S-Functionalized Tripods with Monomethylene Spacers: Routes to Tetrairon(III) Single-Molecule Magnets with Ultrashort Tethering Groups

 ,
, 
Abstract
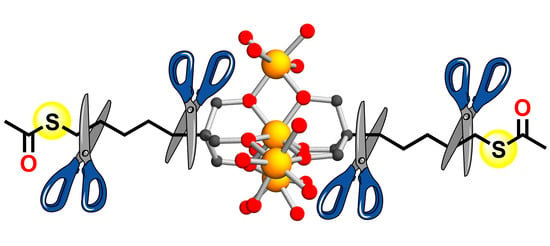
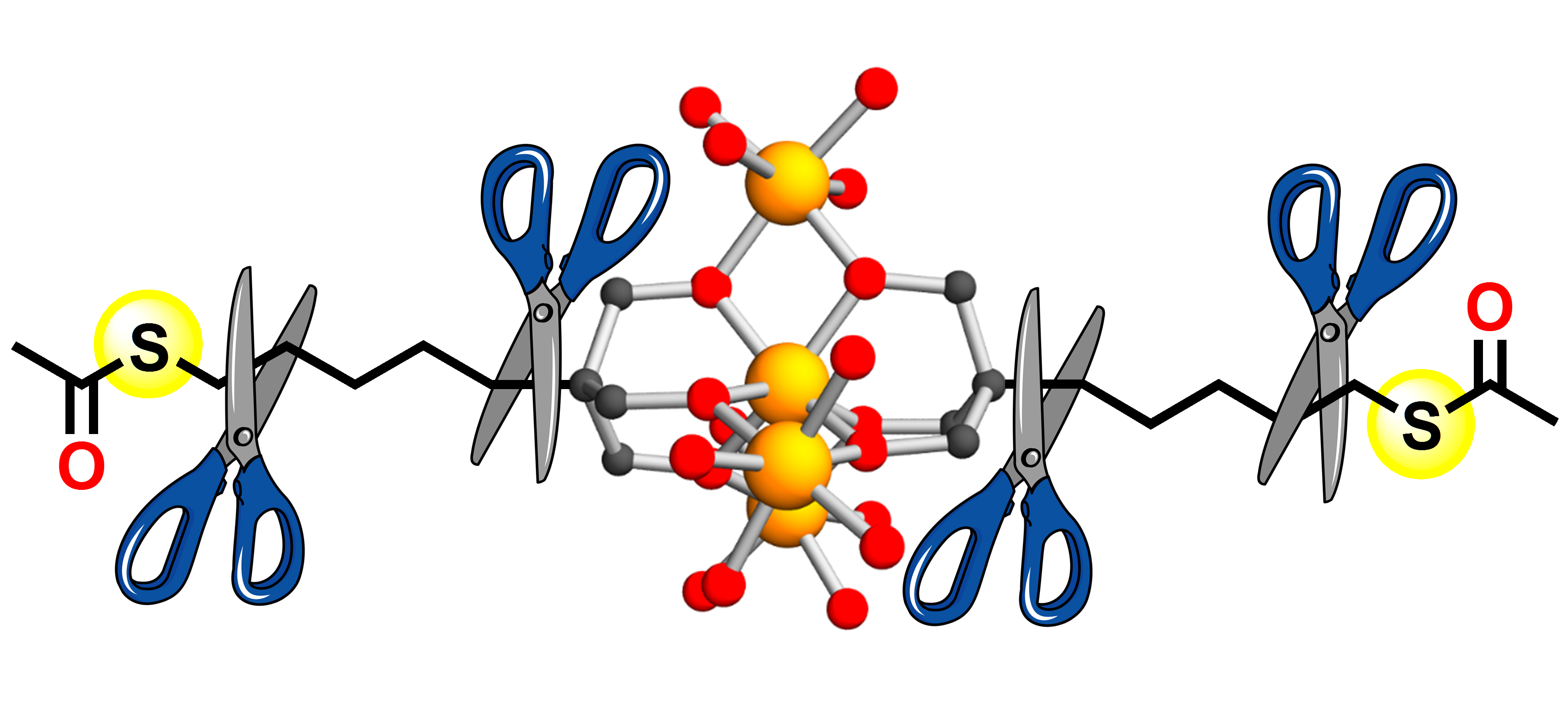
1. Introduction
2. Results and Discussion
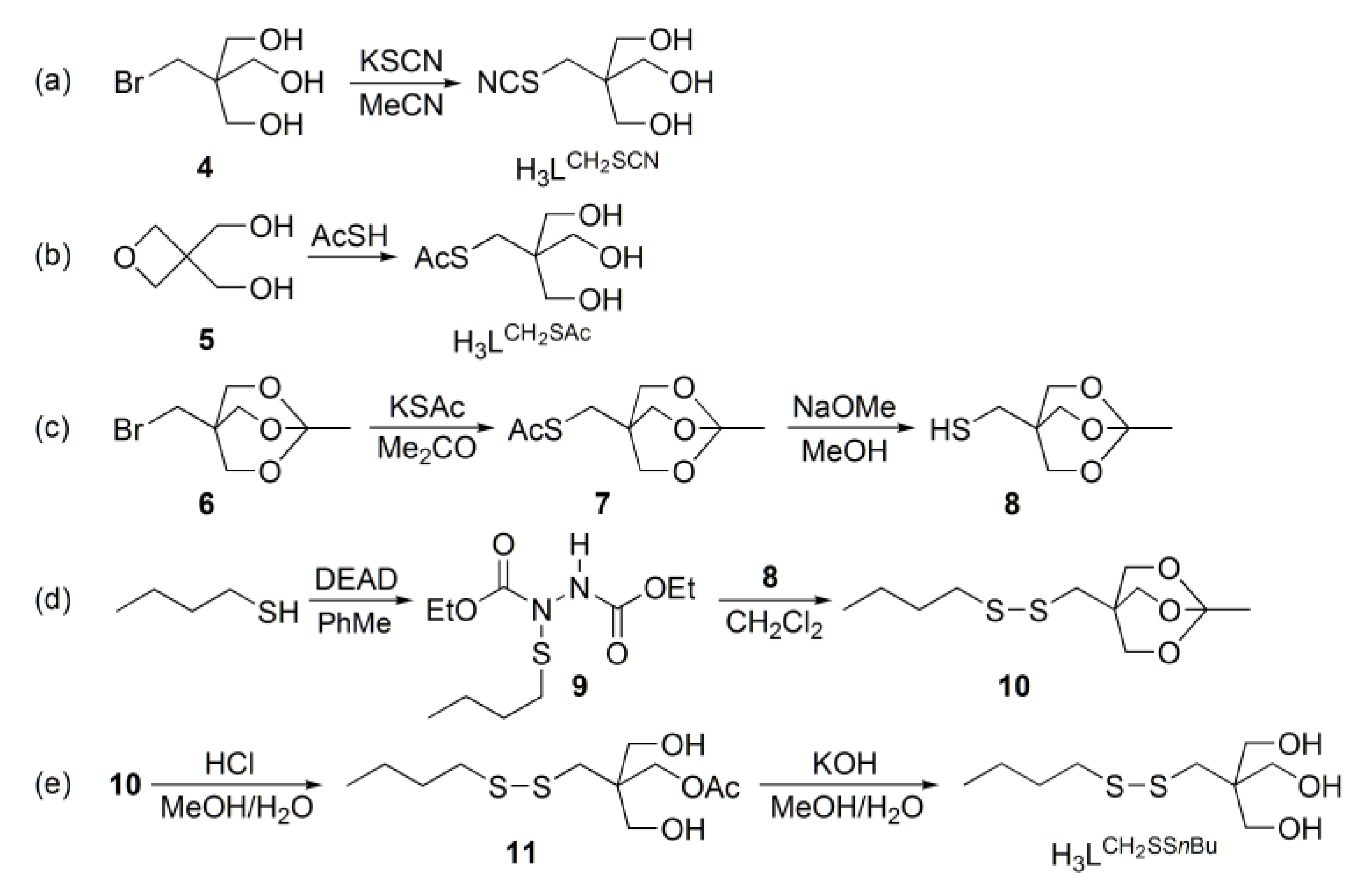
2.1. Synthesis
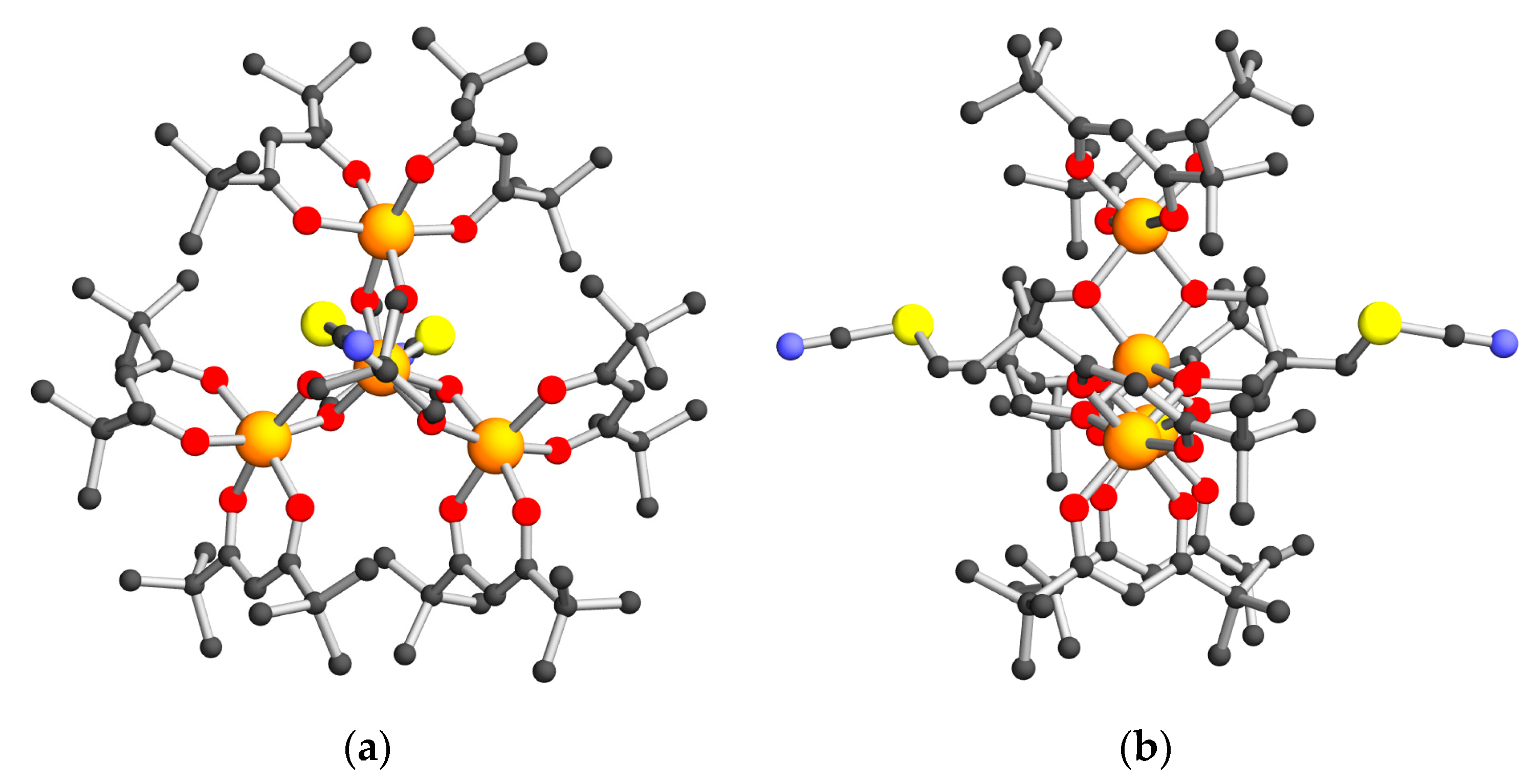
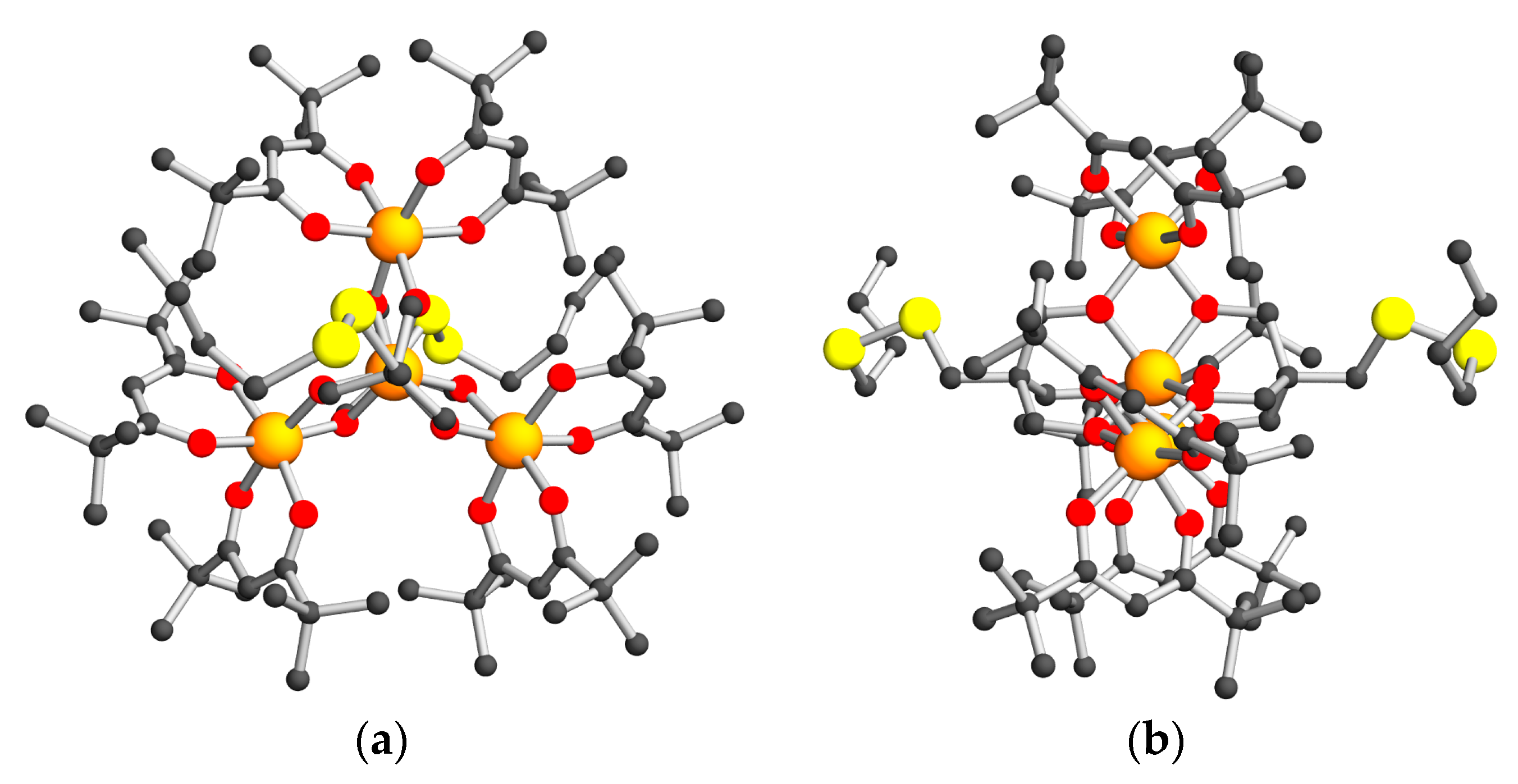
2.2. Structural Descriptions
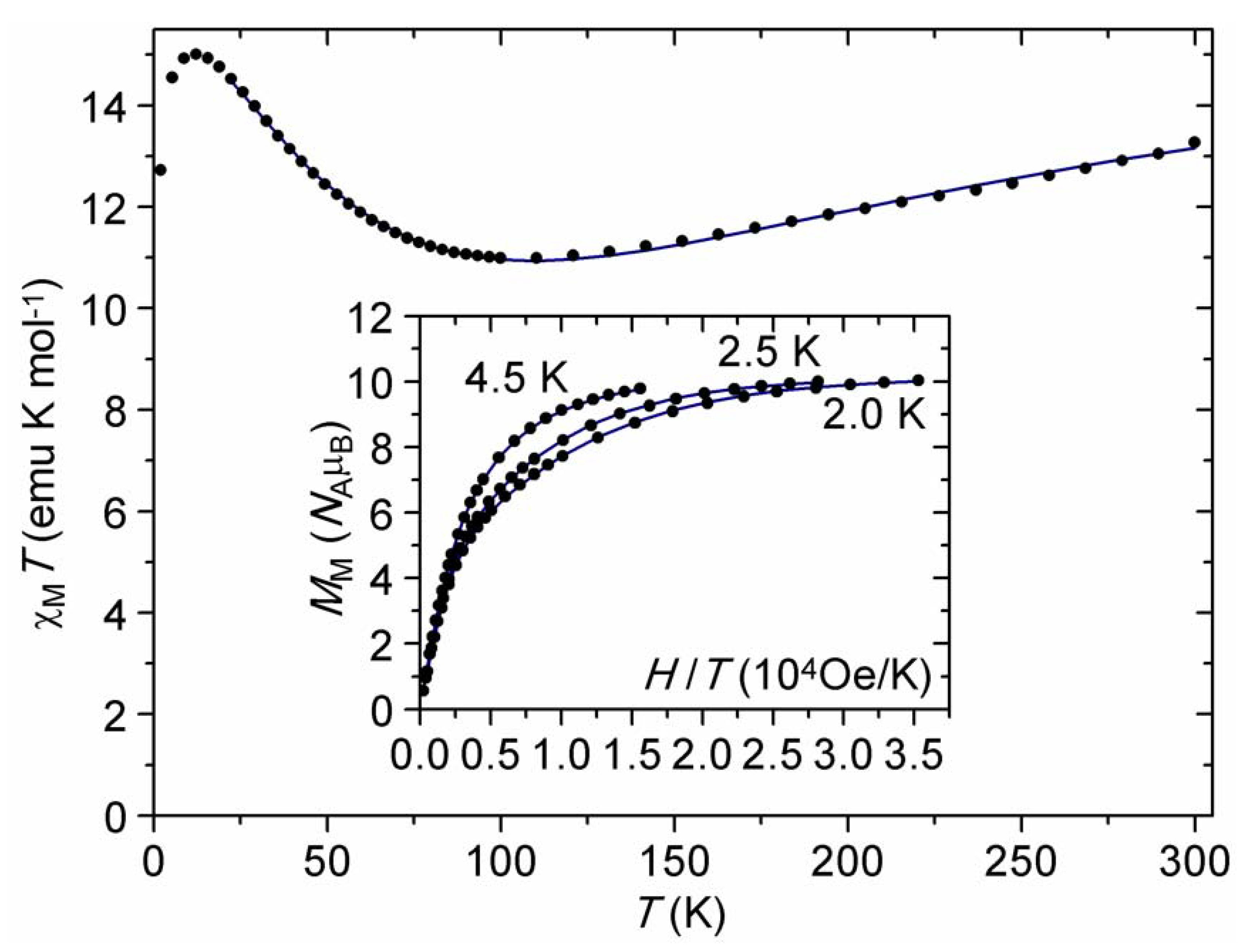
2.3. DC Magnetic Studies
2.4. HF-EPR Spectra
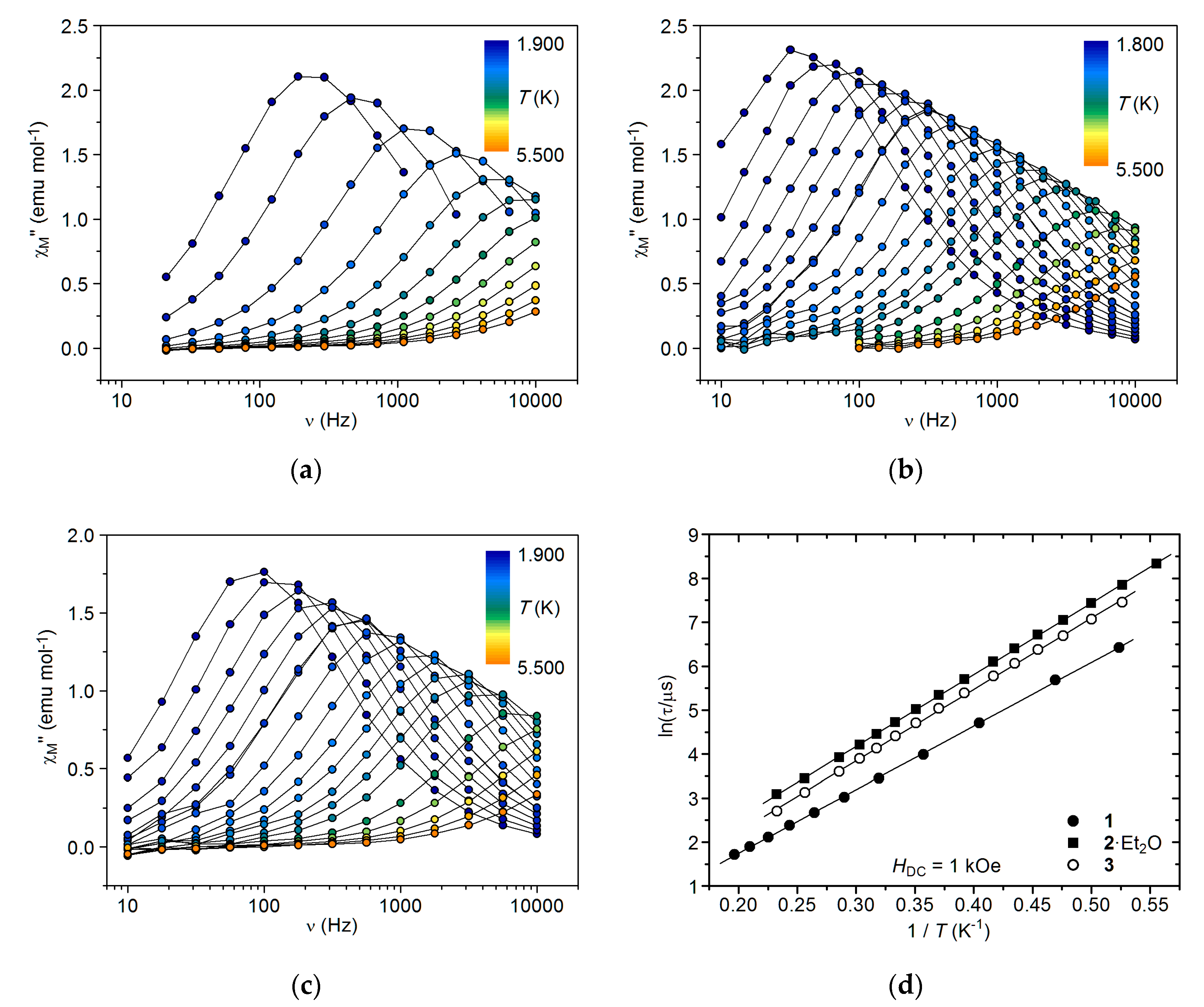
2.5. AC Magnetic Studies
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of 3-hydroxy-2,2-bis(hydroxymethyl)propyl thiocyanate (H3LCH2SCN)
3.3. Synthesis of S-[3-hydroxy-2,2-bis(hydroxymethyl)propyl] ethanethioate (H3LCH2SAc)
3.4. Synthesis of S-[(1-methyl-2,6,7-trioxabicyclo[2.2.2]octan-4-yl)methyl] ethanethioate (7)
3.5. Synthesis of (1-methyl-2,6,7-trioxabicyclo[2.2.2]octan-4-yl)methanethiol (8)
3.6. Synthesis of diethyl 1-(butylsulfanyl)hydrazine-1,2-dicarboxylate (9)
3.7. Synthesis of 2-[(butyldisulfanyl)methyl]-2-(hydroxymethyl)propane-1,3-diol (H3LCH2SSnBu)
3.8. Synthesis of [Fe4(LCH2SCN)2(dpm)6] (1)
3.9. Synthesis of [Fe4(LCH2SAc)2(dpm)6]·xEt2O (2·xEt2O)
3.10. Synthesis of [Fe4(LCH2SSnBu)2(dpm)6] (3)
3.11. Single-Crystal X-ray Diffraction
3.12. Magnetic Measurements
3.13. HF-EPR Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Guo, F.-S.; Day, B.M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet. Science 2018, 362, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Cornia, A.; Talham, D.R.; Affronte, M. Thin Layers of Molecular Magnets. In Molecular Magnetic Materials: Concepts and Applications; Sieklucka, B., Pinkowicz, D., Eds.; Wiley-VCH: Weinheim, Germany, 2017; pp. 187–229. ISBN 9783527694228. [Google Scholar]
