Estimation of Absolute Spin Counts in Nitronyl Nitroxide-Bearing Graphene Nanoribbons



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
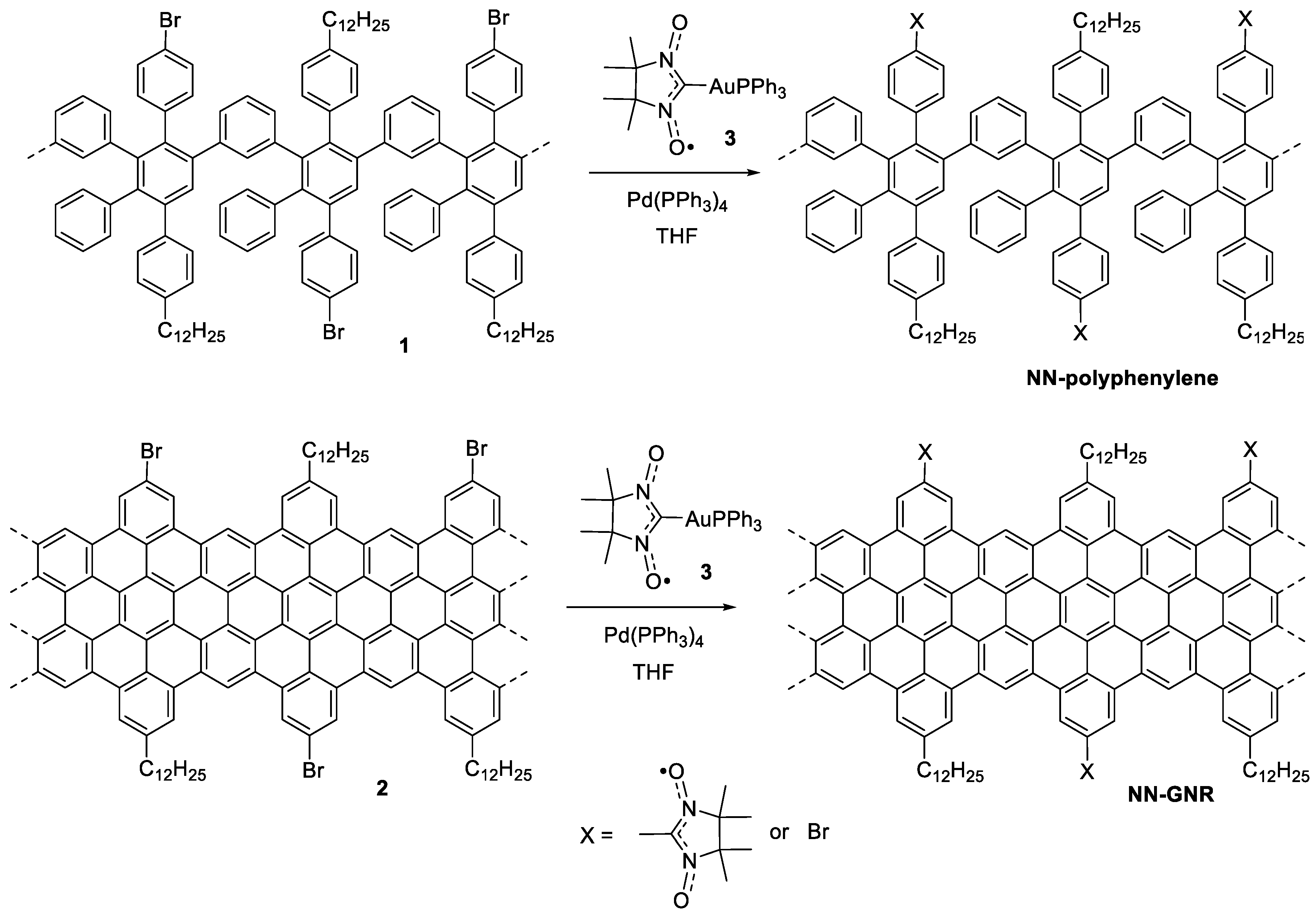
2.1. Compounds Under Study
