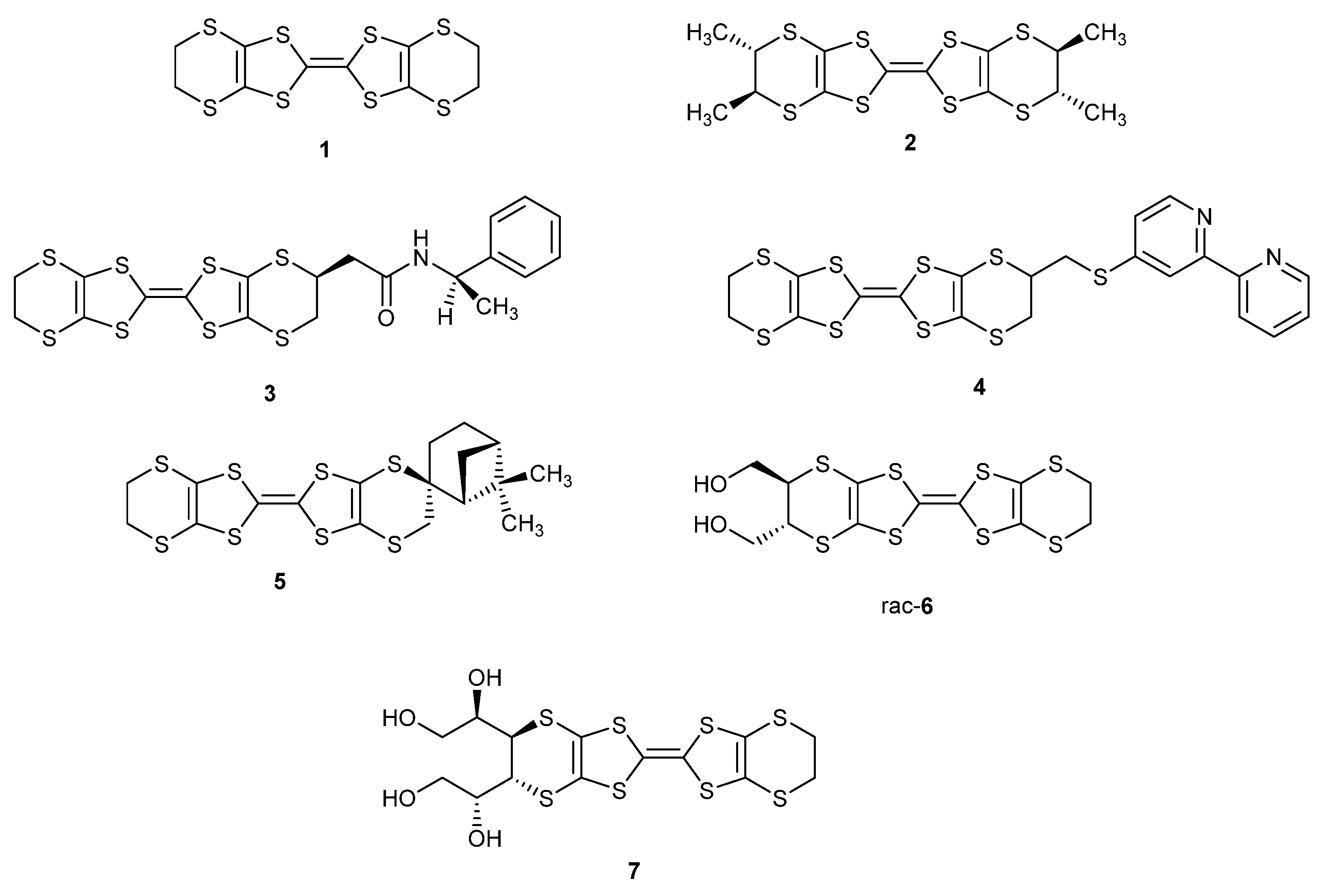
Following these studies on BEDT-TTF, a range of substituted BEDT-TTF derivatives that include compounds 2–5 have been reported (Scheme 1) [15]. Particularly notable examples are the enantiopure tetramethyl derivative 2 which forms a range of radical cation salts [16,17,18,19], the enantiopure amide 3 which forms a 4:1 TCNQ complex which changes from an insulator to an organic metal at 283 K and stays metallic down to ca. 4 K [20], the racemic bipyridylthiomethyl derivative 4 which forms capsular structures with metal ions [21], and the spiro chiral derivative 5 [22]. BEDT-TTF donors with one amino-methyl or -ethyl side chain [23], one hydroxy-methyl or -ethyl side chain [24,25], or with multiple hydroxymethyl or 1,2-dihydroxyethyl chains such as 6 and 7 have also been reported [26,27,28], as well as systems where the BEDT-TTF unit is fused to a thiophene or furan ring [29].
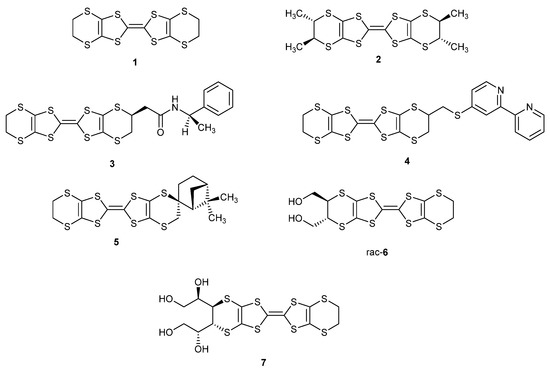
Scheme 1.
Structures of donors 1–7.
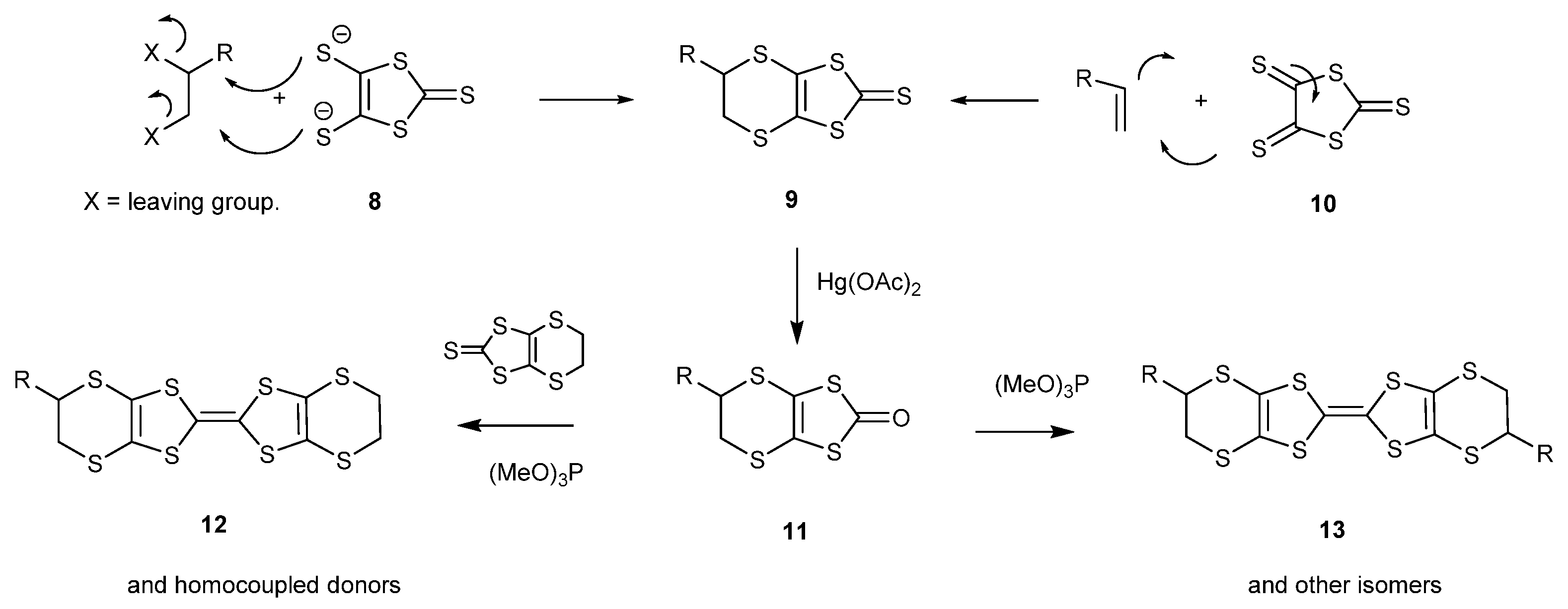
Two general synthetic routes have been used to prepare the aforementioned BEDT-TTF derivatives (Scheme 2) [15]. The first is via the dithiolate 8, available from carbon disulphide and sodium, by double substitutions with dihalides or cyclic sulphate esters to give the bicyclic thione 9 [16,24]. The second is via the trithione 10, which reacts with alkenes in a 4 + 2 electrocyclic reaction to also give the bicyclic thione 9 [22,23,24,25,26,27,28,30,31,32,33,34]. In both cases, the synthesis is completed by conversion of the thione to the oxo compound 11 by treatment with mercuric acetate, and then reaction with triethyl or trimethyl phosphite to form the homo-coupled BEDT-TTF system 13. Cross-coupling of an oxo compound with a different thione is used to prepare an unsymmetrical derivative BEDT-TTF 12.
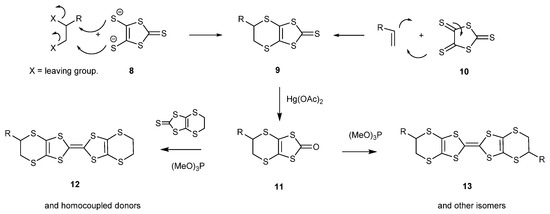
Scheme 2.
Two general synthetic routes to BEDT-TTF derivatives.
In recent work, we have expanded our library of BEDT-TTF donors and now report the syntheses of a range of new racemic BEDT-TTF derivatives, prepared by the second synthetic strategy using the trithione intermediate 10. This includes BEDT-TTFs substituted with an ethynyl side chain, 38, or with metal binding groups, 48 and 53, in addition to molecules bearing three BEDT-TTF units around a benzene ring core, 26–28. Preliminary magnetic results for complexes of donor 53 with 3D transition metals are presented. Stereochemical aspects of substituted donors are also discussed. Full synthetic details are provided in Section 4 and in the Supplementary Information.