- Krylov, D.S.; Schimmel, S.; Dubrovin, V.; Liu, F.; Nguyen, T.T.N.; Spree, L.; Chen, C.-H.; Velkos, G.; Bulbucan, C.; Westerström, R.; et al. Substrate-Independent Magnetic Bistability in Monolayers of the Single-Molecule Magnet Dy2ScN@C80 on Metals and Insulators. Angew. Chem. Int. Ed. 2020, 59, 5756–5764. [Google Scholar] [CrossRef] [PubMed]
- Studniarek, M.; Wäckerlin, C.; Singha, A.; Baltic, R.; Diller, K.; Donati, F.; Rusponi, S.; Brune, H.; Lan, Y.; Klyatskaya, S.; et al. Understanding the Superior Stability of Single-Molecule Magnets on an Oxide Film. Adv. Sci. 2019, 6, 1901736. [Google Scholar] [CrossRef] [PubMed]
- Ara, F.; Oka, H.; Sainoo, Y.; Katoh, K.; Yamashita, M.; Komeda, T. Spin properties of single-molecule magnet of double-decker Tb(III)-phthalocyanine (TbPc2) on ferromagnetic Co film characterized by spin polarized STM (SP-STM). J. Appl. Phys. 2019, 125, 183901. [Google Scholar] [CrossRef]
- Mannini, M.; Pineider, F.; Danieli, C.; Totti, F.; Sorace, L.; Sainctavit, P.; Arrio, M.-A.; Otero, E.; Joly, L.; Cezar, J.C.; et al. Quantum tunnelling of the magnetization in a monolayer of oriented single-molecule magnets. Nature 2010, 468, 417–421. [Google Scholar] [CrossRef]
- Mannini, M.; Pineider, F.; Sainctavit, P.; Danieli, C.; Otero, E.; Sciancalepore, C.; Talarico, A.M.; Arrio, M.-A.; Cornia, A.; Gatteschi, D.; et al. Magnetic memory of a single-molecule quantum magnet wired to a gold surface. Nat. Mater. 2009, 8, 194–197. [Google Scholar] [CrossRef]
- Malavolti, L.; Lanzilotto, V.; Ninova, S.; Poggini, L.; Cimatti, I.; Cortigiani, B.; Margheriti, L.; Chiappe, D.; Otero, E.; Sainctavit, P.; et al. Magnetic bistability in a submonolayer of sublimated Fe4 single-molecule magnets. Nano Lett. 2015, 15, 535–541. [Google Scholar] [CrossRef]
- Cornia, A.; Mannini, M.; Sessoli, R.; Gatteschi, D. Propeller-Shaped Fe4 and Fe3M Molecular Nanomagnets: A Journey from Crystals to Addressable Single Molecules. Eur. J. Inorg. Chem. 2019, 2019, 552–568. [Google Scholar] [CrossRef]