2.2. Sample Preparation, ESR Measurements and Evaluation of Results
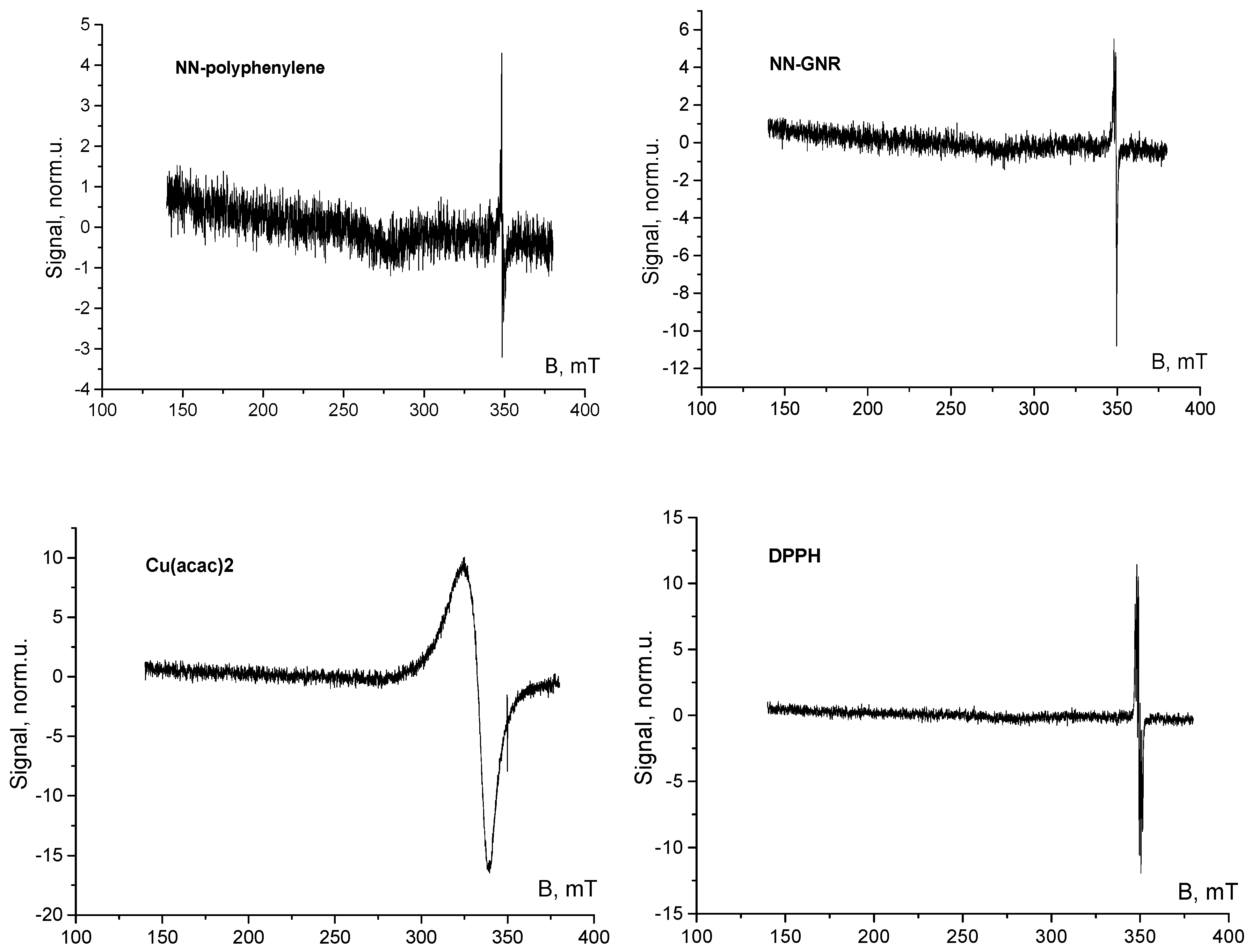