2. Results and Discussion
2.1. BEDT-TTF with Alkyl Chains
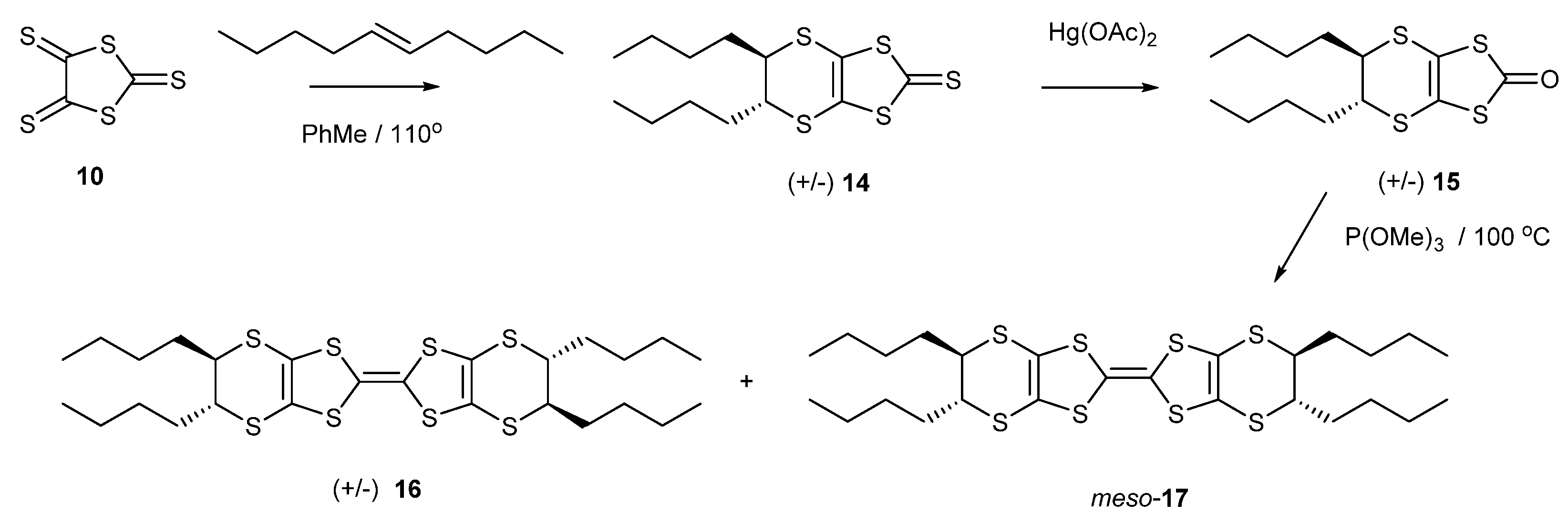
The installation of alkyl chains on a BEDT-TTF molecule should increase the solubility, which is very low for BEDT-TTF itself. Thus, the reaction of trithione 10 with trans-dec-5-ene gave the racemic thione 14 in 95% yield (Scheme 3). The treatment of this thione with mercuric acetate afforded the oxo compound 15 in an almost quantitative yield, which was homo-coupled using trimethyl phosphite to give the substituted BEDT-TTF in 57% yield as a mixture of stereoisomers. The configurations of the two stereogenic centres at one end of the molecule are the same but can be opposite to, or the same as, those at the other end. Thus, there are three stereoisomers: racemic (R,R,R,R)- and (S,S,S,S)-16 and the meso compound (R,R,S,S)-17. Unfortunately, these are not separable by chromatography, but the structures are expected to have very similar shapes. In this respect, the conformation of the dithiin ring is typically an envelope or a half chair. Thus, the R,R and S,S configurations can position their side chains in similar pseudo-equatorial positions by adopting opposite conformations of their dithiin rings.
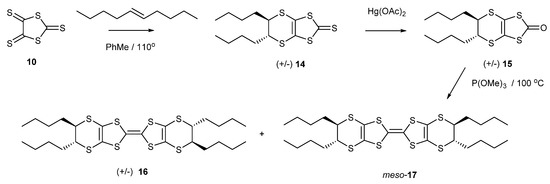
Scheme 3.
Synthetic route to tetra-n-butyl-BEDT-TTF derivatives 16 and 17.
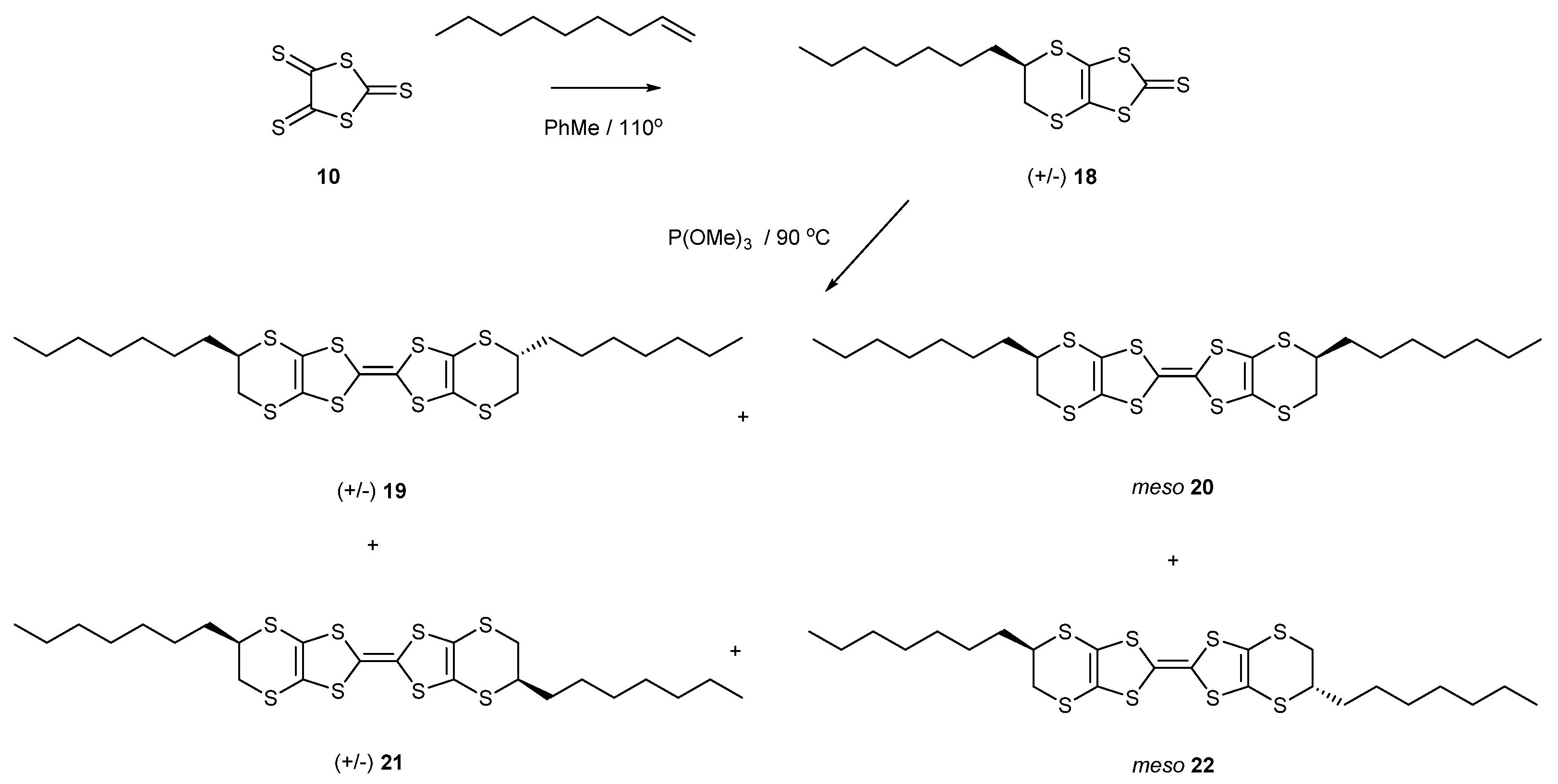
Following the same synthetic strategy, the trithione 10 was reacted with non-1-ene to provide the bicyclic thione 18 with one heptyl side chain in 57% yield (Scheme 4). The thione was directly homo-coupled using trimethyl phosphite to give a mixture of two disubstituted BEDT-TTF donors (51%), each one having two diastereomers. The locations of the side chains could be at the top edge of the molecule, or one can be at the bottom edge, leading to the two differently substituted donors, each one of which has a racemic pair and a meso stereoisomer, 19–22. Again, these were found to be inseparable. Both the 16–17 and 19–22 mixtures showed the expected two reversible oxidation peaks in their cyclic voltammograms (Table 1) at 0.47 and 0.89 V for 16–17 and 0.49 and 0.89 V for 19–22 (relative to Ag/AgCl).
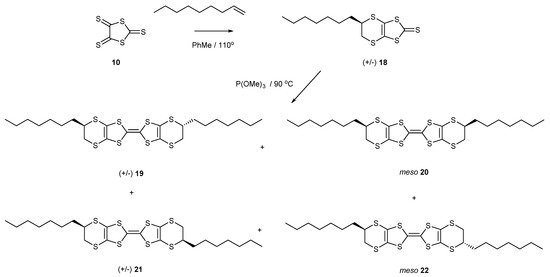
Scheme 4.
Synthetic routes to di-n-heptyl-BEDT-TTF derivatives 19–22.

Table 1.
Cyclic voltammetry data for selected BEDT-TTF derivatives a.
2.2. Tris-(BEDT-TTF) Donors
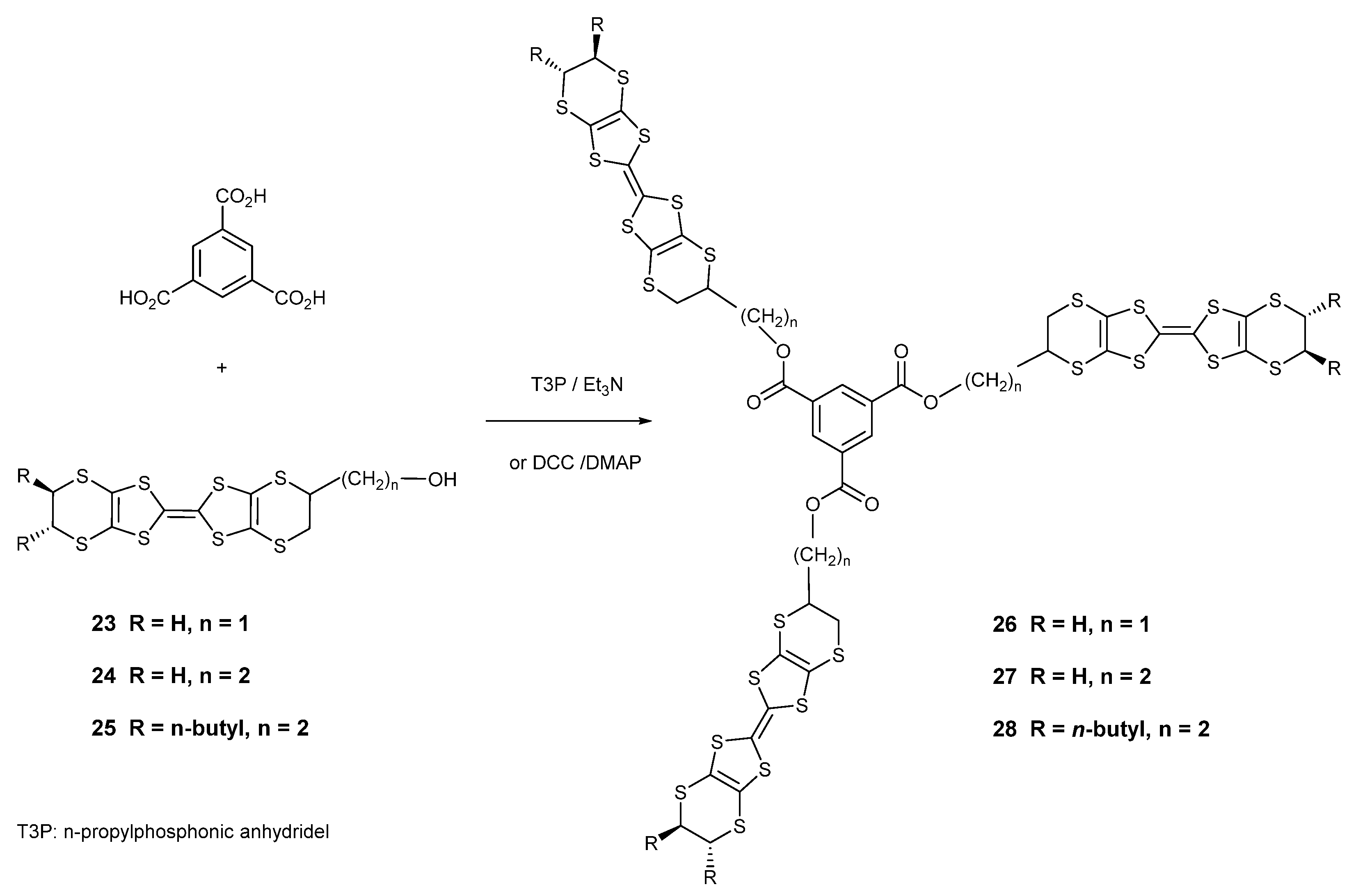
To provide systems which will show different packing arrangements versus the classical BEDT-TTF systems and which could also act as supramolecular synthons, e.g., for forming charge transfer complexes with fullerenes, three tris-donor systems 26–28 were prepared containing three donors appended to a benzene core. The synthetic route to these compounds involved the coupling of benzene-1,3,5-tricarboxylic acid with three equivalents of a BEDT-TTF donor bearing a hydroxyalkyl side chain using n-propyl phosphonic anhydride (“T3P”) or DCC (Scheme 5). Thus, donors 23 and 24 with a hydroxy-methyl or hydroxyethyl side chain afforded the tris-donors 26 and 27 in 22 and 33% yields, respectively. Interestingly, there are two racemic disastereomers for these two donors, one with the same configuration at all three stereogenic centres (R,R,R) and one with the same configuration at just two centres (R,R,S). However, the products proved to have low solubilities in common organic solvents, so the hydroxyethyl-BEDT-TTF donor 25, functionalised with two trans oriented n-butyl side chains on the other ethylene bridge, was prepared and was coupled with the triacid to give the more soluble tris-donor 28 in 45% yield. The number of stereoisomers is increased over 26–27 because the three sets of butyl groups can have all six or just four configurations be the same. However, these stereochemical issues are unlikely to have a profound influence on the behaviour of the donor, due to the flexibilities of the dithiin rings, as discussed earlier. The structures were supported by their chemical analyses and spectral data, e.g., 26 showed a molecular ion in the mass spectrum, and 27–28 showed the three aromatic H atoms at ca. 8.8 ppm. The more soluble tris derivative 28 showed a reversible cyclic voltammogram with peaks at 0.50 and 0.88 V as expected of a BEDT-TTF derivative, but tris donors 26 and 27 showed irreversible volt-ammograms probably due to the binding of the donor to the electrode, with two oxidation peaks between 0.44 and 0.56 V and another in the range 0.86–0.88 V on the outward scan.
