Redox-Active Dysprosium Single-Molecule Magnet: Spectro-Electrochemistry and Theoretical Investigations

 ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
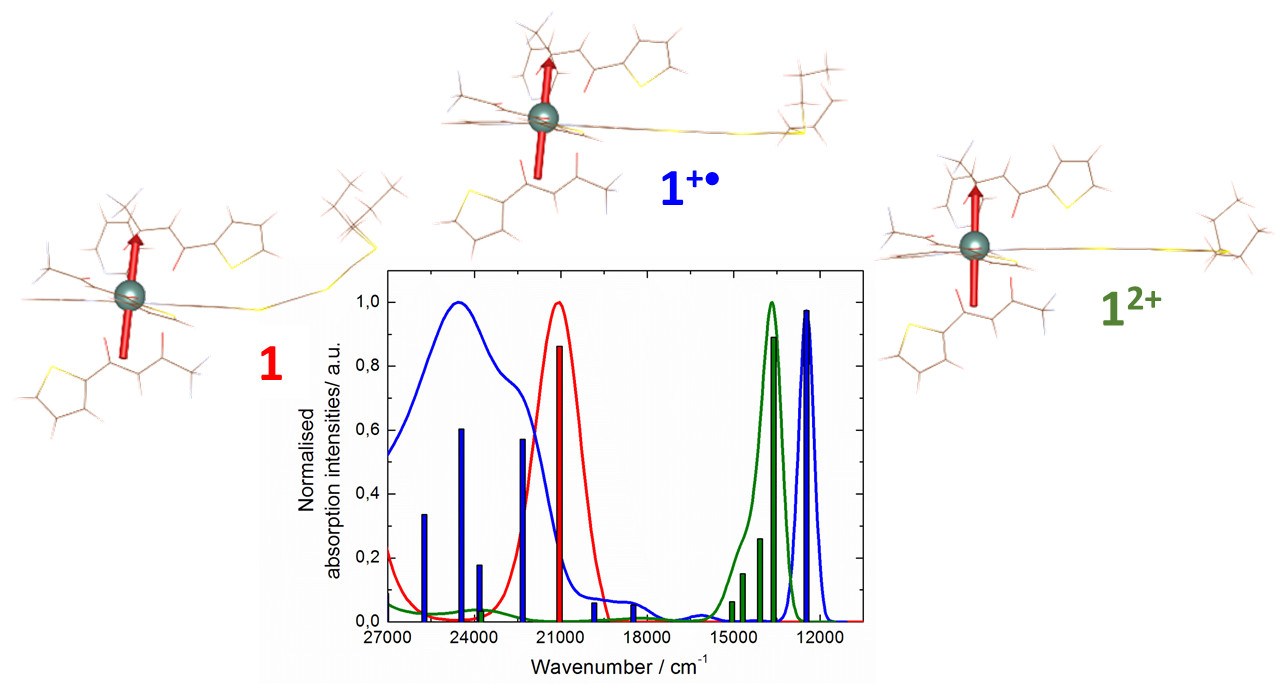
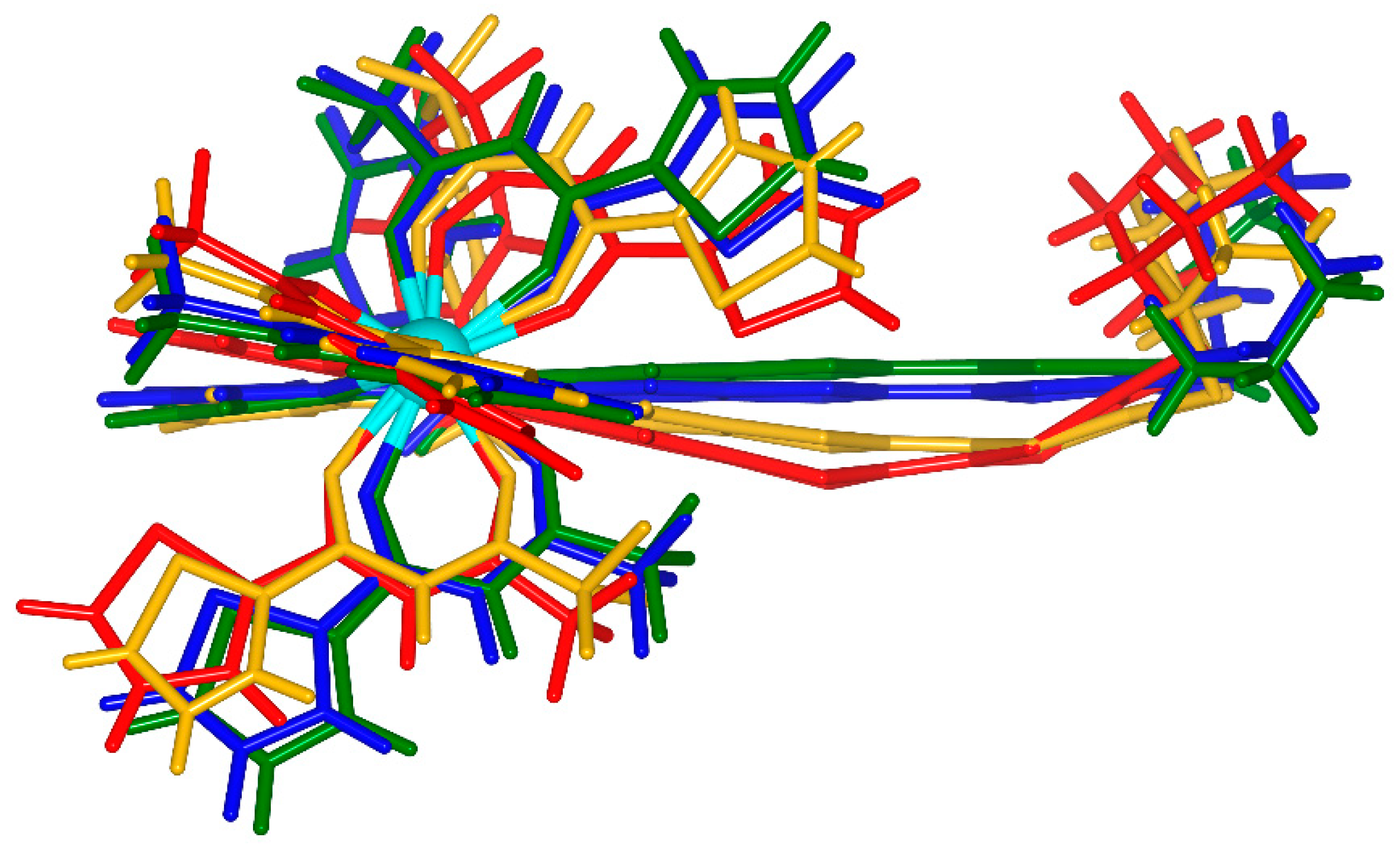
2.1. X-ray Structures of [Dy(tta)3(L)]⋅C6H14 (1)⋅C6H14, and DFT Optimized Structures of [Dy(tta)3(L)] (1opt), [Dy(tta)3(L+∙)] (1+∙opt) and [Dy(tta)3(L2+)] (12+opt)
2.2. Electrochemical Properties
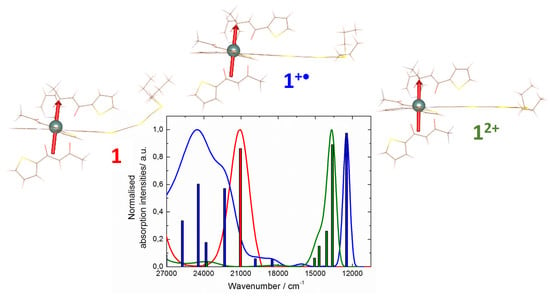
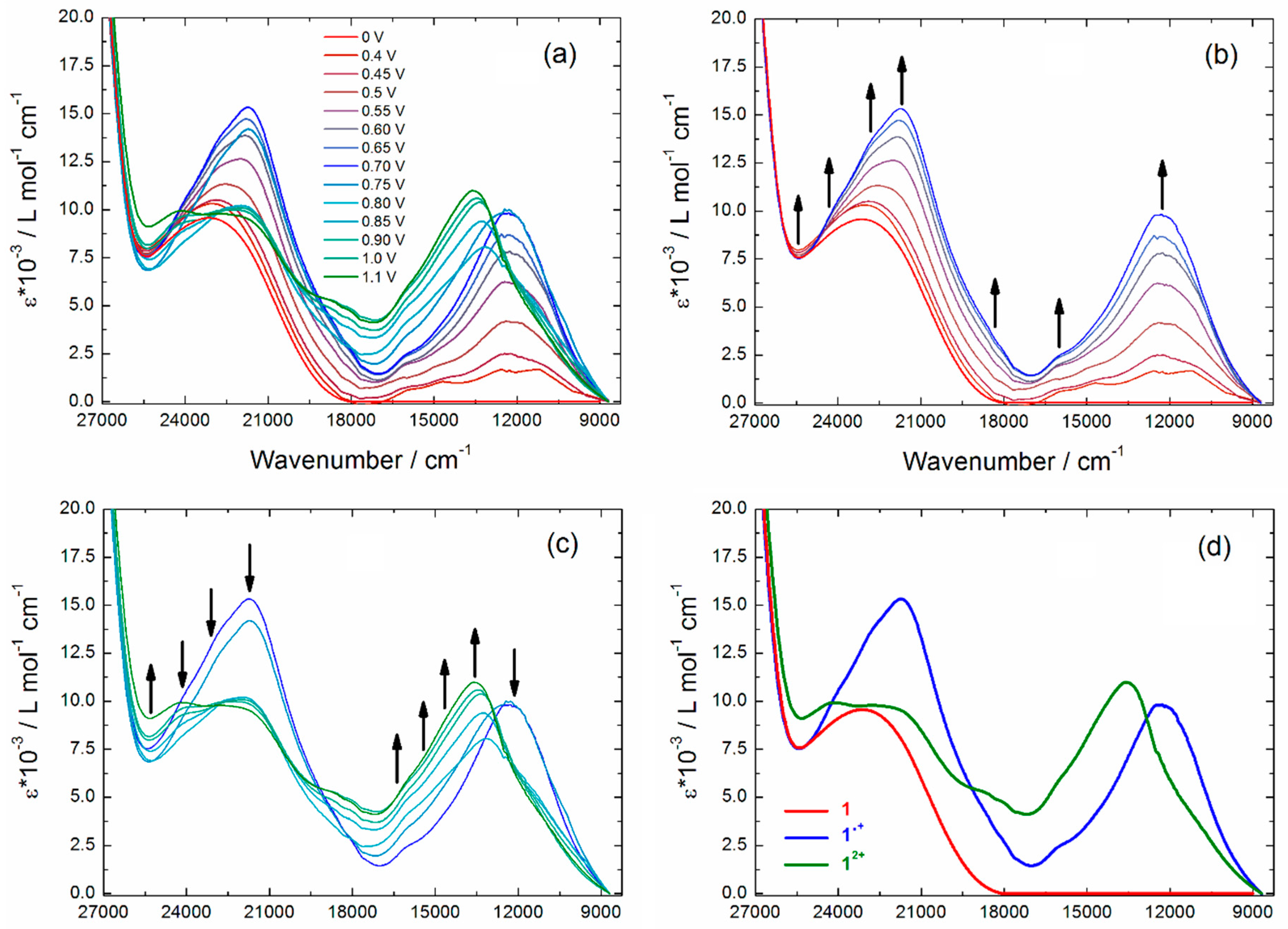
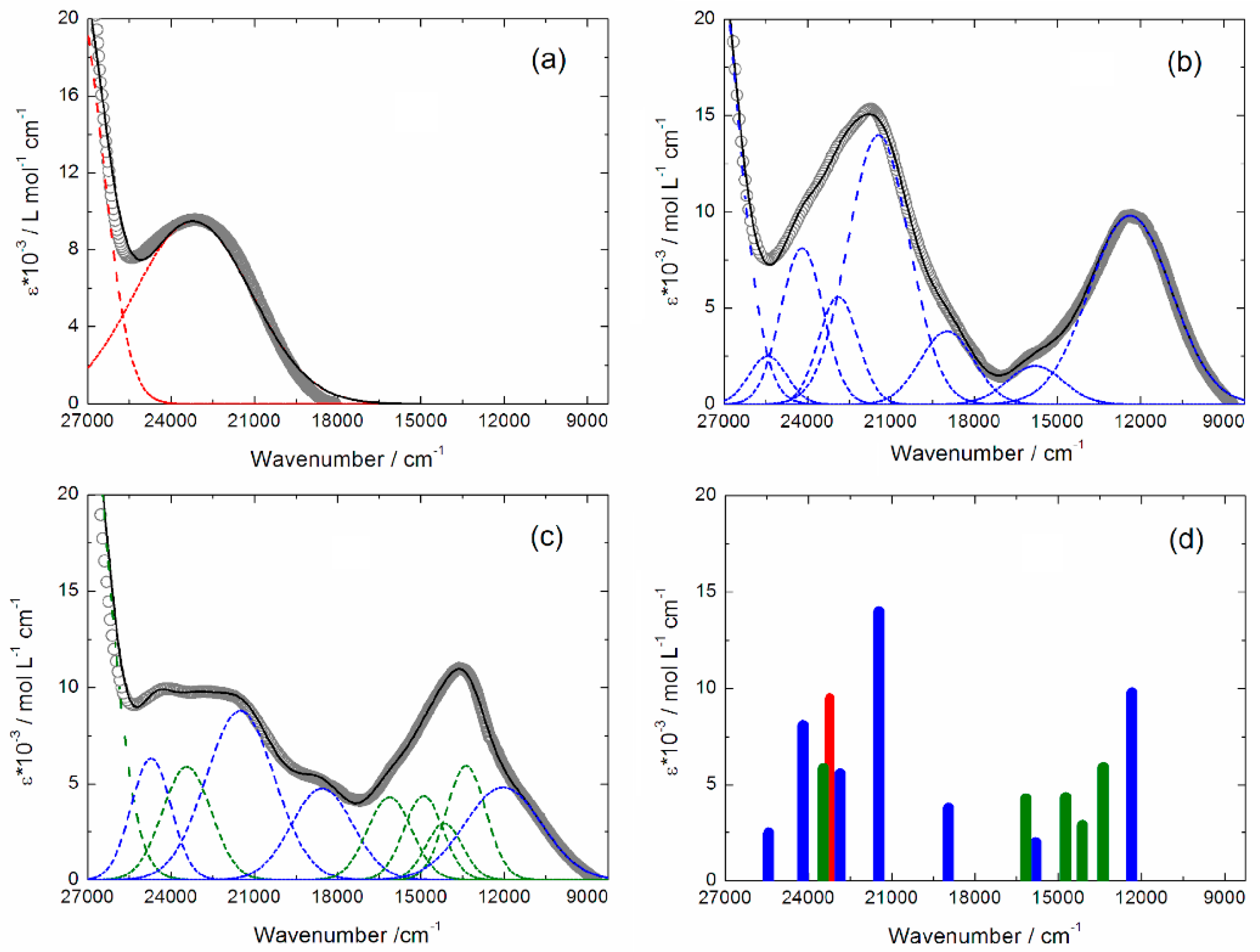
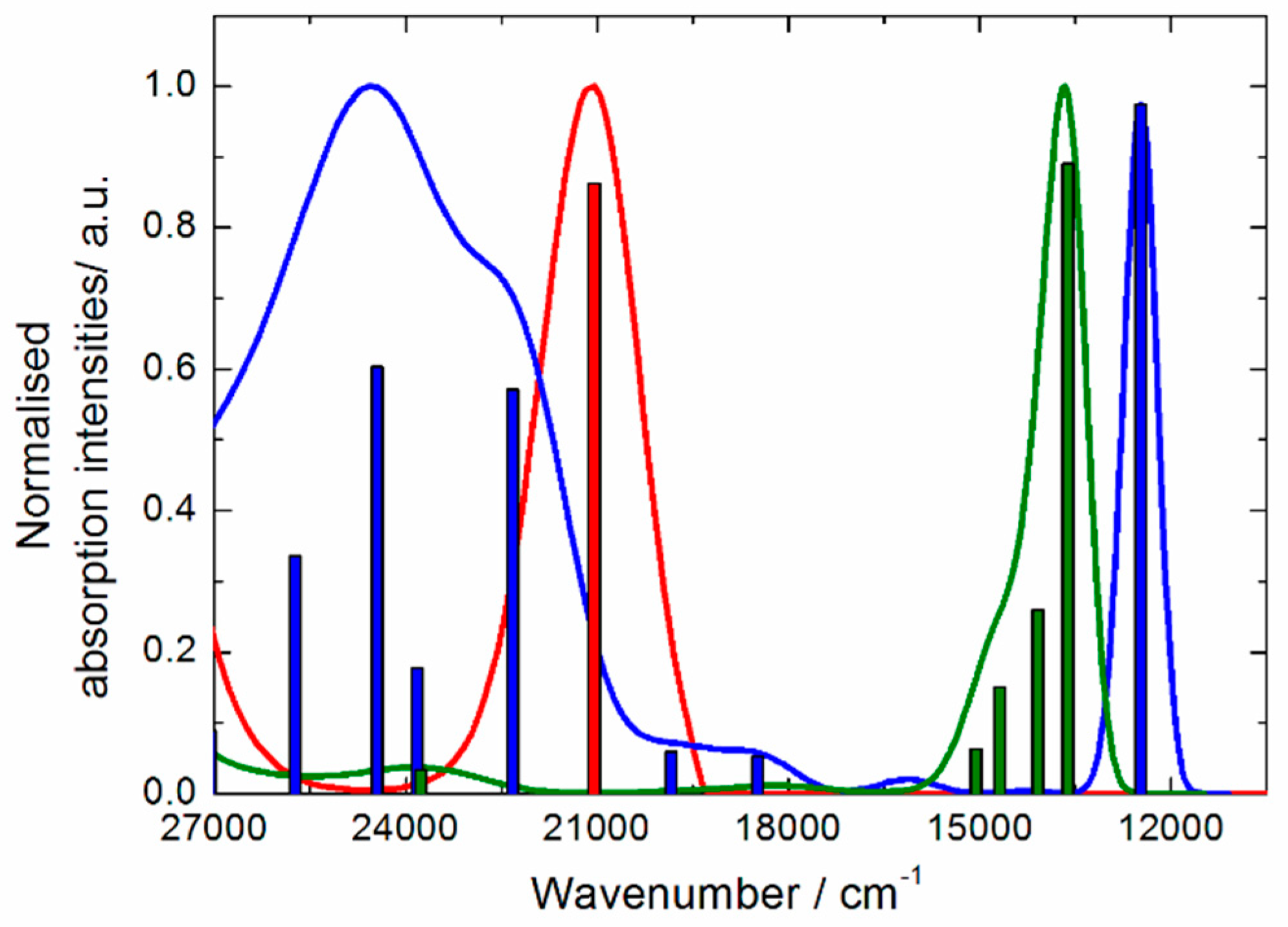
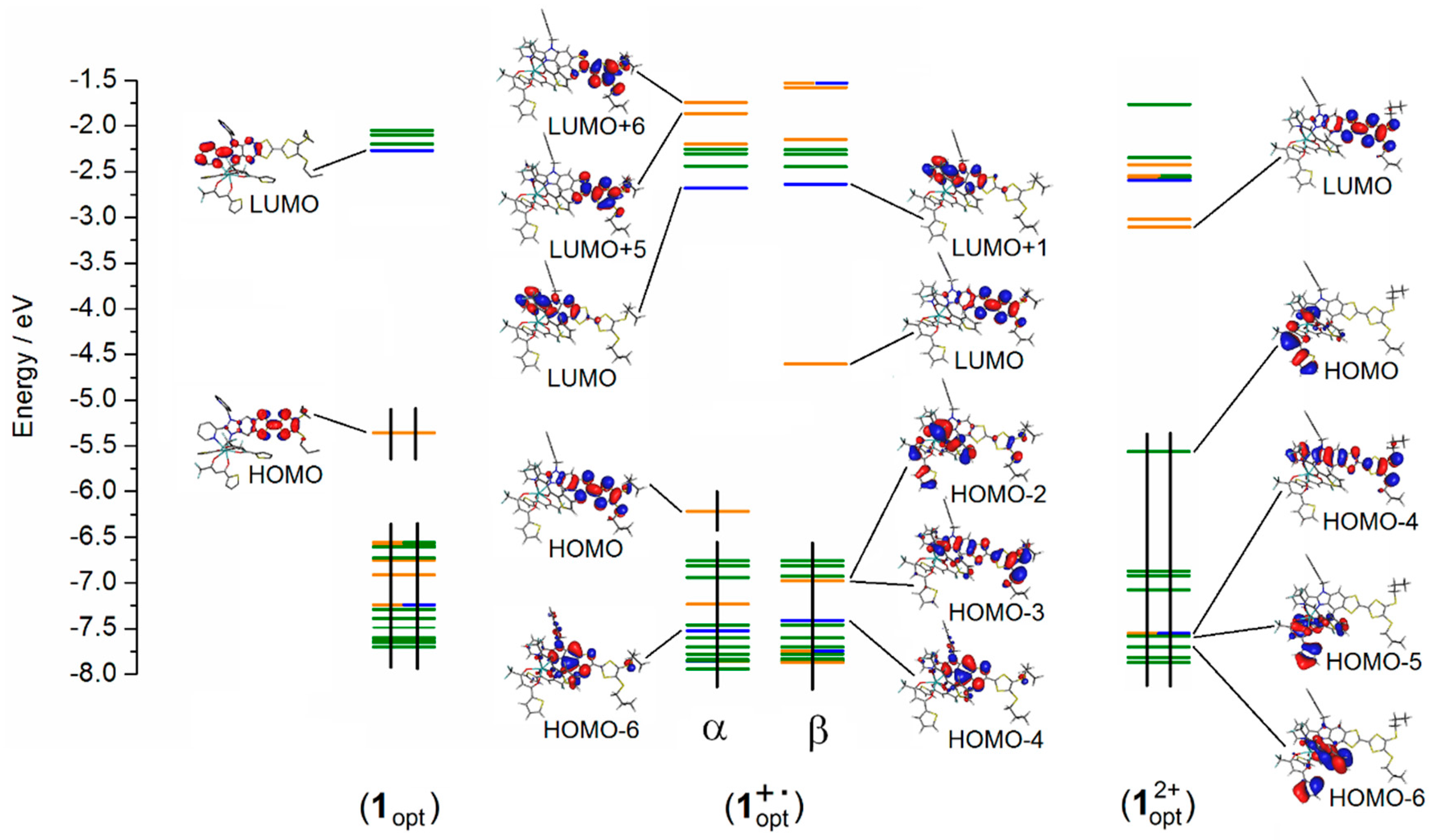
2.3. Photo-Physical and Spectro-Plectrochemical Properties
2.4. Magnetic Properties
3. Materials and Methods
3.1. Synthesis. General Procedures and Materials
3.2. Physical Measurements
3.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SMM | Single Molecule Magnet |
| CH2Cl2 | dichloromethane |
| tta− | 2-thenoyltrifluoroacetonate |
| TTF | Tetrathiafulvalene |
| HOMO | Highest Occupied Molecular Orbital |
| LUMO | Lowest Unoccupied Molecular Orbital |
| ILCT | Intra-Ligand Charge Transfer |
| DFT | Density Functional Theory |
| TD-DFT | Time-Dependent Density Functional Theory |
| CASSCF | Complete Active Space Self-Consistent Field |
| RASSI-SO | Restricted Active Space State Interaction—Spin-Orbit |
References
- Bryce, M.R. Tetrathiafulvalene as π-electron donors for intramolecular charge-transfer Materials. Adv. Mater. 1999, 11, 11–23. [Google Scholar] [CrossRef]
- Yamada, J.; Sugimoto, T. TTF Chemistry: Fundamentals and Applications of Tetrathiafulvalene; Springer: Berlin, Germany, 2004. [Google Scholar]
- Otsubo, T.; Takimiya, K. Recent synthetic advances of tetrathiafulvalene-based organic conductors. Bull. Chem. Soc. Jpn. 2004, 77, 43–58. [Google Scholar] [CrossRef]
- Coronado, E.; Day, P. Magnetic molecular conductors. Chem. Rev. 2004, 104, 5419–5448. [Google Scholar] [CrossRef]
- Lorcy, D.; Bellec, N.; Fourmigué, M.; Avarvari, N. Tetrathiafulvalene-based group XV ligands: Synthesis, coordination chemistry and radical cation salts. Coord. Chem. Rev. 2009, 253, 1398–1438. [Google Scholar] [CrossRef]
- Shatruk, M.; Ray, L. Ligands derived from tetrathiafulvalene: Building blocks for multifunctional materials. Dalton Trans. 2010, 39, 11105–11121. [Google Scholar] [CrossRef] [PubMed]
- Batail, P. Introduction: Molecular conductors. Chem. Rev. 2004, 104, 4887–4890. [Google Scholar] [CrossRef]
- Hansen, T.K.; Jørgensen, T.; Stein, P.C.; Becher, J. Crown ether derivatives of tetrathiafulvalene. J. Org. Chem. 1992, 57, 6403–6409. [Google Scholar] [CrossRef]
- Beer, P.D.; Gale, P.A.; Chen, G.Z. Electrochemical molecular recognition: Pathways between complexation and signalling. J. Chem. Soc. Dalton Trans. 1999, 12, 1897–1910. [Google Scholar] [CrossRef]
- Bernhardt, P.V.; Moore, E.G. Functionalized macrocyclic compounds: Potential sensors of small molecules and ions. Aust. J. Chem. 2003, 56, 239–258. [Google Scholar] [CrossRef]