- Erler, P.; Schmitt, P.; Barth, N.; Irmler, A.; Bouvron, S.; Huhn, T.; Groth, U.; Pauly, F.; Gragnaniello, L.; Fonin, M. Highly ordered surface self-assembly of Fe4 single molecule magnets. Nano Lett. 2015, 15, 4546–4552. [Google Scholar] [CrossRef]
- Paschke, F.; Erler, P.; Enenkel, V.; Gragnaniello, L.; Fonin, M. Bulk-Like Magnetic Signature of Individual Fe4H Molecular Magnets on Graphene. ACS Nano 2019, 13, 780–785. [Google Scholar] [CrossRef]
- Gragnaniello, L.; Paschke, F.; Erler, P.; Schmitt, P.; Barth, N.; Simon, S.; Brune, H.; Rusponi, S.; Fonin, M. Uniaxial 2D Superlattice of Fe4 Molecular Magnets on Graphene. Nano Lett. 2017, 17, 7177–7182. [Google Scholar] [CrossRef] [PubMed]
- Serrano, G.; Poggini, L.; Briganti, M.; Sorrentino, A.L.; Cucinotta, G.; Malavolti, L.; Cortigiani, B.; Otero, E.; Sainctavit, P.; Loth, S.; et al. Quantum dynamics of a single molecule magnet on superconducting Pb(111). Nat. Mater. 2020, 19, 546–551. [Google Scholar] [CrossRef]
- Barra, A.-L.; Bianchi, F.; Caneschi, A.; Cornia, A.; Gatteschi, D.; Gorini, L.; Gregoli, L.; Maffini, M.; Parenti, F.; Sessoli, R.; et al. New single-molecule magnets by site-specific substitution: Incorporation of “alligator clips” into Fe4 complexes. Eur. J. Inorg. Chem. 2007, 4145–4152. [Google Scholar] [CrossRef]
- Kappler, J.-P.; Otero, E.; Li, W.; Joly, L.; Schmerber, G.; Muller, B.; Scheurer, F.; Leduc, F.; Gobaut, B.; Poggini, L.; et al. Ultralow-temperature device dedicated to soft X-ray magnetic circular dichroism experiments. J. Synchrotron Radiat. 2018, 25, 1727–1735. [Google Scholar] [CrossRef]
- Cornia, A.; Barra, A.-L.; Poneti, G.; Tancini, E.; Sessoli, R. Unbiased evaluation of zero-field splitting D parameter in high-spin molecules from DC magnetic data with incomplete powder averaging. J. Magn. Magn. Mater. 2020, 510, 166713. [Google Scholar] [CrossRef]