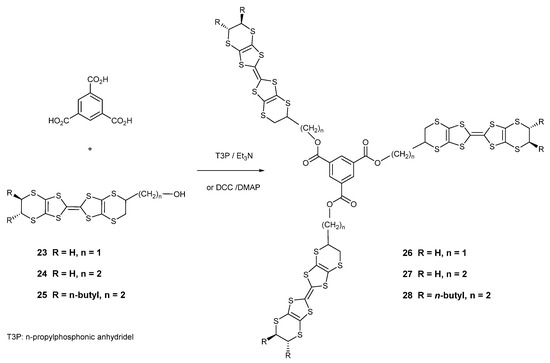
Scheme 5.
Synthetic route to tris-BEDT-TTF derivatives 26–28.
2.3. New Functionalised BEDT-TTFs: Ethynyl and Acetal Substituted Derivatives
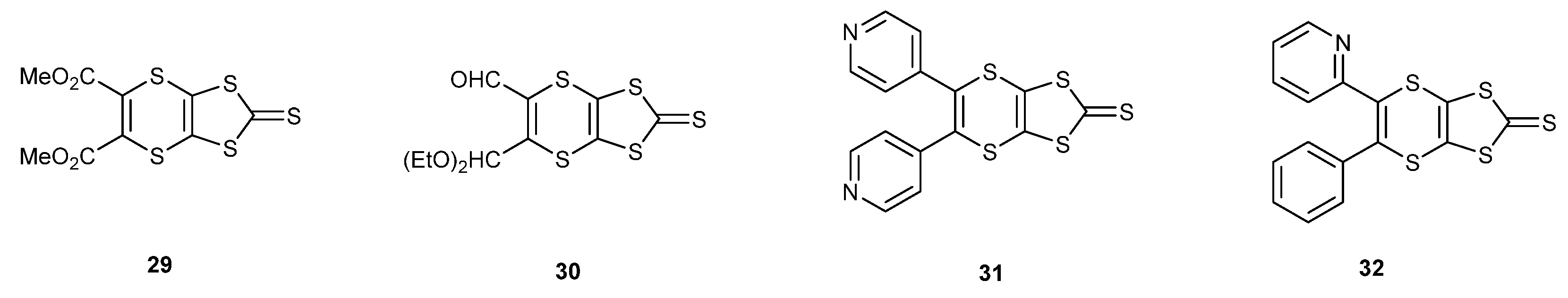
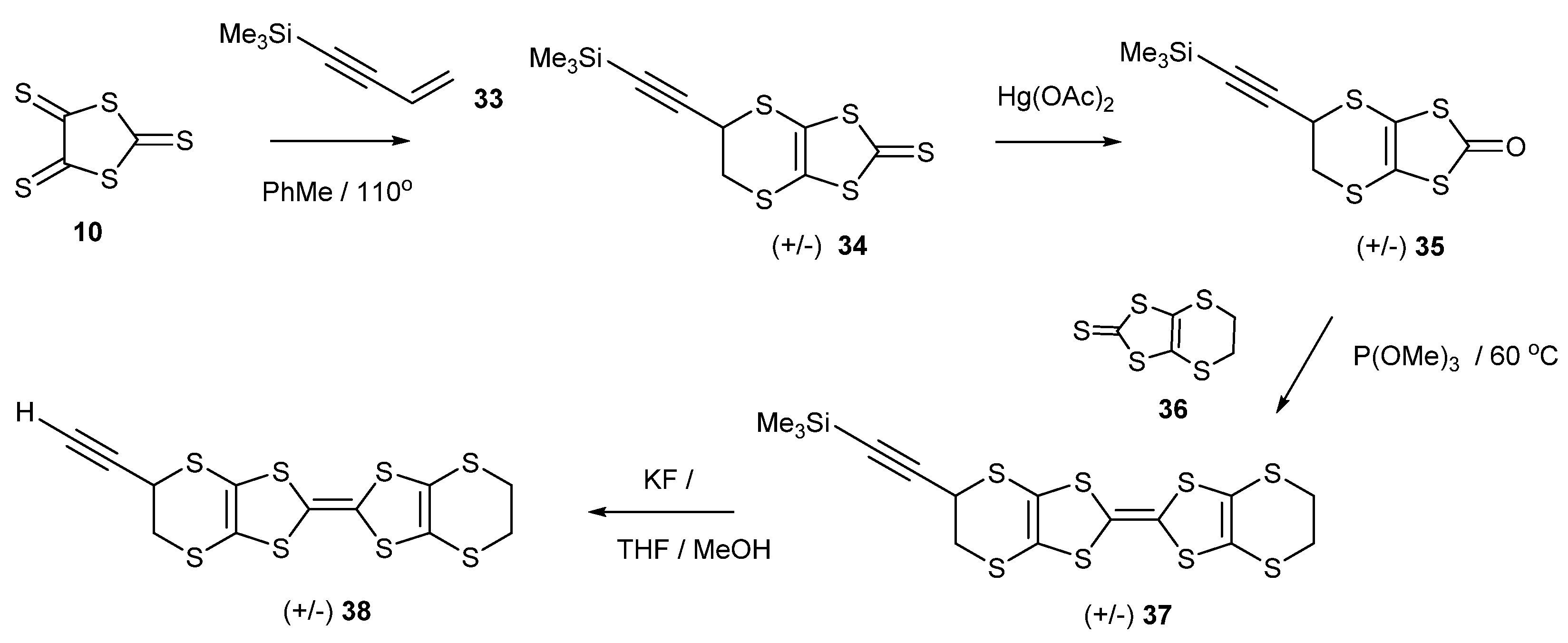
There are several examples of the cyclisation of the trithione 10 with alkynes (Scheme 6), in particular with those where the triple bond bears two carbonyl or acetal functionalities, affording thiones such as 29 and 30 [35,36,37]. The electron deficient diarylethynes also react with the trithione 10, giving access to thiones such as 31 and 32 [38,39]. An interesting case, therefore, would be the reaction with 4-trimethylsilyl-1-buten-3-yne 33, which has both an alkene and an alkyne group, to determine which group preferentially reacts with the trithione 10 (Scheme 7). Refluxing the two materials together gave a 55% yield of the bicyclic trithione 34 in which the alkene, and not the alkyne, had reacted. The reaction of the trimethylsilylethynyl thione with mercuric acetate gave the oxo compound 35 in an almost quantitative yield, without affecting the triple bond, and this material was cross-coupled with the unsubstituted thione 36 to give the trimethylsilylethynyl substituted BEDT-TTF donor 37 in 45% yield. Treatment with potassium fluoride in THF/methanol gave the deprotected ethynyl-BEDT-TTF donor 38 in 88% yield. This shows the greater reactivity of the alkene over the alkyne with the trithione. This has provided an interesting donor which has the potential for connecting to larger molecular systems, either preserving the triple bond by substitution of the ethynyl H atom, e.g., by a Sonogashira reaction, or using the triple bond as a substrate for click chemistry with azide-functionalised materials.
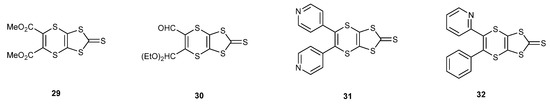
Scheme 6.
Structures of thiones prepared from trithione 10 and various disubstituted alkynes.
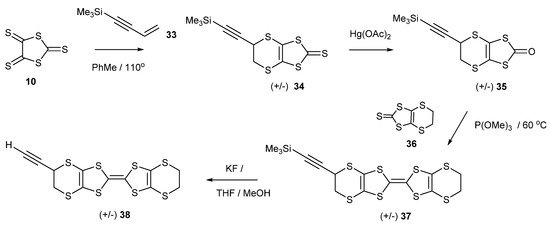
Scheme 7.
Synthetic route to ethynyl-BEDT-TTF 38.