- Lu, H.Y.; Xu, W.; Zhang, D.Q.; Chen, C.F.; Zhu, D.B. A novel multisignaling optical-electrochemical chemosensor for anions based on tetrathiafulvalene. Org. Lett. 2005, 7, 4629–4632. [Google Scholar] [CrossRef]
- McCall, K.L.; Morandeira, A.; Durrant, J.; Yellowlees, L.J.; Robertson, N. Characterisation of a ruthenium bipyridyl dye showing a long-lived charge-separated state on TiO2 in the presence of I−/I3−. Dalton Trans. 2010, 39, 4138–4145. [Google Scholar] [CrossRef][Green Version]
- Wenger, S.; Bouit, P.-A.; Chen, Q.L.; Teuscher, J.; Censo, D.D.; Humphry-Baker, R.; Moser, J.-E.; Delgado, J.L.; Martín, N.; Zakeeruddin, S.M.; et al. Efficient electron transfer and sensitizer regeneration in stable π-extended tetrathaifulvalene-sensitized solar cells. J. Am. Chem. Soc. 2010, 132, 5164–5169. [Google Scholar] [CrossRef]
- Spaldin, N.A.; Fiebig, M. The renaissance of magnetoelectric multiferroics. Science 2005, 309, 391–392. [Google Scholar] [CrossRef]
- Eerenstein, W.; Mathur, N.D.; Scott, J.F. Multiferroic and magnetoelectric materials. Nature 2006, 442, 759–765. [Google Scholar] [CrossRef]
- Train, C.; Gheorghe, R.; Krstic, V.; Chamoreau, L.-M.; Ovanesyan, N.S.; Rikken, G.L.J.A.; Gruselle, M.; Verdaguer, M. Strong magneto-chiral dichroism in enantiopure chiral ferromagnets. Nat. Mater. 2008, 7, 729–734. [Google Scholar] [CrossRef]
- Ouahab, L.; Enoki, T. Multiproperty molecular materials: TTF-based conducting and magnetic molecular materials. Eur. J. Inorg. Chem. 2004, 933–941. [Google Scholar] [CrossRef]
- Kobayashi, A.; Fujiwara, E.; Kobayashi, H. Single-component molecular metals with extended-TTF dithiolate ligands. Chem. Rev. 2004, 104, 5243–5264. [Google Scholar] [CrossRef]
- Enoki, T.; Miyasaki, A. Magnetic TTF-based charge-transfer complexes. Chem. Rev. 2004, 104, 5449–5478. [Google Scholar] [CrossRef]
- Pointillart, F.; Golhen, S.; Cador, O.; Ouahab, L. 3d and 4d coordination complexes and coordination polymers involving electroactive tetrathiafulvalene containing ligands. Comptes Rendus Chimie 2013, 16, 679–687. [Google Scholar] [CrossRef]
- Pointillart, F.; Le Gal, Y.; Golhen, S.; Cador, O.; Ouahab, L. First paramagnetic 4d transition-metal complex with a redox-active tetrathiafulvalene derivative, [Ru(salen)(PPh3)(TTF-CH=CH-Py)]BF4 [salen2− = N,N’-Ethan−1,2-diylbis(salicylidenamine), PPh3 = triphenylphosphine, TTF-CH=CH-Py = 4-(2-tetrathiafulvalenylethenyl)pyridine]. Inorg. Chem. 2008, 47, 9730–9732. [Google Scholar]
- Rabaca, S.; Almeida, M. Dithiolene complexes containing N coordinating groups and corresponding tetrathiafulvalene donors. Coord. Chem. Rev. 2010, 254, 1493–1508. [Google Scholar] [CrossRef]
- Pointillart, F.; Golhen, S.; Cador, O.; Ouahab, L. Paramagnetic 3d coordination complexes involving redox-active tetrathiafulvalene derivatives: An efficient approach to elaborate multi-properties materials. Dalton Trans. 2013, 42, 1949–1960. [Google Scholar] [CrossRef]