- Cini, A.; Mannini, M.; Totti, F.; Fittipaldi, M.; Spina, G.; Chumakov, A.; Rüffer, R.; Cornia, A.; Sessoli, R. Mössbauer spectroscopy of a monolayer of single molecule magnets. Nat. Commun. 2018, 9, 480. [Google Scholar] [CrossRef]
- Totaro, P.; Poggini, L.; Favre, A.; Mannini, M.; Sainctavit, P.; Cornia, A.; Magnani, A.; Sessoli, R. Tetrairon(III) single-molecule magnet monolayers on gold: Insights from ToF-SIMS and isotopic labeling. Langmuir 2014, 30, 8645–8649. [Google Scholar] [CrossRef]
- Rodriguez-Douton, M.J.; Mannini, M.; Armelao, L.; Barra, A.-L.; Tancini, E.; Sessoli, R.; Cornia, A. One-step covalent grafting of Fe4 single-molecule magnet monolayers on gold. Chem. Commun. 2011, 47, 1467–1469. [Google Scholar] [CrossRef]
- Rodriguez-Douton, M.J.; Sessoli, R.; Cornia, A. A novel tripodal ligand with organosulfur alligator clips for deposition of tetrairon(III) single-molecule magnets on gold. Polyhedron 2011, 30, 2960–2964. [Google Scholar] [CrossRef]
- Ciszek, J.W.; Stewart, M.P.; Tour, J.M. Spontaneous Assembly of Organic Thiocyanates on Gold Sufaces. Alternative Precursors for Gold Thiolate Assemblies. J. Am. Chem. Soc. 2004, 126, 13172–13173. [Google Scholar] [CrossRef]
- Ciszek, J.W.; Keane, Z.K.; Cheng, L.; Stewart, M.P.; Yu, L.H.; Natelson, D.; Tour, J.M. Neutral Complexes of First Row Transition Metals Bearing Unbound Thiocyanates and Their Assembly on Metallic Surfaces. J. Am. Chem. Soc. 2006, 128, 3179–3189. [Google Scholar] [CrossRef] [PubMed]
- Béthencourt, M.I.; Srisombat, L.; Chinwangso, P.; Lee, T.R. SAMs on Gold Derived from the Direct Adsorption of Alkanethioacetates Are Inferior to Those Derived from the Direct Adsorption of Alkanethiols. Langmuir 2009, 25, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.H.A.; Huang, C.; Yakovlev, N.; Chen, Z.K.; O’Shea, S.J. Direct Adsorption and Monolayer Self-Assembly of Acetyl-Protected Dithiols. Langmuir 2006, 22, 2968–2971. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, O.P.H.; Turner, M.; Williams, F.J.; Hille, A.; Sanders, J.K.M.; Lambert, R.M. Direct Observation of Surface-Mediated Thioacetyl Deprotection: Covalent Tethering of a Thiol-Terminated Porphyrin to the Ag(100) Surface. J. Am. Chem. Soc. 2006, 128, 9578–9579. [Google Scholar] [CrossRef]
- Biebuyck, H.A.; Bain, C.D.; Whitesides, G.M. Comparison of Organic Monolayers on Polycrystalline Gold Spontaneously Assembled from Solutions Containing Dialkyl Disulfides or Alkanethiols. Langmuir 1994, 10, 1825–1831. [Google Scholar] [CrossRef]
- Biebuyck, H.A.; Whitesides, G.M. Interchange between monolayers on gold formed from unsymmetrical disulfides and solutions of thiols: Evidence for sulfur-sulfur bond cleavage by gold metal. Langmuir 1993, 9, 1766–1770. [Google Scholar] [CrossRef]
- Tollens, B.; Wiegand, P. Über den Penta-Erythrit, einen aus Formaldehyd und Acetaldehyd synthetisch hergestellten vierwerthigen Alkohol. Liebigs Ann. der Chemie 1891, 265, 316–340. [Google Scholar] [CrossRef]
- Dermer, O.C.; Solomon, P.W. Extensions of the Tollens Condensation. J. Am. Chem. Soc. 1954, 76, 1697–1699. [Google Scholar] [CrossRef][Green Version]
- Tancini, E.; Mannini, M.; Sainctavit, P.; Otero, E.; Sessoli, R.; Cornia, A. On-Surface Magnetometry: The Evaluation of Superexchange Coupling Constants in Surface-Wired Single-Molecule Magnets. Chem. Eur. J. 2013, 19, 16902–16905. [Google Scholar] [CrossRef]
- Singh, R.; Whitesides, G.M. Comparisons of rate constants for thiolate-disulfide interchange in water and in polar aprotic solvents using dynamic 1H NMR line shape analysis. J. Am. Chem. Soc. 1990, 112, 1190–1197. [Google Scholar] [CrossRef]
- Lothian-Tomalia, M.K.; Hedstrand, D.M.; Tomalia, D.A.; Padias, A.B.; Hall, H.K. A contemporary survey of covalent connectivity and complexity. The divergent synthesis of poly(thioether) dendrimers. Amplified, genealogically directed synthesis leading to the de Gennes dense packed state. Tetrahedron 1997, 53, 15495–15513. [Google Scholar] [CrossRef]