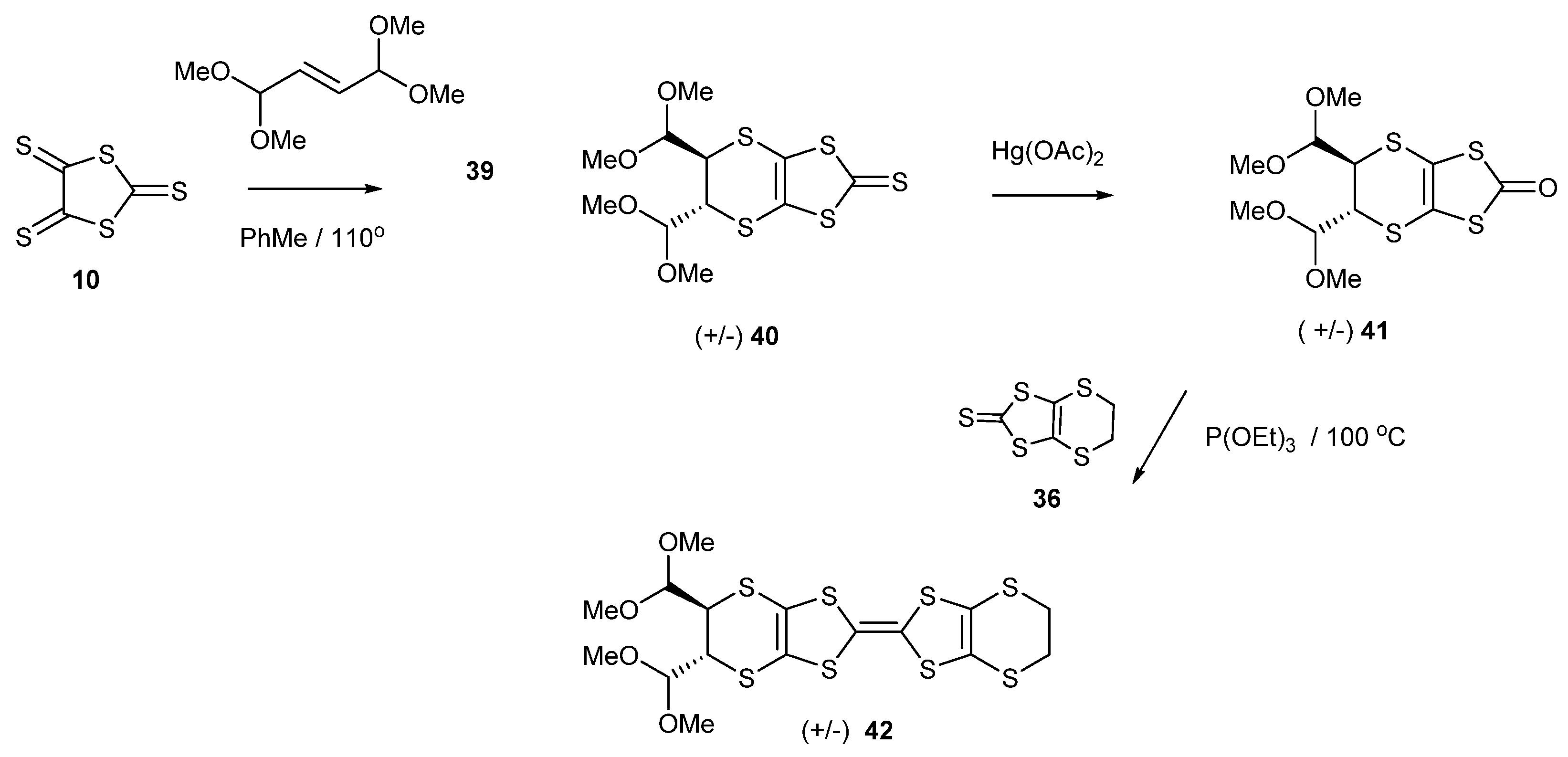
We were interested in making BEDT-TTF with two aldehyde groups attached, which could facilitate further ring constructions or the attachment of side chains. Thus, we targeted the corresponding bis(dimethylacetal) containing donor 42. The reaction of the corresponding alkene, the bis-dimethylacetal of fumaraldehyde 39, with trithione 10 gave the disubstituted thione 40 in a low yield of 17%, which was converted to the oxo compound 41 in almost quantitative yield (Scheme 8). The donor 42 was obtained by the cross-coupling of oxo compound 41 with the unsubstituted thione 36 in 49% yield. However, all attempts to hydrolyse the acetal groups with HCl or tosic acid in a range of concentrations failed to give the corresponding dialdehyde. Similar experiences have been reported on organo-sulphur donors containing acetal or ketal functionality [24,40]. It may be that these donor molecules stack together in clumps and isolate themselves from the acidic environment. Nevertheless, it is quite remarkable that these groups are resistant to hydrolysis, when dilute acid is usually quite sufficient. The cyclic voltammograms of BEDT-TTF derivatives 38 and 42 showed the expected two reversible oxidation peaks (Table 1).
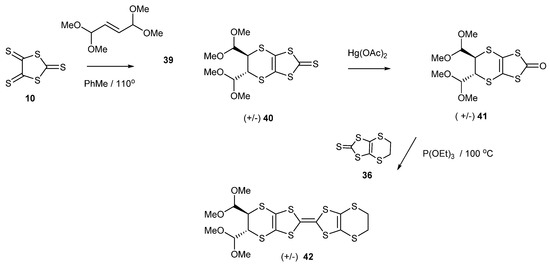
Scheme 8.
Synthetic route to the BEDT-TTF derivative functionalised with two trans-oriented, dimethylacetal groups 42.
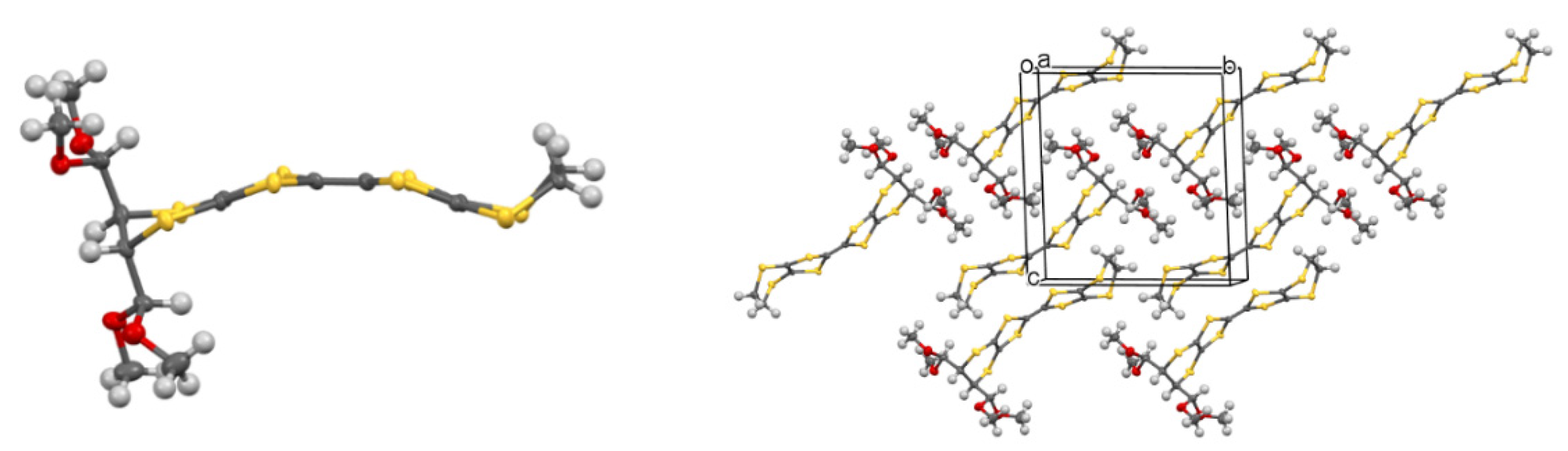
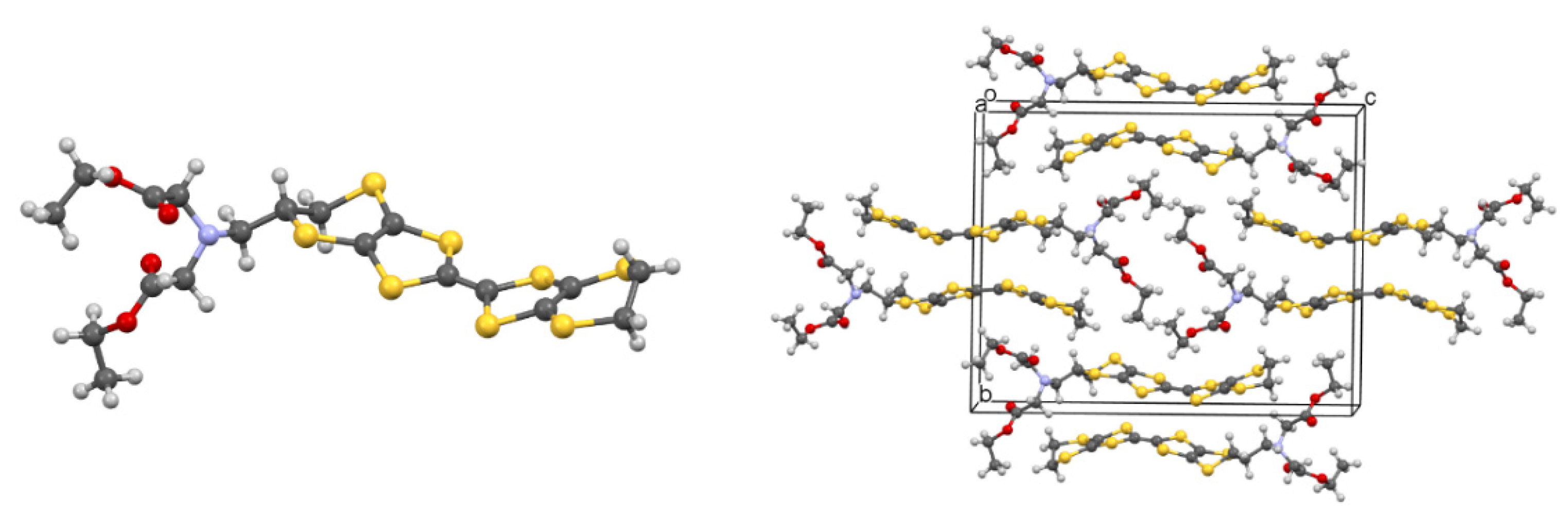
Single crystals suitable for X-ray diffraction of 42 were grown from CH3CN, and the molecular structure of the donor was determined (Figure 1). The substituted dithiin ring adopts a half-chair conformation and directs the acetal groups into pseudo-axial positions. The organosulphur moiety adopts a bowed shape with inflexions about the S···S vectors across the two dithiole rings of 19.6 and 17.2°. In the crystal structure, the donors pack in centrosymmetric slipped pairs to accommodate the side chains, with the shortest C···C contact of 3.546 Å between the TTF cores.
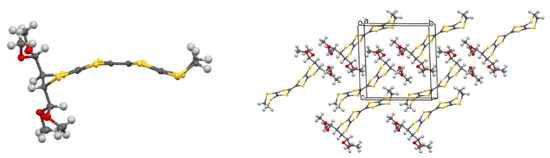
Figure 1.
Molecular structure of donor 42 (left) and its crystal packing arrangement (right).
2.4. Donors with Metal Ion Binding Potential and the First Salts and Their Magnetic Properties
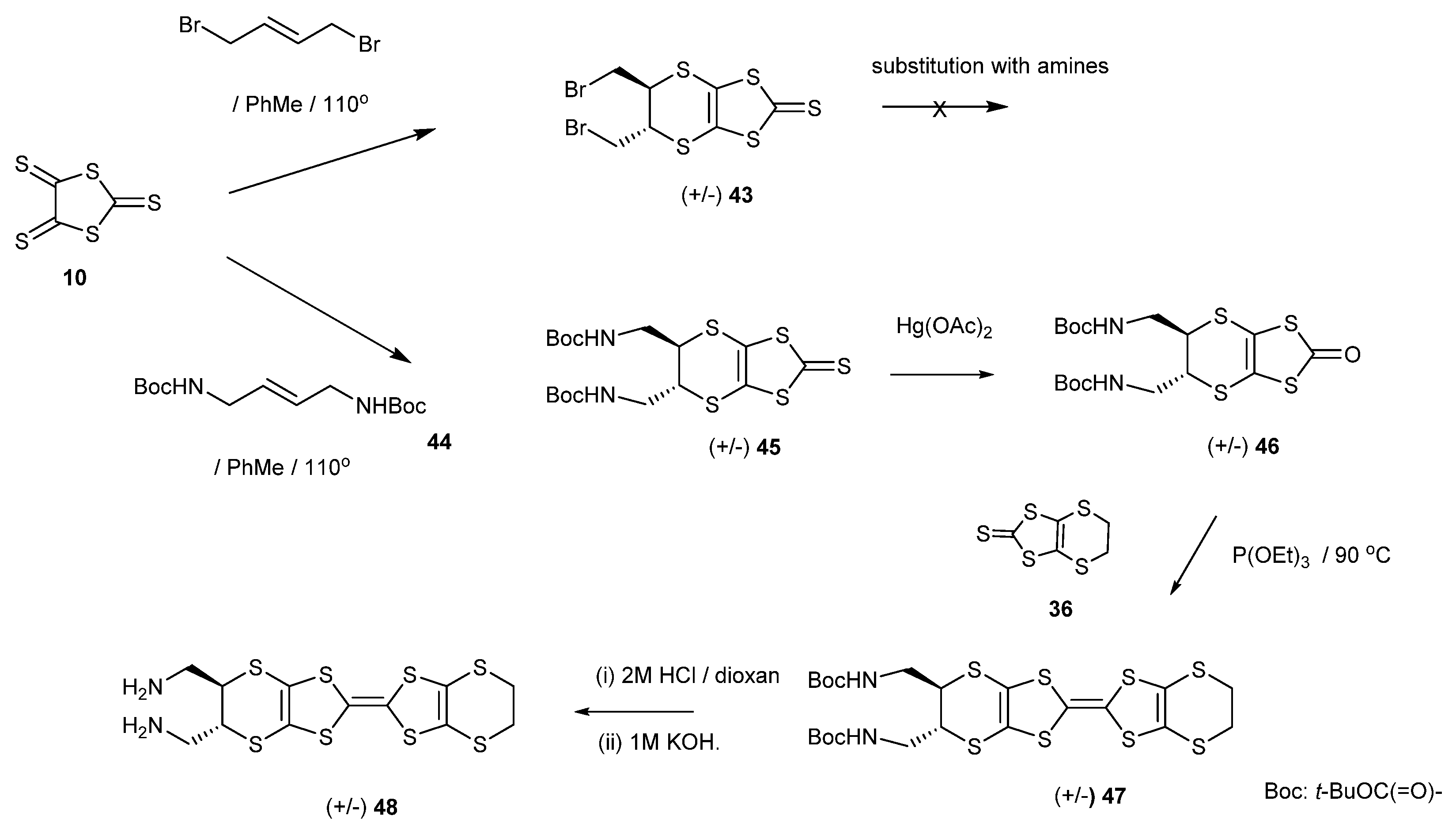
The BEDT-TTF donor 48 with trans aminomethyl groups was an interesting target, both for further synthetic elaborations and for the binding of metal ions. In a first approach, which we hoped would have a wide scope, thione 10 was reacted successfully with trans-1,4-dibromobut-2-ene to give the trans bis(bromomethyl)thione 43. However, attempts to substitute the bromide groups of this material with amines were unsuccessful, with the amine probably causing the elimination of HBr. Therefore, to synthesize the donor with two aminomethyl groups 48, the trithione 10 was reacted with the bis-N-boc derivative of trans-but-2-en-1,4-diamine 44 to give the thione 45 in 53% yield (Scheme 9). The corresponding reaction of the trithione with the readily obtained bis-phthalimido derivative of trans-but-2-en-1,4-diamine was unsuccessful. Following the established procedure, the thione 45 was converted to the oxo compound 46 which was cross-coupled with the unsubstituted thione 36 to give the bis-N-Boc donor 47 in 63% yield. Deprotection of this donor with HCl in dioxane followed by basification gave the bis(aminomethyl)BEDT-TTF 48 in 78% yield. The cyclic voltammogram of 48 in THF showed broad irreversible peaks, probably due to the donor binding to the electrode.