- Huang, Y.-D.; Huo, P.; Shao, M.-Y.; Yin, J.-X.; Shen, W.-C.; Zhu, Q.-Y.; Dai, J. A new type of charge-transfer salts based on tetrathiafulvalene-tetracarboxylate coordination polymers and methyl viologen. Inorg. Chem. 2014, 53, 3480–3487. [Google Scholar] [CrossRef]
- Faulkner, S.; Burton-Pye, B.P.; Khan, T.; Martin, L.R.; Wray, S.D.; Skabara, P.J. Interaction between tetrathiafulvalene carboxylic acid and ytterbium DO3A: Solution state self-assembly of a ternary complex which is luminescent in the near IR. Chem. Commun. 2002, 16, 1668–1669. [Google Scholar] [CrossRef]
- Pope, S.J.A.; Burton-Pye, B.P.; Berridge, R.; Khan, T.; Skabara, P.; Faulkner, S. Self-assembly of luminescent ternary complexes between seven-coordinate lanthanide(III) complexes and chromophore bearing carboxylates and phosphonates. Dalton Trans. 2006, 2907–2912. [Google Scholar] [CrossRef]
- Pointillart, F.; Le Gal, Y.; Golhen, S.; Cador, O.; Ouahab, L. 4f Gadolinium(III) complex involving tetrathiafulvalene-amido-2-pyrimidine−1-oxide as a ligand. Inorg. Chem. 2009, 48, 4631–4633. [Google Scholar] [CrossRef]
- Pointillart, F.; Le Guennic, B.; Golhen, S.; Cador, O.; Maury, O.; Ouahab, L. High nuclearity complexes of lanthanide involving tetrathiafulvalene ligands: Structural, magnetic, and photophysical properties. Inorg. Chem. 2013, 52, 1610–1620. [Google Scholar] [CrossRef]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide single-molecule magnets. Chem. Rev. 2013, 113, 5110–5148. [Google Scholar] [CrossRef]
- Pointillart, F.; Cador, O.; Le Guennic, B.; Ouahab, L. Uncommon lanthanide ions in purely 4f single molecule magnets. Coord. Chem. Rev. 2017, 346, 150–175. [Google Scholar] [CrossRef]
- Zhang, P.; Guo, Y.-N.; Tang, J. Recent advances in dysprosium-based single molecule magnets: Structural overview and synthetic strategies. Coord. Chem. Rev. 2013, 257, 1728–1763. [Google Scholar] [CrossRef]
- Liddle, S.T.; van Slageren, J. Improving f-element single molecule magnets. Chem. Soc. Rev. 2015, 44, 6655–6669. [Google Scholar] [CrossRef]
- Meng, Y.-S.; Jiang, S.-D.; Wang, B.-W.; Gao, S. Understanding the magnetic anisotropy toward single-ion magnets. Acc. Chem. Res. 2016, 49, 2381–2389. [Google Scholar] [CrossRef]
- Liu, J.-L.; Chen, Y.-C.; Tong, M.-L. Symmetry strategies for high performance lanthanide-based single-molecule magnets. Chem. Soc. Rev. 2018, 47, 2431–2453. [Google Scholar] [CrossRef]
- Gupta, S.K.; Murugavel, R. Enriching lanthanide single-ion magnetism through symmetry and axiality. Chem. Commun. 2018, 54, 3685–3696. [Google Scholar] [CrossRef]
- Zhu, Z.; Guo, M.; Li, X.-L.; Tang, J. Molecular magnetism of lanthanide: Advances and perspectives. Coord. Chem. Rev. 2019, 378, 350–364. [Google Scholar] [CrossRef]
- Guo, F.-S.; Bar, A.K.; Layfield, R.A. Main group chemistry at the interface with molecular magnetism. Chem. Rev. 2019. [Google Scholar] [CrossRef]