- Stellenboom, N.; Hunter, R.; Caira, M.R. One-pot synthesis of unsymmetrical disulfides using 1-chlorobenzotriazole as oxidant: Interception of the sulfenyl chloride intermediate. Tetrahedron 2010, 66, 3228–3241. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Takahashi, K. A convenient method for the preparation of unsymmetrical disulfides by the use of diethyl azodicarboxylate. Tetrahedron Lett. 1968, 9, 5907–5908. [Google Scholar] [CrossRef]
- Le Coustumer, G.; Catel, J.-M. Synthese Et Etude Electrochimique De Disulfures Thiopheniques. Phosphorus. Sulfur. Silicon Relat. Elem. 2006, 181, 191–217. [Google Scholar] [CrossRef]
- Duchenet, V.; Andrieu, C.G.; Catel, J.-M.; Le Coustumer, G.; Penneau, J.-F.; Le Guillanton, G.; Hapiot, P.; Audebert, P. Thienyl and thenyl alkyl disulfides: Monomers and polymers. J. Chim. Phys. Physico Chimie Biol. 1998, 95, 1229–1233. [Google Scholar] [CrossRef]
- Accorsi, S.; Barra, A.-L.; Caneschi, A.; Chastanet, G.; Cornia, A.; Fabretti, A.C.; Gatteschi, D.; Mortalò, C.; Olivieri, E.; Parenti, F.; et al. Tuning anisotropy barriers in a family of tetrairon(III) single-molecule magnets with an S = 5 ground state. J. Am. Chem. Soc. 2006, 128, 4742–4755. [Google Scholar] [CrossRef]
- Gregoli, L.; Danieli, C.; Barra, A.-L.; Neugebauer, P.; Pellegrino, G.; Poneti, G.; Sessoli, R.; Cornia, A. Magnetostructural correlations in tetrairon(III) single-molecule magnets. Chem. Eur. J. 2009, 15, 6456–6467. [Google Scholar] [CrossRef]
- Lunghi, A.; Totti, F. DFT magnetic characterization of a Fe4 SMMs series: From isotropic exchange interactions to multi-spin zero field splitting. J. Mater. Chem. C 2014, 2, 8333–8343. [Google Scholar] [CrossRef]
- Cole, K.S.; Cole, R.H. Dispersion and Absorption in Dielectrics I. Alternating Current Characteristics. J. Chem. Phys. 1941, 9, 341–351. [Google Scholar] [CrossRef]