Scheme 9.
Synthetic route to trans-bis-(aminomethyl)-BEDT-TTF 48.
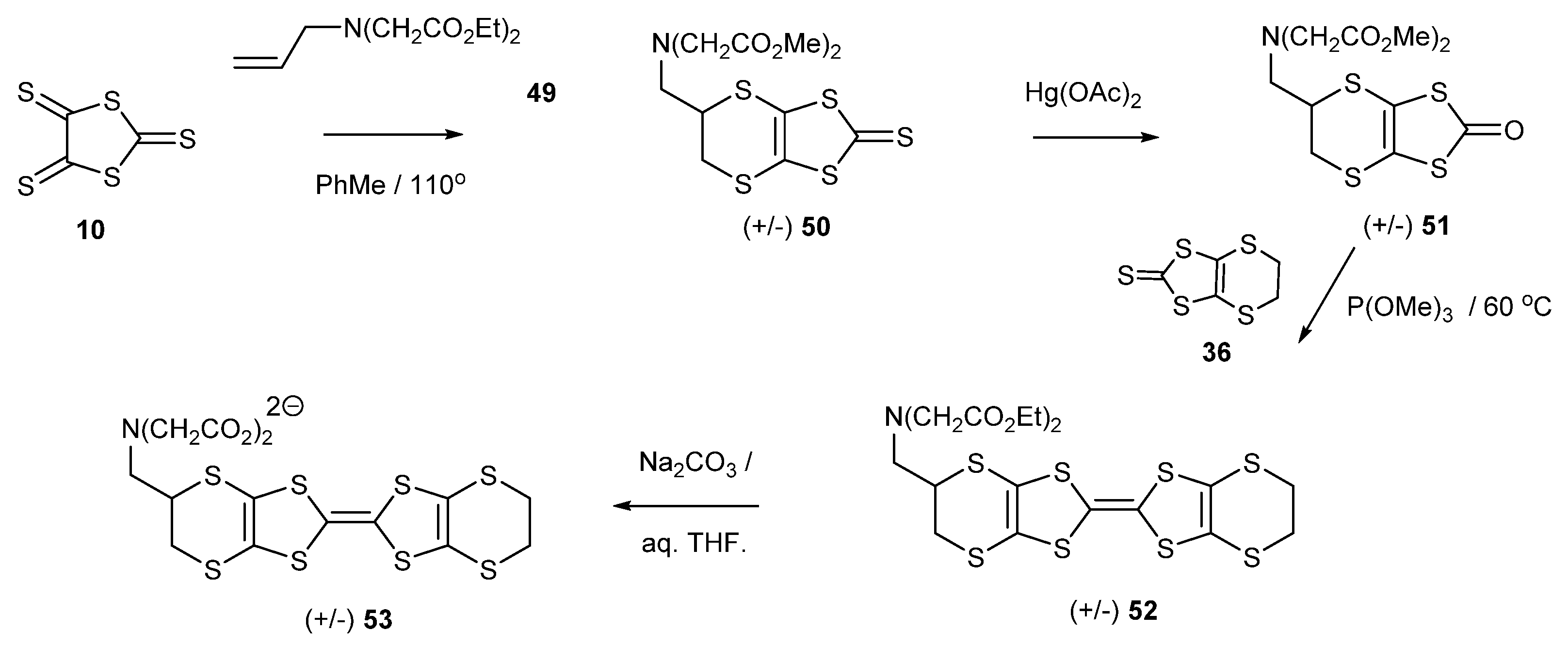
Installing an iminodicarboxylate dianion group on the side chain of BEDT-TTF could provide another BEDT-TTF donor with potential metal binding properties, so donor 53 with just a methylene between the donor and the metal binding group was prepared. This was synthesized following the synthetic strategy outlined in Scheme 10. The attachment of an allyl group to diethyl iminodiacetate gave alkene 49 which reacted with trithione 10 to give the substituted thione 50 in high yield. Conversion to the corresponding oxo compound 51 and subsequent cross-coupling with the unsubstituted thione 36 afforded the BEDT-TTF donor 52 with an imino diethyl ester group in the side chain in 34% yield.
Scheme 10.
Synthetic route to the disodium salt of BEDT-TTF-methylamino-N,N-dicarboxylate 53.
Single crystals of 52 were grown from DCM, and the crystal structure was determined by single crystal X-ray diffraction (Figure 2). The BEDT-TTF moiety adopts a bowed structure, with inflexions about the two S···S vectors in the dithiole rings of 14.6 and 23.3°. The unsubstituted ethylene bridge is disordered between two half-chair conformations (62:38). On the other bridge the carbon bearing the side chain and its attached hydrogen atom are disordered between two positions (82:18) which correspond to the structures of opposite enantiomers of the donor. This illustrates how the bridge can flex to accommodate both enantiomers without affecting the positions of either the main parts of the organosulphur system or the side chain. The donors are packed in centrosymmetric pairs with the shortest C···C distance between the TTF cores of 3.42 Å.
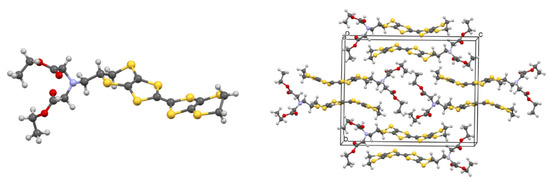
Figure 2.
Crystal structure of donor 52, showing the main molecular conformation (left) and the crystal packing arrangement (right).
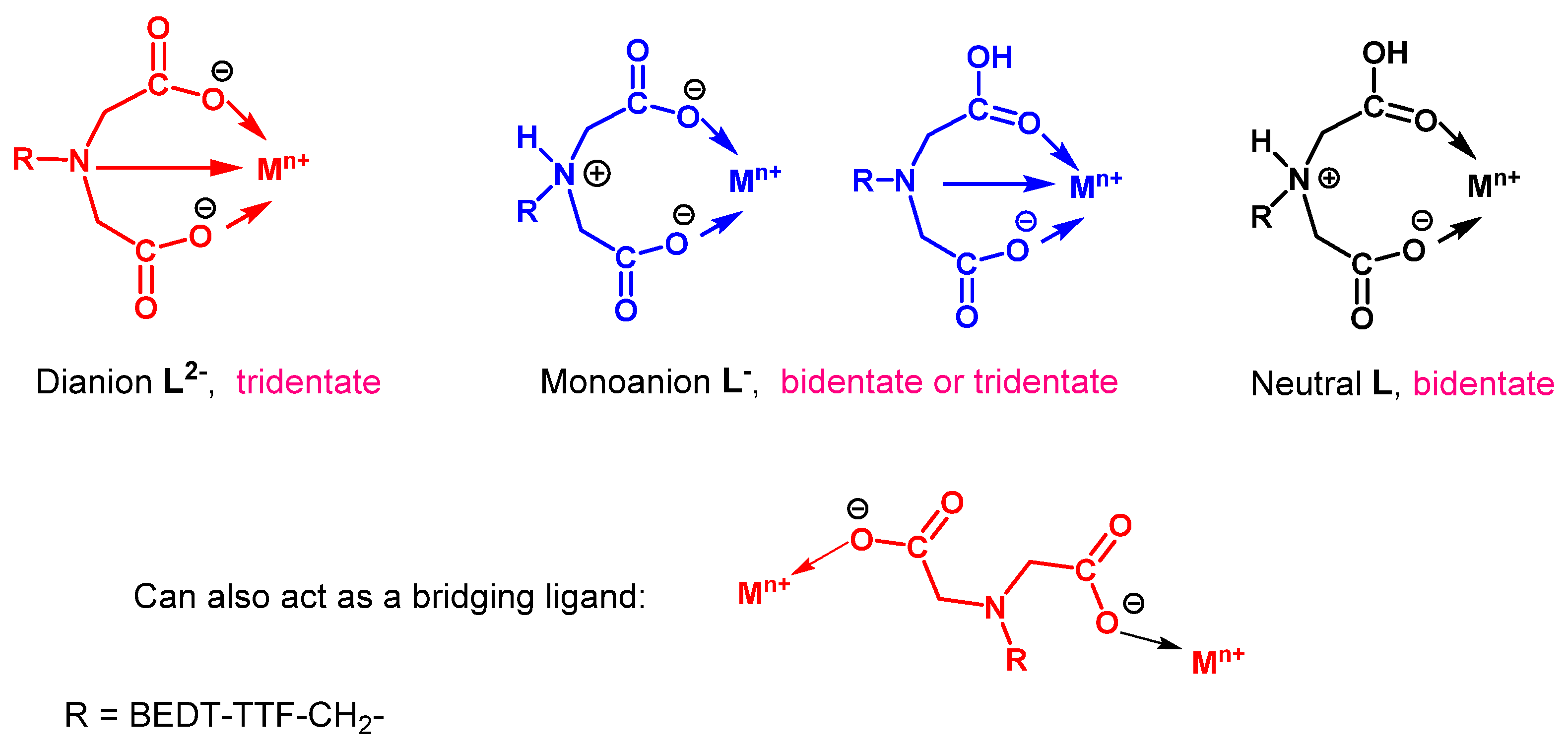
Hydrolysis of the imino diethyl ester substituted donor 52 with sodium hydroxide gave the dianion 53 as a disodium salt in high yield. The reaction of this donor with a range of transition metal salts afforded precipitates which were all insoluble in a wide range of solvents, and from which single crystals could not be obtained. Thus, assignments of compositions are tentative, based only on chemical analysis, magnetic measurements, and infrared spectra, and we acknowledge that the structural topologies may be more complex. Ligand 53 can show a number of coordination modes (Figure 3). It can act as tridentate dianion (L2−) binding by two oxygens and nitrogen; be protonated once (HL−), on nitrogen or oxygen, and act as a bidentate or tridentate monoanion, respectively; or be protonated twice and coordinate as a neutral ligand (H2L). Such behaviours are seen with simpler N-alkylated iminodiacetate ligands [41,42,43]. It can also act as just a bridging ligand [44]. Furthermore, binding to the outer set of the BEDT-TTF unit’s sulphur atoms cannot be excluded, as has been observed with copper(I) and silver(I) [45,46,47]. Details of three of the coordination complexes obtained are provided in Table 2. With zinc triflate, the CHN data are consistent with the product 54 containing two equivalents of the HL− monoanion coordinated to Zn(II). With MnCl2, the product 55 contains two neutral H2L ligands along with MnCl2 and two waters. For the MnCl2 complex, at room temperature, the value of the χT product (4.4 cm3 K mol−1) is in excellent agreement with the theoretical expected value for an isolated HS octahedral Mn(II) ion (4.375 cm3 K mol−1) with a g value of 2.00. Similarly, for the second Mn(II) complex 56 prepared from Mn(hfac)2, the room temperature χT product of 4.5 cm3 K mol−1 is again very reasonable for an isolated HS Mn(II) ion in octahedral geometry, where compared to complex 55, the hfac ions replace the chlorides, though with one less H2L ligand and one less water molecule. For both Mn(II) complexes, dc magnetic studies reveal that the χT product is temperature independent down to ca. 50 K, after which time it rapidly decreases, which is consistent with the presence of antiferromagnetic interactions.