- Gatteschi, D.; Sessoli, R.; Villain, J. Molecular Nanomagnets; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Bogani, L.; Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 2008, 7, 179–186. [Google Scholar] [CrossRef]
- Mannini, M.; Pineider, F.; Sainctavit, P.; Danieli, C.; Otero, E.; Sciancalepore, C.; Talarico, A.-M.; Arrio, M.-A.; Cornia, A.; Gatteschi, D.; et al. Magnetic memory of a single-molecule quantum magnet wired to a gold surface. Nat. Mater. 2009, 8, 194–197. [Google Scholar] [CrossRef]
- Leuenberger, M.N.; Loss, D. Quantum computing in molecular magnets. Nature 2001, 410, 789–793. [Google Scholar] [CrossRef]
- Lehmann, J.; Gaita-Arino, A.; Coronado, E.; Loss, D. Spin qubits with electrically gated polyoxometallate molecules. Nat. Nanotechnol. 2007, 2, 312–317. [Google Scholar] [CrossRef]
- Ganzhorn, M.; Klyatskaya, S.; Ruben, M.; Wernsdorfer, W. Strong spin-phonon coupling between a single-molecule magnet and a carbon nanotube nanoelectromechanical system. Nat. Nanotechnol. 2013, 8, 165–169. [Google Scholar] [CrossRef]
- Gao, F.; Cui, L.; Liu, W.; Hu, L.; Zhong, Y.-W.; Li, Y.-Z.; Zuo, J.-L. Seven-coordinate lanthanide sandwich-type complexes with a tetrathiafulvalene-fused schiff base ligand. Inorg. Chem. 2013, 52, 11164–11172. [Google Scholar] [CrossRef]
- Pointillart, F.; Le Guennic, B.; Maury, O.; Golhen, S.; Cador, O.; Ouahab, L. Lanthnaide dinuclear complexes involving tetrathiafulvalene-3-pyridine-N-oxide ligand: Semiconductor radical salt, magnetic, and photophysical studies. Inorg. Chem. 2013, 52, 1398–1408. [Google Scholar] [CrossRef]
- Pointillart, F.; Jung, J.; Berraud-Pache, R.; Le Guennic, B.; Dorcet, V.; Golhen, S.; Cador, O.; Maury, O.; Guyot, Y.; Decurtins, S.; et al. Luminescence and single-molecule magnet behavior in lanthanide complexes invlving a tetrathiafulvalene-fused dipyridophenazine ligand. Inorg. Chem. 2015, 54, 5384–5397. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, X.-M.; Cui, L.; Deng, K.; Zeng, Q.-D.; Zuo, J.-L. Tetrathiafulvalene-supported triple-decker phthalocyaninato dysprosium(III) complex: Synthesis, properties and surface assembly. Sci. Rep. 2014, 4, 5928–5935. [Google Scholar] [CrossRef]
- Pointillart, F.; Le Guennic, B.; Golhen, S.; Cador, O.; Maury, O.; Ouahab, L. A redox-active luminescent ytterbium based single molecule magnet. Chem. Commun. 2013, 49, 615–617. [Google Scholar] [CrossRef]
- Soussi, K.; Jung, J.; Pointillart, F.; Le Guennic, B.; Lefeuvre, B.; Golhen, S.; Cador, O.; Guyot, Y.; Maury, O.; Ouahab, L. Magnetic and photo-physical inverstigations into DyIII and YbIII complexes involving tetrathiafulvalene ligand. Inorg. Chem. Front. 2015, 2, 1105–1117. [Google Scholar] [CrossRef]
- Pointillart, F.; Le Guennic, B.; Cador, O.; Maury, O.; Ouahab, L. Lanthanide ion and tetrathiafulvalene-based ligand as a “magic” couple toward luminescence, single molecule magnets, and magnetostructural correlations. Acc. Chem. Res. 2015, 48, 2834–2842. [Google Scholar] [CrossRef]