- Dekker, C.; Arts, A.F.M.; de Wijn, H.W.; van Duyneveldt, A.J.; Mydosh, J.A. Activated dynamics in a two-dimensional Ising spin glass: Rb2Cu1−xCoxF4. Phys. Rev. B 1989, 40, 11243–11251. [Google Scholar] [CrossRef]
- Prasad, T.K.; Poneti, G.; Sorace, L.; Rodriguez-Douton, M.J.; Barra, A.-L.; Neugebauer, P.; Costantino, L.; Sessoli, R.; Cornia, A. Magnetic and optical bistability in tetrairon(III) single molecule magnets functionalized with azobenzene groups. Dalton Trans. 2012, 41, 8368–8378. [Google Scholar] [CrossRef]
- Rodriguez-Douton, M.J.; Cornia, A.; Sessoli, R.; Sorace, L.; Barra, A.-L. Introduction of ester and amido functions in tetrairon(III) single-molecule magnets: Synthesis and physical characterization. Dalton Trans. 2010, 39, 5851–5859. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: Burlington, VT, USA, 2003; ISBN 0-7506-7571-3. [Google Scholar]
- Issidorides, C.H.; Matar, A.I. Pentaerythritol Derivatives. I. The Preparation of Pentaerythritol Monomethyl Ether. J. Am. Chem. Soc. 1955, 77, 6382–6383. [Google Scholar] [CrossRef]
- Ishizone, T.; Tominaga, T.; Kitamura, K.; Hirao, A.; Nakahama, S. Protection and Polymerization of Functional Monomers. 25. Synthesis of Well-Defined Polystyrene Bearing a Triol Functionality by Means of Anionic Living Polymerization of 4-[(4-(4-Vinylphenyl)butoxy)methyl]-1-methyl-2,6,7-trioxabicyclo[2.2.2]octane. Macromolecules 1995, 28, 4829–4836. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Completion and refinement of crystal structures with SIR92. J. Appl. Crystallogr. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Thorn, A.; Dittrich, B.; Sheldrick, G.M. Enhanced rigid-bond restraints. Acta Crystallogr. Sect. A Found. Crystallogr. 2012, 68, 448–451. [Google Scholar] [CrossRef]
- Persistence of Vision Raytracer; Version 3.5; Persistence of Vision Pty. Ltd.: Williamstown, Victoria, Australia, 2002.