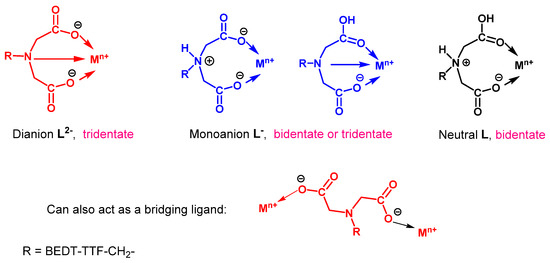
Figure 3.
Possible binding modes of donor 53 to a metal ion, depending on its degree of protonation.
Table 2.
Proposed compositions, magnetic data, and chemical analysis for the reaction of ligand 53 with Zn(II) and Mn(II) salts.
3. Conclusions
The synthesis of a range of new BEDT-TTF derivatives, which are substrates for the formation of charge transfer salts as well as for incorporation in more complex systems, has been described. The development of conducting and hybrid materials is highly dependent on the availability of new donor systems, and such syntheses are often not straightforward. For example, the trithione 10 does not react with all alkenes, and the presence of the BEDT-TTF unit can disrupt apparently standard synthetic manipulations to side chain functionalities. The possibility of forming hybrid materials between the donor bearing an iminodiacetate function 53 and metal ions has been demonstrated and will be extended in the future to the bis(diaminomethyl) donor 48, though methods for forming crystalline products, e.g., by hydrothermal synthesis, need to be developed. Furthermore, to produce conducting materials, an oxidation step needs to be included. Of particular note is that the flexibility of the ethylene bridges of BEDT-TTF donors results in both enantiomers of the substituted BEDT-TTF being close to superimposable, as shown in the crystal structure of 52. This indicates that to prepare systems with chiral packing arrangements, the stereogenic feature might be more effective in the side chain. However, we note that the first observation of magnetochiral anisotropy in organic conductors was in the perchlorate salts of enantiomers of dimethyl EDT-TTF, where the chirality was on the donor [48].
4. Materials and Methods
General. Solution NMR spectra were measured on either a Jeol ECLIPSE ECX or ECZ spectrometer operating at 400 MHz for 1H and at 100.6 MHz for 13C, using CDCl3 as solvent and tetramethylsilane (TMS) as standard unless otherwise stated, and measured in p.p.m. downfield from TMS with coupling constants reported in hertz. IR spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR Spectrometer using attenuated total reflection sampling on solids or oils and are reported in cm−1. Mass spectra were recorded at the EPSRC Mass Spectrometry Centre at the University of Swansea or on a Waters Xevo QTOF G2 XS using an ESI source with a Waters Acquity UPLC system at NTU. Chemical analysis data were obtained from London Metropolitan University and Nottingham University, UK. Flash chromatography was performed on 40–63 μm silica gel obtained from Fluorochem Ltd. (Hadfield, Glossop, Derbyshire, UK).
Magnetic Measurements: Variable-temperature dc magnetic measurements were performed on a Quantum Design SQUID MPMS magnetometer in an applied field of 0.1 T, from 2 to 300 K. The experimental data were corrected for the diamagnetism and signal of the sample holder.
Full details of the synthetic procedures and further details of the magnetic data are provided in the ESI. The syntheses of 49–56 are given below to illustrate the general methods.
Diethyl allylamino-N,N-diacetate, 49: To a stirred solution of diethyl amino-N,N-diacetate (3.04 g, 16.1 mmol) in THF (50 mL), allyl bromide (2.20 mL, 25.3 mmol) was slowly added followed by addition of K2CO3 (3.42 g, 24.7 mmol), and the resulting suspension was warmed up to reflux and left to stir for 20 h. The reaction mixture was cooled to room temperature, and THF was evaporated under reduced pressure. DCM was added to the residue (100 mL) and the organic layer was washed with water (100 mL) and brine (100 mL) and dried over Na2SO4. Evaporation of DCM afforded diethyl allyl-amino-N,N-diacetate 49 (3.16 g, 86%) as a pale yellow oil; δH (400 MHz, CDCl3): 5.88 (1H, m, -CH=CH2), 5.22 (1H, dd, J = 18.8, 1.7 Hz, -CH=CHtransH), 5.17 (1H, dd, J = 10.1, 1.8 Hz, -CH=CHHcis), 4.17 (4H, q, J = 7.2 Hz, -O-CH2-CH3), 3.57 (4H, s, 2 × –N(CH2COOEt)2), 3.38 (2H, d, J = 6.7 Hz, -CH2-CH=), 1.27 (6H, t, J = 7.0 Hz, 2 × -CH3); δC: (100 MHz, CDCl3): 170.6 (2 × -C=O), 134.9 (=CH), 118.0 (=CH2), 60.0 (2 × -O-CH2-CH3), 57.0 (=CH-CH2-N-), 53.8 (2 × -N-CH2CO), 13.8 (2 x-CH3); νmax: 2981, 1737, 1674, 1399, 1333, 1190, 714; HRMS: (ASAP) found: 230.1383 (100%), C11H20NO4 +H: requires: 230.1387; found C, 57.69; H, 8.36; N, 6.19%; C11H20NO4 requires C, 57.64; H, 8.29; N, 6.11%.
Diethyl (+/−)-5,6-dihydro-2-thioxo-1,3-dithiolo[4,5-b]1,4-dithiin-5-methylamine-N,N-diacetate, 50: Diethyl allyl-amino-N,N-diacetate 49 (1.17 g, 5.0 mmol) was added to a suspension of trithione 10 (3.25 g, 17.0 mmol) in toluene (100 mL), and the resulted suspension was warmed up to reflux and left to stir overnight under a nitrogen atmosphere. The solid formed during the reaction was filtered off and washed with CHCl3 until washes ran clear. The combined filtrates were evaporated under reduced pressure to give the desired thione 50 as a dark red oil (2.09 g, 96%). δH (400 MHz, CDCl3): 4.10 (4H, q, J = 7.0 Hz, -O-CH2-CH3), 3.72 (1H, m, 5-H), 3.51 (4H, s, 2 × (-N-CH2-C=O), 3.43 (1H, dd, J = 13.4, 6.1 Hz, 5-(CHαH)-N), 3.29 (1H, dd, J = 13.3, 2.8 Hz, 5-(CHHβ)-N)), 3.13 (1H, dd, J = 13.8, 8.7 Hz, 6-Hα), 3.05 (1H, dd, J = 13.8, 6.5 Hz, 6-Hβ), 1.20 (6H, t, J = 7.2 Hz, 2 × -CH3); δC: (100 MHz, CDCl3): 208.0 (C=S), 170.9 (2 × C=O), 123.6, 122.2 (3a-, 7a-C), 60.8 (2 x-O-CH2), 58.7 (5-CH2-N), 56.2 (2 x-N-CH2-C=O), 42.6 (5-C), 31.9 (6-C), 14.2 (2 x-CH3); νmax: 2975, 1729, 1483, 1368, 1259, 1180, 1140, 1054, 883, 799, 512; HRMS: (ASAP) found: 425.9995 (100%), C14H19NO4S5 + H: requires: 425.9990; found C, 39.55; H, 4.51; N, 3.35%, C14H19NO4S5 requires C, 39.53; H, 4.47; N, 3.29%.
Diethyl (+/−)-5,6-dihydro-2-oxo-1,3-dithiolo[4,5-b]1,4-dithiin-5-methylamine-N,N-diacetate,51: To a solution of thione 50 (1.88 g, 4.40 mmol) in CHCl3 (100 mL), mercury (II) acetate (3.54 g, 11.0 mmol) was added, and the suspension was left stirring for 3 h at room temperature under a nitrogen atmosphere. The solid formed during the reaction was filtered off and washed with CHCl3 until washes ran clear. The combined filtrates were washed with saturated sodium hydrogen carbonate solution (5 × 50 mL).The organic layer was washed with water (50 mL) and brine (50 mL) and dried over MgSO4. Evaporation of the chloroform yielded the desired oxo-compound 51 as a brown oil (1.71 g, 91%). δH (400 MHz, CDCl3): 4.17 (4H, q, J = 7.1 Hz, -O-CH2CH3), 3.82 (1H, m, 5-H), 3.59 (4H, s, 2 × -N-CH2-C=O), 3.49 (1H, dd, J = 13.3, 6.2 Hz, 5-(CHα)N-), 3.39 (1H, dd, J = 13.3, 3.1 Hz, 5-(CHβH)N-), 3.23 (1H, dd, J = 13.8, 8.7 Hz, 6-Hα), 3.13 (1H, dd, J = 13.8, 6.4 Hz, 6-Hβ), 1.28 (6H, t, 2 × -CH3); δC (100 MHz, CDCl3): 188.8 (C=O), 171.0 (2 × -C=O(OEt)), 113.7, 111.8 (3a-, 7a-C), 60.8 (2 × -O-CH2-CH3), 58.8 (-CH2-N-), 56.2 (2 × -N-CH2C=O), 44.1 (5-C), 32.9 (6-C), 14.2 (2 × -CH3); νmax: 2978, 1731, 1670, 1443, 1411, 1368, 1186, 1139, 1024, 762, 463; HRMS: (ASAP) found: 410.0208, C14H19NO5S4+H: requires: 410.0219; found C, 40.94; H, 4.72; N, 3.50%, C14H19NO5S4 requires C, 41.07; H, 4.64; N, 3.42%.