- Feng, M.; Pointillart, F.; Lefeuvre, B.; Dorcet, V.; Golhen, S.; Cador, O.; Ouahab, L. Multiple single-molecule magnet behaviors in dysprosium dinuclear complexes involving a multiple functionalized tetrathiafulvalene-based ligand. Inorg. Chem. 2015, 54, 4021–4028. [Google Scholar] [CrossRef]
- Speed, S.; Feng, M.; Fernandez-Garcia, G.; Pointillart, F.; Lefeuvre, B.; Riobé, F.; Golhen, S.; Le Guennic, B.; Totti, F.; Guyot, Y.; et al. Lanthanide complexes involving multichelating TTF-based ligands. Inorg. Chem. Front. 2017, 4, 604–617. [Google Scholar] [CrossRef]
- Lefeuvre, B.; Galangau, O.; Flores Gonzalez, J.; Montigaud, V.; Dorcet, V.; Ouahab, L.; Le Guennic, B.; Cador, O.; Pointillart, F. Field-induced dysprosium single-molecule magnet based on a redox-active fused 1,10-phenantroline bridging triad. Front. Chem. 2018, 6, 552–562. [Google Scholar] [CrossRef]
- Pointillart, F.; Ou-Yang, J.-K.; Fernandez Garcia, G.; Montigaud, V.; Flores Gonzalez, J.; Marchal, R.; Favereau, L.; Totti, F.; Crassous, J.; Cador, O.; et al. Tetrathiafulvalene-based helicene ligand in the design of a dysprosium field-induced single-molecule magnet. Inorg. Chem. 2019, 58, 52–56. [Google Scholar] [CrossRef]
- Pointillart, F.; Le Gal, Y.; Golhen, S.; Cador, O.; Ouahab, L. Binuclear gadolinium(III) coordination complex based on bridging tetrathiafulvalenecarboxylate radical cations. Chem. Commun. 2009, 3777–3779. [Google Scholar] [CrossRef]
- Pointillart, F.; Le Guennic, B.; Golhen, S.; Cador, O.; Ouahab, L. Slow magnetic relaxation in radical cation tetrathiafulvalene-based lanthanide(III) dinuclear complexes. Chem. Commun. 2013, 49, 11632–11634. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Ge, J.-Y.; Hua, C.; Jiao, C.-Q.; Wu, Y.; Leong, C.F.; D’Alessandro, D.M.; Liu, T.; Zuo, J.-L. Photo-and electronically switchable spin-crossover iron(II) metal-organic frameworks based on a tetrathiafulvalene ligand. Angewandte Chemie Int. Ed. 2017, 56, 5465–5470. [Google Scholar] [CrossRef]
- Su, J.; Hu, T.-H.; Murase, R.; Wang, H.-Y.; D’Alessandro, D.M.; Kurmoo, M.; Zuo, J.-L. Redox activities of metal-organic frameworks incorporating rare-earth metal chains and tetrathiafulvalene linkers. Inorg. Chem. 2019, 58, 3698–3706. [Google Scholar] [CrossRef]
- Da Cunha, T.T.; Jung, J.; Boulon, M.-E.; Campo, G.; Pointillart, F.; Pereira, C.L.M.; Le Guennic, B.; Cador, O.; Bernot, K.; Pineider, F.; et al. Magnetic poles determinations and robustness of memory effect upon solubilization in a DyIII-based single ion magnet. J. Am. Chem. Soc. 2013, 135, 16332–16335. [Google Scholar] [CrossRef]
- Pointillart, F.; Bernot, K.; Golhen, S.; Le Guennic, B.; Guizouarn, T.; Ouahab, L.; Cador, O. Magnetic Memory in an Isotopically Enriched and Magnetically Isolated Mononuclear Dysprosium Complex. Angewandte Chemie Int. Ed. 2015, 54, 1504–1507. [Google Scholar] [CrossRef]
- Tesi, L.; Salman, Z.; Cimatti, I.; Pointillart, F.; Bernot, K.; Mannini, M.; Sessoli, R. Isotope effects on the spin dynamics of single-molecule magnets probed using muon spin spectroscopy. Chem. Commun. 2018, 54, 7826–7829. [Google Scholar] [CrossRef]