- Bain, G.A.; Berry, J.F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532–536. [Google Scholar] [CrossRef]
- E04FCF, E04YCF and F02ABF, NAG Fortran Library Routines (Mark 17); NAG Ltd.: Oxford, UK, 1996.
- Jacobsen, C.J.H.; Pedersen, E.; Villadsen, J.; Weihe, H. ESR characterization of trans-VII(py)4X2 and trans-MnII(py)4X2 (X = NCS, Cl, Br, I; py = pyridine). Inorg. Chem. 1993, 32, 1216–1221. [Google Scholar] [CrossRef]
- Mossin, S.; Weihe, H.; Barra, A.-L. Is the Axial Zero-Field Splitting Parameter of Tetragonally Elongated High-Spin Manganese(III) Complexes Always Negative? J. Am. Chem. Soc. 2002, 124, 8764–8765. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2·Et2O | 3 | 12 1 |
|---|---|---|---|---|
| J1 (cm−1), J2 (cm−1) 2 | 15.69(8), −0.03(6) | 15.800(16), −0.121(14) | 14.36(7), −0.19(6) | 14.64(8), −0.12(7) 3 |
| g2 | 2.008(3) | 2.0057(6) | 1.972(3) | 2.000(3) 3 |
| g, D (cm−1) 4 | 2.0545(12), −0.3961(18) | 2.021(2), −0.426(3) | 1.978(3), −0.419(3) | 2.013(2), −0.423(3) |
| a24,5 | −0.0547(8) | −0.0236(12) | −0.0666(13) | −0.0548(10) |
| gx = gy, gz 6 | 2.00(1), 2.00(1) | 1.995(10), 2.00(1) | not available | 2.00(1), 2.001(5) |
| D (cm−1) 6 | −0.412(1) | −0.438(2) | not available | −0.430(1) |
| E (cm−1), (cm−1) 6 | 0.010(2), 1.6(3)·10−5 | 0.009(4), 1.5(3)·10−5 | not available | 0.041(2), 1.2(1)·10−5 |
| Ueff/kB (K), 7 U/kB (K) 8 | 14.48(7), 14.8 | 16.32(5), 15.8 | 16.22(10), 15.1 | 13.78(8), 15.5 |
| τ0 (s) 7 | 3.15(7)·10−7 | 4.87(10)·10−7 | 3.63(15)·10−7 | 5.2(2)·10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornia, A.; Danieli, C.; Meglioli, F.; Tancini, E.; Nicolini, A.; Rodriguez-Douton, M.J.; Barra, A.-L.; Affronte, M.; Sessoli, R. S-Functionalized Tripods with Monomethylene Spacers: Routes to Tetrairon(III) Single-Molecule Magnets with Ultrashort Tethering Groups. Magnetochemistry 2020, 6, 55. https://doi.org/10.3390/magnetochemistry6040055
Cornia A, Danieli C, Meglioli F, Tancini E, Nicolini A, Rodriguez-Douton MJ, Barra A-L, Affronte M, Sessoli R. S-Functionalized Tripods with Monomethylene Spacers: Routes to Tetrairon(III) Single-Molecule Magnets with Ultrashort Tethering Groups. Magnetochemistry. 2020; 6(4):55. https://doi.org/10.3390/magnetochemistry6040055
Chicago/Turabian StyleCornia, Andrea, Chiara Danieli, Fabio Meglioli, Erik Tancini, Alessio Nicolini, Maria Jesus Rodriguez-Douton, Anne-Laure Barra, Marco Affronte, and Roberta Sessoli. 2020. "S-Functionalized Tripods with Monomethylene Spacers: Routes to Tetrairon(III) Single-Molecule Magnets with Ultrashort Tethering Groups" Magnetochemistry 6, no. 4: 55. https://doi.org/10.3390/magnetochemistry6040055
APA StyleCornia, A., Danieli, C., Meglioli, F., Tancini, E., Nicolini, A., Rodriguez-Douton, M. J., Barra, A.-L., Affronte, M., & Sessoli, R. (2020). S-Functionalized Tripods with Monomethylene Spacers: Routes to Tetrairon(III) Single-Molecule Magnets with Ultrashort Tethering Groups. Magnetochemistry, 6(4), 55. https://doi.org/10.3390/magnetochemistry6040055