Diethyl (+/−)-BEDT-TTF-methylamino-N,N-diacetate, 52: Oxo compound 51 (2.01 g, 5.0 mmol) and 5,6-dihydro-dithiolo[4,5-b]dithiin-2-thione 36 (1.79 g, 8.0 mmol) were heated together in freshly distilled triethyl phosphite (50 mL) at 90 °C for 6.5 h under a nitrogen atmosphere. The solid formed was filtered and washed with CHCl3, and the combined filtrates were concentrated under reduced pressure. The triethyl phosphite was distilled off using a Kugelrohr apparatus. The residue was purified by flash chromatography (6:1 = cyclohexane: ethyl acetate) to give the substituted BEDT-TTF 52 as an orange solid (0.99 g, 34%), m.p. 107–108 °C; δH (400 MHz, CDCl3): 4.09 (4H, q, J = 7.1 Hz, 2 × -O-CH2-CH3), 3.64 (1H, m, 5-H), 3.50 & 3.51 (2 × 2H, 2 × s, 2 × (-N-CH2-C(O)), 3.33 (1H, dd, J = 13.0, 6.0 Hz, 5-(CHαH)-N, 3.21 (4H, s, (5′-, 6′-CH2)), 3.20 (1H, dd, J = 13.0, 3.1 Hz, 5-(CHβH)-N), 3.03 (2H, m, 6-H2), 1.20 (6H, t, J = 7.2 Hz, 2 × -CH3); δC (100 MHz, CDCl3): 171.0 (2 × C=O), 113.8, 113.7, 112.5, 111.5, 109.8 (2-, 2′-, 3a-, 7a-, 3′a-, 7′a-C), 60.6 (2 x-O-CH2-CH3), 58.7 (5-CH2-N-), 56.1 (2 x-N-CH2C=O), 42.6 (5-C), 32.5 (6-C), 30.1 (5′-,6′-C), 14.1 (2 x-CH3); νmax: 2921, 1733, 1449, 1412, 1372, 1190, 1022, 770; found C, 39.16; H, 4.00; N, 2.29%; C19H23NO4S8 requires C, 38.97; H, 3.93; N, 2.39%.
Disodium (+/−)-BEDT-TTF-methylamino-N,N-dicarboxylate 53as a tetrahydrate, Na2L.4H2O: The di-ester donor 52 (0.37 g, 6.40 mmol) was dissolved in THF (10 mL), and an aqueous solution of NaOH (5 mL, 0.256 M, 12.8 mmol) was added. The suspension was warmed to 50 °C and left stirring overnight. The THF was evaporated, and the solid filtered and washed successively with DCM (3 × 5 mL), water (3 × 5 mL), and ether (3 × 10 mL). Filtration gave 53 (0.41 g, quantitative) as a highly insoluble red solid, m.p. 240 °C (dec). νmax: 2917, 1582, 1400, 1326, 1122, 995, 904, 771, 669; found C, 28.16; H, 3.03; N, 2.38%, C15H13NO4S8Na2.4H2O required C, 27.90; H, 3.28; N, 2.17%.
Preparation of metal salts of53: (a) with zinc(II) triflate to give54: To a suspension of sodium salt 53 (77 mg, 0.12 mmol) in dry MeOH (8 mL) at room temperature under a nitrogen atmosphere, zinc triflate (25 mg, 0.069 mmol) was added, and immediately after the addition, a brown suspension was formed. The reaction mixture was left to stir for 1 h, and then the solid was filtered off, washed with diethyl ether, and left to dry in air to give Zn(HL)2, 54, (45 mg, 58%); m.p. 243–244 °C (dec.), νmax: 2950, 2854, 1596 (C=O), 1422, 1398, 1342, 1143, 996, 893, 766, 748; found C, 31.94; H, 2.41; N, 2.64%; C30H28N2O8S16Zn requires C, 32.09; H, 2.51; N, 2.49%.
(b) with manganese(II) chloride to give55: To a suspension of sodium salt 53 (0.212 g, 0.33 mmol) in distilled water (5 mL) at room temperature, MnCl2.4H2O (0.035 g, 0.18 mmol) was added, and immediately after the addition, a red precipitate was formed. The obtained solid was filtered and washed with diethyl ether and left to dry in air to give Mn(H2L)2Cl2.2H2O, 55, (147 mg, 68%), m.p. 206–207 °C (dec). νmax: 3321, 2914, 1595 (C=O), 1399, 1364, 1194, 1143, 996, 892; found C, 29.28; H, 2.57; N, 2.58%; C30H30O8S16N2Cl2Mn.2H2O required C, 29.50; H, 2.81; H, 2.29%.
(c) with manganese(II) (hfac)2 to give56: To a suspension of sodium salt 53 (26.0 mg, 0.052 mmol) in dry MeOH (6 mL) at room temperature under a nitrogen atmosphere, Mn(hfac)2.3H2O (24 mg, 0.046 mmol) was added. Immediately after the addition, a brown solid precipitated out from the mixture. The reaction was left to stir for 1 h, and then the solid was filtered. The brown precipitate was collected, washed with diethyl ether, and left to dry in air to give Mn(H2L)(hfac)2.H2O, 56, (22 mg, 48%), m.p. 137–140 °C; νmax: 3296, 1601, 1401, 1319, 1054, 994, 902, 771; found C, 29.61; H, 1.76; N, 1.81%; C25H17NO8F12S8Mn.H2O requires C, 29.53; H, 1.88; N, 1.38%.
X-ray Crystallography. X-ray diffraction data (MoKα) were measured at low temperature for 42 (120 K) and 52 (150 K). Structures were solved and refined using the SHELXS and SHELXL suite of programs [49,50] using the XSEED interface [51]. Molecular illustrations and geometric analysis were made with Mercury [52]. Data are deposited at the Cambridge Crystallographic Data Centre with code numbers CCDC 2083578–2083579.
Crystal data for 42: C16H20O4S8, Mr = 532.80, triclinic, a = 6.3985(4), b = 12.6825(11), c = 13.4735(10) Å, α = 89.198(4), β = 81.424(5), γ = 87.025(5)°, V = 1079.66(14) Å3, Z = 2, P-1, Dc = 1.64 g cm−3, μ = 0.849 mm−1, T = 120(2) K, 4995 unique reflections (Rint = 0.078), 3422 with F2 > 2σ, R(F, F2 > 2σ) = 0.059, Rw (F2, all data) = 0.13. Crystal from acetonitrile.
Crystal data for 52: C19H23NO4S8, Mr = 585.8, monoclinic, a = 6.5481(4), b = 17.6090(16), c = 22.2441(14) Å, β = 97.298(5)°, V = 2544.1(3) Å3, Z = 4, P21/c, Dc = 1.53 g cm−3, μ = 0.729 mm−1, T = 150(2) K, 5831 unique reflections (Rint = 0.090), 4196 with F2 > 2σ, R(F, F2 > 2σ) = 0.085, Rw (F2, all data) = 0.23. Crystal from dichloromethane.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/magnetochemistry7080110/s1, S1: full experimental details for the syntheses not described in Section 4; S2: magnetic data for 55 and 56; S3: references for S1–S2.
Author Contributions
Conceptualization, J.D.W.; synthetic methodology, S.Y., M.Z., A.C.B., S.J.K., D.M.D., and A.M.M.; magnetic measurements, E.L.S. and M.P.; crystallography J.D.W. and A.C.B.; manuscript preparation, J.D.W. and M.P. All authors have read and agreed to the published version of the manuscript.
Funding
NSERC (DG 2018-04255) (MP).
Institutional Review Board Statement
Not relevant.
Informed Consent Statement
Not applicable.
Data Availability Statement
Crystallographic information files for 42 and 52 are available at The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK, with numbers: CCDC 2083578-2083579.
Acknowledgments
J.D.W. thanks Nottingham Trent University for PhD scholarships (M.Z., A.C.B.) and for financial support. J.D.W. also thanks the EPSRC Mass Spectrometry Service, Cardiff University, Wales, UK, for data. M.P. acknowledges financial support from NSERC (DG-2018-04255).
Conflicts of Interest
The authors declare no conflict of interest.
Dedication
This work was stimulated by our interaction with Peter Day FRS whom we first met when he was the Director of the Royal Institution in London. Most of the authors have benefited from contact with Professor Day, and some have worked closely with him. We very much appreciated his great interest and strong encouragement to synthesize new BEDT-TTF derivatives for study and his continuing interest over the years after his “retirement”.
References
- Singleton, J.; Mielke, C. Quasi-Two-Dimensional Organic Superconductors. Contemp. Phys. 2002, 43, 63–96. [Google Scholar] [CrossRef] [Green Version]
- Singleton, J.; Mielke, C. Superconductors Go Organic. Phys. World 2002, 15, 35–39. [Google Scholar] [CrossRef]
- Day, P. BEDT-TTF Charge Transfer Salts: New Structures, New Functionalities. Compt. Rend. Chim. 2003, 6, 301–308. [Google Scholar] [CrossRef]
- Mori, H. Introduction to Organic Superconducting Materials. Opt. Sci. Eng. 2008, 133, 263–285. [Google Scholar]
- Mori, T. Structural Genealogy of BEDT-TTF-Based Organic Conductors. I. Parallel Molecules: β and β” Phases. Bull. Chem. Soc. Jpn. 1998, 71, 2509–2526. [Google Scholar] [CrossRef]
- Mori, T.; Mori, H.; Tanaka, S. Structural Genealogy of BEDT-TTF-Based Organic Conductors. II. Inclined Molecules: θ, α and κ Phases. Bull. Chem. Soc. Jpn. 1999, 72, 179–197. [Google Scholar] [CrossRef]
- Mori, T. Structural Genealogy of BEDT-TTF-Based Organic Conductors. III. Twisted Molecules: δ and α’ Phases. Bull. Chem. Soc. Jpn. 1999, 72, 2011–2027. [Google Scholar] [CrossRef]
- Williams, J.M.; Wang, H.H.; Kini, A.M.; Carlson, K.D.; Beno, M.A.; Geiser, U.; Whangbo, M.-H.; Jung., D.; Evain, M.; Novoa, J.J. Recent Progress in the Development of Structure-Property Correlations for κ-Phase Organic Superconductors. Mol. Crys. Liq. Crys. 1999, 181, 59–64. [Google Scholar] [CrossRef]
- Ishiguro, T.; Yamaji, K.; Saito, G. Organic Superconductors; Springer: Berlin, Germany, 1998. [Google Scholar]
- Mori, H. Materials Viewpoint of Organic Superconductors. J. Phys. Soc. Jpn. 2006, 75, 051003-15. [Google Scholar] [CrossRef]
- Kurmoo, M.; Graham, A.W.; Day, P.; Coles, S.J.; Hursthouse, M.B.; Caulfield, J.L.; Singleton, J.; Pratt, F.L.; Hayes, W.; Ducasse, L.; et al. Superconducting and Semiconducting Magnetic Charge Transfer Salts: (BEDT-TTF)4AFe(C2O4)3·C6H5CN (A = H2O, K, NH4). J. Am. Chem. Soc. 1995, 117, 12209–12217. [Google Scholar] [CrossRef]
- Coronado, E.; Day, P. Magnetic molecular conductors. Chem. Rev. 2004, 104, 5419–5448. [Google Scholar] [CrossRef]
- Martin, L. Molecular Conductors of BEDT-TTF with Tris(oxalato)metallate Anions. Coord. Chem. Rev. 2018, 376, 277–291. [Google Scholar] [CrossRef] [Green Version]
- Sahadevan, S.A.; Abherve, A.; Monni, N.; Auban-Senzier, P.; Cano, J.; Lloret, F.; Julve, M.; Cui, H.; Kato, R.; Canadell, E.; et al. Magnetic Molecular Conductors Based on Bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF) with the Tris(chlorocyananilato)ferrate(III) Complex. Inorg. Chem. 2019, 58, 15359–15370. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.-P.; Wallis, J.D. Substituted BEDT-TTF Derivatives: Synthesis, Chirality, Properties and Potential Applications. J. Mater. Chem. 2005, 15, 347–365. [Google Scholar]