- Flores Gonzalez, J.; Pointillart, F.; Cador, O. Hyperfine coupling and slow magnetic relaxation in isotopically enriched DyIII mononuclear single-molecule magnets. Inorg. Chem. Front. 2019, 6, 1081–1086. [Google Scholar] [CrossRef]
- Llunell, M.; Casanova, D.; Cirera, J.; Bofill, J.M.; Alemany, P.; Alvarez, S. SHAPE (Version 2.1); Electronic Structure Group: Barcelona, Spain, 2013. [Google Scholar]
- Lu, W.; Zhang, Y.; Dai, J.; Zhu, Q.-Y.; Bian, G.-Q.; Zhang, D.-Q. A Radical-radical and metal-metal coupling tetrathiafulvalene derivative in which organic radicals directly coordinate to CuII ions. Eur. J. Inorg. Chem. 2006, 1629–1634. [Google Scholar] [CrossRef]
- Inoue, M.B.; Inoue, M.; Bruck, M.A.; Fernando, Q. Structure of bis(ethylenedithio)tetrathiafulvalenium tribromodicuprate(I), (BEDT-TTF+)CuI2Br3: Coordination of the organic radical cation to the metal ions. J. Chem. Soc. Chem. Commun. 1992, 515. [Google Scholar] [CrossRef]
- Liu, S.-X.; Ambrus, C.; Dolder, S.; Neels, A.; Decurtins, S. A dinuclear Ni(II) complex with two types of intramolecular magnetic couplings: Ni(II)-Ni(II) and Ni(II)-TTF∙+. Inorg. Chem. 2006, 45, 9622–9624. [Google Scholar] [CrossRef]
- Jia, C.; Liu, S.-X.; Tanner, C.; Leiggener, C.; Neels, A.; Sanguinet, L.; Levillain, E.; Leutwyler, S.; Hauser, A.; Decurtins, S. An experimental and computational study on intramolecular charge transfer: A tetrathiafulvalene-fused dipyridophenazine molecule. Chem. Eur. J. 2007, 13, 3804–3812. [Google Scholar] [CrossRef]
- Cosquer, G.; Pointillart, F.; Le Guennic, B.; Le Gal, Y.; Golhen, S.; Cador, O.; Ouahab, L. 3d4f heterobimetallic dinuclear and tetranuclear complexes involving tetrathiafulvalene as ligands: X-ray structures and magnetic and photophysical investigations. Inorg. Chem. 2012, 51, 8488–8501. [Google Scholar] [CrossRef]
- Takamatsu, S.; Ishikawa, T.; Koshihara, S.-Y.; Ishikawa, N. Significant increase of the barrier energy for magnetization reversal of a single-4f-ionic single-molecule magnet by a longitudinal contraction of the coordination space. Inorg. Chem. 2007, 46, 7250–7252. [Google Scholar] [CrossRef]
- Gonidec, M.; Stephen Davies, E.; McMaster, J.; Amabilino, D.B.; Veciana, J. Probing the magnetic properties of three interconvertible redox states of a single-molecule magnet with magnetic circular dichroism spectroscopy. J. Am. Chem. Soc. 2010, 132, 1756–1757. [Google Scholar] [CrossRef]
- Dolinar, B.S.; Gomez-Coca, S.; Alexandropoulos, D.I.; Dunbar, K.R. An air stable radical-bridged dysprosium single molecule magnet and its neutral counterpart: Redox switching of magnetic relaxation dynamics. Chem. Commun. 2017, 53, 2283–2286. [Google Scholar] [CrossRef]
- Cosquer, G.; Pointillart, F.; Golhen, S.; Cador, O.; Ouahab, L. Slow magnetic relaxation in condensed versus dispersed dysprosium(III) mononuclear complexes. Chem. Eur. J. 2013, 19, 7895–7903. [Google Scholar] [CrossRef]
- Vooshin, A.I.; Shavaleev, N.M.; Kazakov, V.P. Chemiluminescence of praseodymium (III), neodymium (III) and ytterbium (III) β-diketonates in solution excited from 1,2-dioxetane decomposition and singlet-singlet energy transfer from ketone to rare-earth β-diketonate. J. Lumin. 2000, 91, 49–58. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision, A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H. A combination of quasirelativistic pseudopotential and ligand field calculations for lanthanoid compounds. Theor. Chim. Acta. 1993, 85, 441–450. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V. Time-dependent density functional theory for molecules in liquid solutions. J. Chem. Phys. 2001, 115, 4708–4717. [Google Scholar] [CrossRef]
- Improta, R.; Barone, V.; Scalmani, G.; Frisch, M.J.A. A state-specific polarizable continuum model time dependent density functional theory method for excited state calculations in solution. J. Chem. Phys. 2006, 125, 054103–054109. [Google Scholar] [CrossRef]
- Allouche, A.R. Gabedit-a graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Fernández Galván, I.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. MOLCAS 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef]
- Roos, B.O.; Taylor, P.R.; Siegbahn, P.E.M. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Malmqvist, P.Å.; Roos, B.O.; Schimmelpfennig, B. The restricted active space (RAS) state interaction approach with spin-orbit coupling. Chem. Phys. Lett. 2002, 357, 230–240. [Google Scholar] [CrossRef]
- Malmqvist, P.-Å.; Roos, B.O. The CASSCF state interaction method. Chem. Phys. Lett. 1989, 155, 189–194. [Google Scholar] [CrossRef]
- Chibotaru, L.F.; Ungur, L. Ab initio calculation of anisotropic magnetic properties of complexes I. Unique definition of pseudospin Hamiltonians and their derivation. J. Chem. Phys. 2012, 137, 064112–064122. [Google Scholar] [CrossRef]
- Chibotaru, L.; Ungur, L.; Soncini, A. The origin of nonmagnetic kramers doublets in the ground state of dysprosium triangles: Evidence for a toroidal magnetic moment. Angewandte Chemie Int. Ed. 2008, 47, 4126–4129. [Google Scholar] [CrossRef]
- Aquilante, F.; Malmqvist, P.-Å.; Pedersen, T.B.; Ghosh, A.; Roos, B.O. Cholesky decomposition-based multiconfiguration second-order perturbation theory (CD-CASPT2): Application to the spin-state energetics of CoIII(diiminato)(NPh). J. Chem. Theory Comput. 2008, 4, 694–702. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.A.; Veryazov, V.; Widmark, P.O. Main group atoms and dimers studied with a new relativistic ANO Basis Set. J. Phys. Chem. A 2004, 108, 2851–2858. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.-A.; Veryazov, V.; Widmark, P.-O. New relativistic ANO Basis Sets for transition metal atoms. J. Phys. Chem. A 2005, 109, 6575–6579. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.; Veryazov, V.; Widmark, P.O.; Borin, A.C. New relativistic atomic natural orbital basis sets for lanthanide atoms with applications to the Ce diatom and LuF3. J. Phys. Chem. A 2008, 112, 11431–11435. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp. Energy (cm−1) | Theo Energy (cm−1) | Osc. | Type | Assignment | Transition | |