- Wallis, J.D.; Karrer, A.; Dunitz, J.D. Chiral Metals? A Chiral Substrate for Organic Conductors and Superconductors. Helv. Chim. Acta 1986, 69, 69–70. [Google Scholar] [CrossRef]
- Coronado, E.; Galán-Mascarós, J.R.; Coldea, A.I.; Goddard, P.; Singleton, J.; Wallis, J.D.; Coles, S.J.; Alberola, A. A Chiral Ferromagnetic Molecular Metal. J. Am. Chem. Soc. 2010, 132, 9271–9273. [Google Scholar]
- Pop, F.; Laroussi, S.; Cauchy, T.; Gomez-Garcia, C.J.; Wallis, J.D.; Avarvari, N. Tetramethyl-Bis(ethylenedithio)-Tetrathiafulvalene (TM-BEDT-TTF) Revisited: Crystal Structures, Chiroptical Properties, Theoretical Calculations and a Complete Series of Conducting Radical Cattion Salts. Chirality 2013, 25, 466–474. [Google Scholar] [CrossRef]
- Pop, F.; Meziere, C.; Allain, M.; Auban-Senzier, P.; Tajima, N.; Hirobe, D.; Yamamoto, H.; Canadell, E.; Avarvari, N. Unusual Stoichiometry, Band Structure and Band Filling in Conducting Enantiopure Radical Cation Salts of TM-BEDT-TTF Showing Helical Packing of the Donors. J. Mater. Chem. C. 2021. online. [Google Scholar] [CrossRef]
- Short, J.; Blundell, T.J.; Krivickas, S.J.; Yang, S.; Wallis, J.D.; Akutsu, H.; Nakazawa, Y.; Martin, L. Chiral Molecular Conductor with an Insulator–Metal Transition Close to Room Temperature. Chem. Commun. 2020, 56, 9497–9500. [Google Scholar] [CrossRef]
- Wang, Q.; Martin, L.; Blake, A.J.; Day, P.; Akutsu, H.; Wallis, J.D. Coordination Chemistry of 2,2′-Bipyridyl- and 2,2′: 6′,2″-Terpyridyl-substituted BEDT-TTFs: Formation of a Supramolecular Capsule Motif by the Iron (II) Tris Complex of 2,2′-Bipyridine-4-thiomethyl-BEDT-TTF. Inorg. Chem. 2016, 55, 8543–8551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, J.-P.; Nie, H.; Brown, R.J.; Day, P.; Wallis, J.D. Synthetic Strategies to Chiral Organosulfur Donors related to Bis(ethylenedithio)tetrathiafulvalene. Org. Biomolec. Chem. 2005, 3, 2155–2166. [Google Scholar] [CrossRef]
- Griffiths, J.; Arnal, A.A.; Appleby, G.; Wallis, J.D. Synthesis and Reactivity of Amino-substituted BEDT-TTF Donors as Building Blocks for Bifunctional Materials. Tetrahedron Lett. 2004, 45, 2813–2816. [Google Scholar] [CrossRef]
- Saygili, N.; Brown, R.J.; Day, P.; Hoelzl, R.; Kathirgamanathan, P.; Mageean, E.R.; Ozturk, T.; Pilkington, M.; Qayyum, M.M.B.; Turner, S.S.; et al. Functionalised Organosulfur Donor Molecules: Synthesis of Racemic Hydroxymethyl-, Alkoxymethyl- and Dialkoxymethyl-bis(ethylenedithio)tetrathiafulvalenes. Tetrahedron 2001, 57, 5015–5026. [Google Scholar] [CrossRef]
- Wang, Q.; Zecchini, M.; Wallis, J.D.; Wu, Y.; Rawson, J.M.; Pilkington, M. A Family of Unsymmetrical Hydroxyl-substituted BEDT-TTF Donors: Syntheses, Structures and Preliminary Thin Film Studies. RSC Adv. 2015, 5, 40205–40218. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.J.; Brooks, A.C.; Griffiths, J.-P.; Vital, B.; Day, P.; Wallis, J.D. Synthesis of Bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF) Derivatives with Two, Four or Eight Hydroxyl Groups. Org. Biomolec. Chem. 2007, 5, 3172–3182. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, D.; Zhang, B.; Yao, Y.; Xu, W.; Zhu, D.; Wang, Z. Synthesis of New Electron Donors with Hydroxymethyl Groups and Studies on Their Cation-Radical Salts. J. Mater. Chem. 2000, 10, 2063–2067. [Google Scholar] [CrossRef]
- Li, H.; Zhang, D.; Xu, W.; Fan, L.; Zhu, D. New Electron Donor: Bis(ethylenedithio)tetrathiafulvalene Derivative with Four Hydroxyl Groups. Syn. Met. 1999, 106, 111–114. [Google Scholar] [CrossRef]
- Berridge, R.; Serebryakov, I.M.; Skabara, P.J.; Orti, E.; Viruela, R.; Pou-Amerigo, R.; Coles, S.J.; Hursthouse, M.B. A New Series of π-Extended Tetrathiafulvalene Derivatives Incorporating Fused Furanodithiino and Thienodithiino Units: A Joint Experimental and Theoretical Study. J. Mater. Chem. 2004, 14, 2822–2830. [Google Scholar] [CrossRef]
- Aoyagi, I.; Katsuhara, M.; Mori, T. Synthesis and Structure of Highly Soluble Bis(ethylenedithio)tetrathiafulvalene Molecules with Alkyl Chains. Sci. Tech. Adv. Mater. 2004, 5, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Kini, A.M.; Parakka, J.P.; Geiser, U.; Wang, H.-H.; Rivas, F.; DiNiro, E.; Thomas, S.; Dudek, J.D.; Williams, J.M. Tetra-alkyl and Di-alkyl Substituted BEDT-TTF Derivatives and Their Cation-Radical Salts: Synthesis, Structure and Properties. J. Mater. Chem. 1999, 9, 883–892. [Google Scholar] [CrossRef]
- Troitsky, V.I.; Berzina, T.S.; Katsen, Y.Y.; Neilands, O.Y.; Nicolini, C. Conducting Langmuir-Blodgett Films of Heptadecylcarboxymethyl-BEDT-TTF. Syn. Metals 1995, 74, 1–6. [Google Scholar] [CrossRef]
- Goldenberg, L.M.; Khodorkovsky, V.Y.; Vladimir, Y.; Becker, J.Y.; Lukes, P.J.; Bryce, M.R.; Petty, M.C.; Yarwood, J. Highly Conducting Langmuir-Blodgett Films of an Amphiphilic Bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF) Derivative: BEDT-TTF-C18H37. Chem. Mater. 1994, 6, 1426–1431. [Google Scholar] [CrossRef]
- Khodorkovskii, B.Y.; Pukitis, G.; Puplovskii, A.Y.; Edzina, A.; Neilands, O.Y. Synthesis and Properties of the Hexadecyl Derivative of Bis(ethylenedithio)tetrathiafulvalene. Khim. Geterotsikl. Soedin. 1990, 131–132. [Google Scholar]
- Yang, X.; Rauchfuss, T.B.; Wilson, S. The Chemistry of C6S10: A Channel Structure for C6S10.(CS2)0.5 and Access to the Versatile DMAD-C3S4O. Chem. Commun. 1990, 34–36. [Google Scholar] [CrossRef]
- Leriche, P.; Gorgues, A.; Jubault, M.; Becher, J.; Orduna, J.; Garin, J. Cycloaddition of Acetylenedicarbaldehyde Monoacetal and 2,4,5-Trithioxo-1,3-dithiole: Ready Access to Novel Highly Extended and Sulfur-rich Analogues of Tetrathiafulvalene (TTF). Tetrahedron Lett. 1995, 36, 1275–1278. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Miyamoto, T.; Yoshida, A.; Kawada, Y.; Nakazaki, J.; Izuoka, A.; Sugawara, T. New Synthesis of 2-(1,3-Dithiol-2-ylidene)-5,6-dihydro-1,3-dithiolo[4,5-b][1,4]dithiiins with Formyl Group on Fused Benzene, [1,4]dithiin or Thiophene Ring. Tetrahedron Lett. 1999, 40, 8819–8822. [Google Scholar] [CrossRef]
- Brooks, A.C.; Day, P.; Dias, S.I.G.; Rabaca, S.; Santos, I.C.; Henriques, R.T.; Wallis, J.D.; Almeida, M. Pyridine-functionalised (Vinylenedithio)tetrathiafulvalene (VDT-TTF) Derivatives and Their Dithiolene Analogues. Eur. J. Inorg. Chem. 2009, 3084–3093. [Google Scholar] [CrossRef]
- Niu, Z.-G.; He, L.-R.; Li, L.; Cheng, W.-F.; Li, X.-Y.; Chen, H.-H.; Li, G.-N. Synthesis, Characterisation and DFT Studies of Two New π-Conjugated Pyridine-based Tetrathiafulvalene Derivatives. Acta Chim. Sloven. 2014, 61, 786–791. [Google Scholar]
- Marshallsay, G.J.; Bryce, M.R.; Cooke, G.; Joergensen, T.; Becher, J.; Reynolds, C.D.; Wood, S. Functionalised Tetrathiafulvalene (TTF) Systems Derived from 4, 5-(Propylenedithio)-1, 3-dithiole Units. Tetrahedron 1993, 49, 6849–6862. [Google Scholar] [CrossRef]
- Arenzano, J.A.; Virues, J.O.; Colorado-Peralta, R.; Ramirez-Montes, P.I.; Santillan, R.; Sanchez, M.; Rivera, J.M. Heterometallic Coordination Framework by Sodium Carboxylate Subunits and Cobalt(III) Centres Obtained from a Highly Hydrogen Bonding Stabilized Cobalt(II) Monomeric Complex. Inorg. Chem. Commun. 2015, 51, 55–60. [Google Scholar] [CrossRef]
- Puentes, R.; Torres, J.; Kremer, C.; Cano, J.; Lloret, F.; Capucci, D.; Bacchi, A. Mononuclear and Polynuclear Complexes Ligated by an Iminodiacetic Acid Derivative: Synthesis, Structure, Solution Studies and Magnetic Properties. Dalton Trans. 2016, 45, 5356–5373. [Google Scholar] [CrossRef]
- Yousuf, I.; Zeeshan, M.; Arjmand, F.; Rizvi, M.A.; Tabassum, S. Synthesis, Structural Investigations and DNA Cleavage Properties of a New Water Soluble Cu(II)-iminodiacetate Complex. Inorg. Chem. Commun. 2019, 106, 48–53. [Google Scholar] [CrossRef]
- Puentes, R.; Torres, J.; Faccio, R.; Bacchi, A.; Kremer, C. Lanthanide Coordination Polymers Based on Flexible Ligands Derived from Iminodiacetic acid. Polyhedron 2019, 170, 683–689. [Google Scholar] [CrossRef]
- Kanehama, R.; Umemiya, M.; Iwahori, F.; Miyasaka, H.; Sugiura, K.-I.; Yamashita, M.; Yokochi, Y.; Ito, H.; Kuroda, S.-I.; Kishida, H.; et al. New ET-Coordinated Copper(I) Complexes: Synthesis, Structures and Physical Properties. Inorg. Chem. 2003, 42, 7173–7181. [Google Scholar] [CrossRef]
- Jia, C.; Zhang, D.; Liu, C.-M.; Xu, W.; Hu, H.; Zhu, D. Novel Silver(I) Complexes Derived From Tetrakis(methylthio)tetrathiafulvalene and Bis(ethylenedithio)tetrathiafulvalene with 3D and 1D Structures. New J. Chem. 2002, 26, 490–494. [Google Scholar] [CrossRef]
- Inoue, M.B.; Inoue, M.; Bruck, M.A.; Fernando, Q. Structure of Bis(ethylenedithio)tetrafulvalenium Tribromodicuprate(I), (BEDT-TTF+)Cu(I)2Br3: Coordination of the Organic Radical Cation to the Metal Ions. J. Chem. Soc. Chem. Commun. 1992, 515–516. [Google Scholar] [CrossRef]
- Pop, F.; Auban-Senzier, P.; Canadell, E.; Rikken, G.L.R.A.; Avarvari, N. Electrical Magnetochiral Anisotropy in a Bulk Chiral Molecular Conductor. Nat. Commun. 2014, 5, 3757. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Barbour, L.J. X-Seed—A Software Tool for Supramolecular Crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Wood, P.A. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).