|---|---|---|---|---|---|---|
| 1 | 23,200 | 21,044 | 0.23 | ILCT | πTTF→π*bzip | H→L (91%) |
| 21,356 | 0.04 | πTTF→π*tta | H→L + 1 (72%) | |||
| 1+∙ | 12,300 | 12,475 | 0.35 | ID ILCT | πTTF→π*TTF πtta→π*TTF | H-3β→Lβ (75%) H-2β→Lβ (21%) |
| 15,800 | 18,491 | 0.02 | ID | πTTF→π*TTF | H−12β→Lβ (30%) H-8β→Lβ (54%) | |
| 18,900 | 19,844 | 0.02 | ID | πTTF→π*TTF | H−12β→Lβ (49%) H-8β→Lβ (25%) | |
| 21,500 | 22,325 | 0.20 | ILCT | πTTF→π*bzip | Hα→Lα (49%) | |
| 22,900 | 23,823 | 0.06 | ID | πTTF→π*TTF | Hα→L + 5α (37%) Hα→L + 6α (23%) | |
| 24,200 | 24,449 | 0.22 | IA ID ILCT | πbzip→π*bzip πTTF→π*TTF πTTF→π*bzip | H-6α→Lα (12%) H-4α→L+1α (15%) Hα→L+6α (21%) Hα→Lα (20%) | |
| 25,400 | 25,732 | 0.12 | ID | πTTF→π*TTF | Hα→L+5α (25%) Hα→L+6α (21%) | |
| 12+ | 12,000 * | 12,475 | 0.35 | ID ILCT | πTTF→π*TTF πtta→π*TTF | H-3β→Lβ (75%) H-2β→Lβ (21%) |
| 13,400 | 13,620 | 0.66 | ID ILCT | πL→π*TTF | H-4→L (69%) | |
| 14,100 | 14,085 | 0.19 | ILCT | πtta→π*TTF πL→π*TTF | H-5/-6→L (65%) H-4→L (69%) | |
| 14,900 | 14,689 | 0.11 | ILCT | πtta→π*TTF | H-5/-8→L (65%) | |
| 16,200 | 15,063 | 0.05 | ILCT | πtta→π*TTF | H-5/-6/-9/−10→L (66%) | |
| 18,600 * | 19,844 | 0.02 | ID | πTTF→π*TTF | H−12β→Lβ (49%) H-2β→Lβ (25%) | |
| 21,500 * | 22,325 23,823 | 0.20 0.06 | ILCT ID | πTTF→π*bzip πTTF→π*TTF | Hα→Lα (49%) Hα→L + 5α (37%) Hα→L + 6α (23%) | |
| 23,500 | 23,769 | 0.02 | ID | πTTF→π*TTF | H-20→L (95%) | |
| 24,700 * | 24,449 25,732 | 0.220.12 | IA ID ILCT | πbzip→π*bzip πTTF→π*TTF πTTF→π*bzip | H-6α→Lα (12%) H-4α→L + 1α (15%) Hα→L + 5/ + 6α (42%) Hα→Lα (20%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez Garcia, G.; Montigaud, V.; Norel, L.; Cador, O.; Le Guennic, B.; Totti, F.; Pointillart, F. Redox-Active Dysprosium Single-Molecule Magnet: Spectro-Electrochemistry and Theoretical Investigations. Magnetochemistry 2019, 5, 46. https://doi.org/10.3390/magnetochemistry5030046
Fernandez Garcia G, Montigaud V, Norel L, Cador O, Le Guennic B, Totti F, Pointillart F. Redox-Active Dysprosium Single-Molecule Magnet: Spectro-Electrochemistry and Theoretical Investigations. Magnetochemistry. 2019; 5(3):46. https://doi.org/10.3390/magnetochemistry5030046
Chicago/Turabian StyleFernandez Garcia, Guglielmo, Vincent Montigaud, Lucie Norel, Olivier Cador, Boris Le Guennic, Federico Totti, and Fabrice Pointillart. 2019. "Redox-Active Dysprosium Single-Molecule Magnet: Spectro-Electrochemistry and Theoretical Investigations" Magnetochemistry 5, no. 3: 46. https://doi.org/10.3390/magnetochemistry5030046
APA StyleFernandez Garcia, G., Montigaud, V., Norel, L., Cador, O., Le Guennic, B., Totti, F., & Pointillart, F. (2019). Redox-Active Dysprosium Single-Molecule Magnet: Spectro-Electrochemistry and Theoretical Investigations. Magnetochemistry, 5(3), 46. https://doi.org/10.3390/magnetochemistry5030046