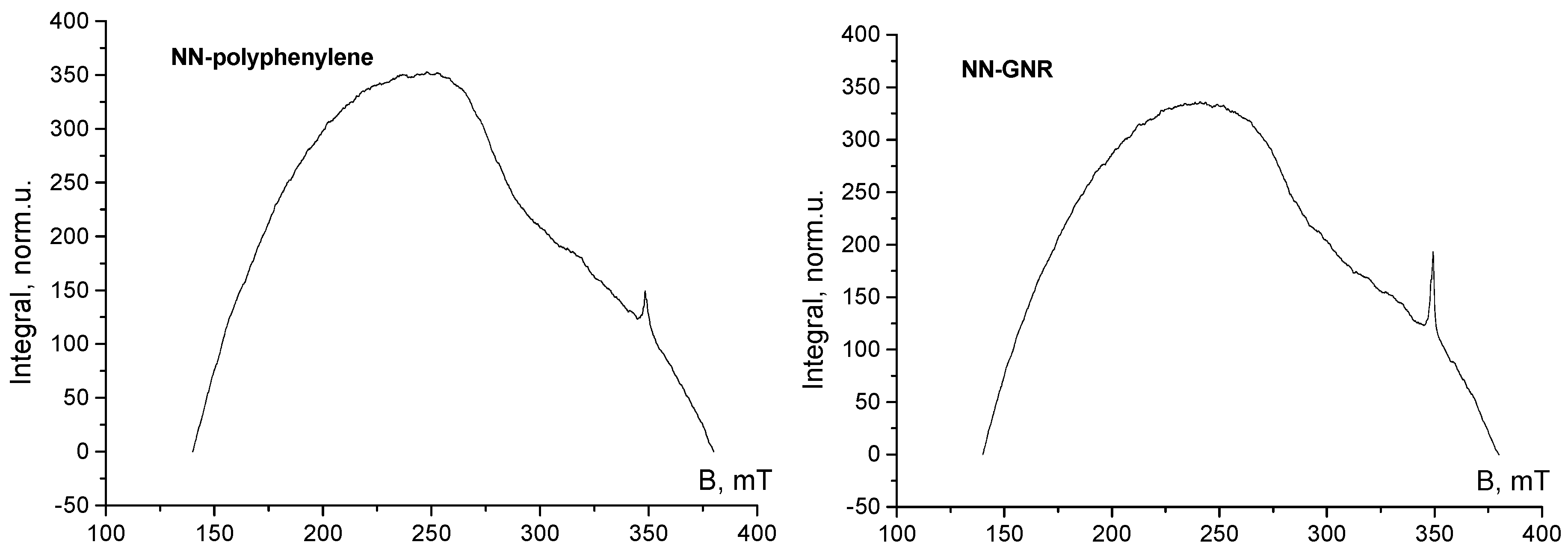
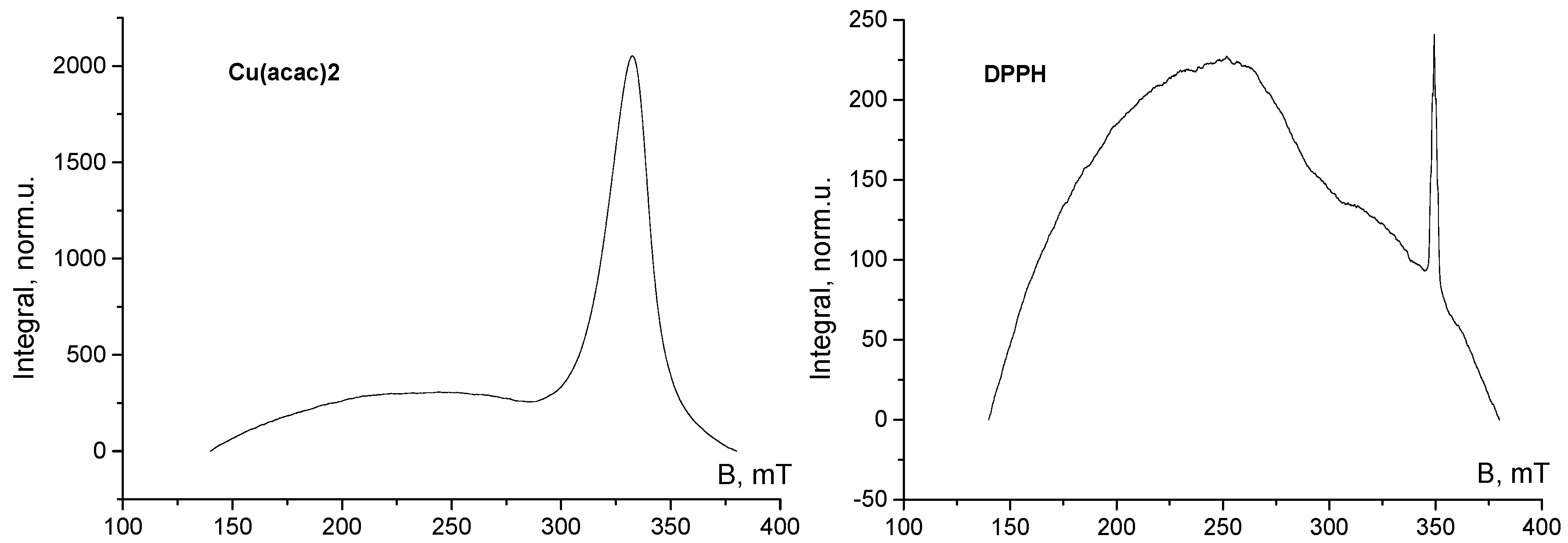
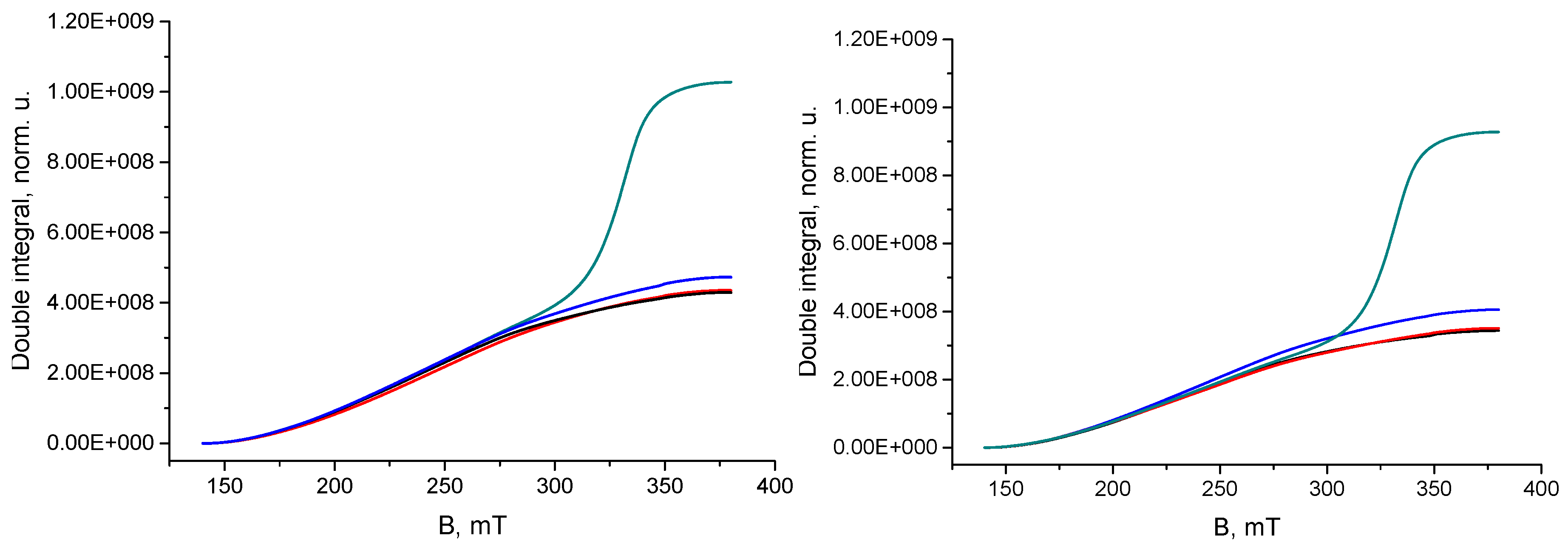
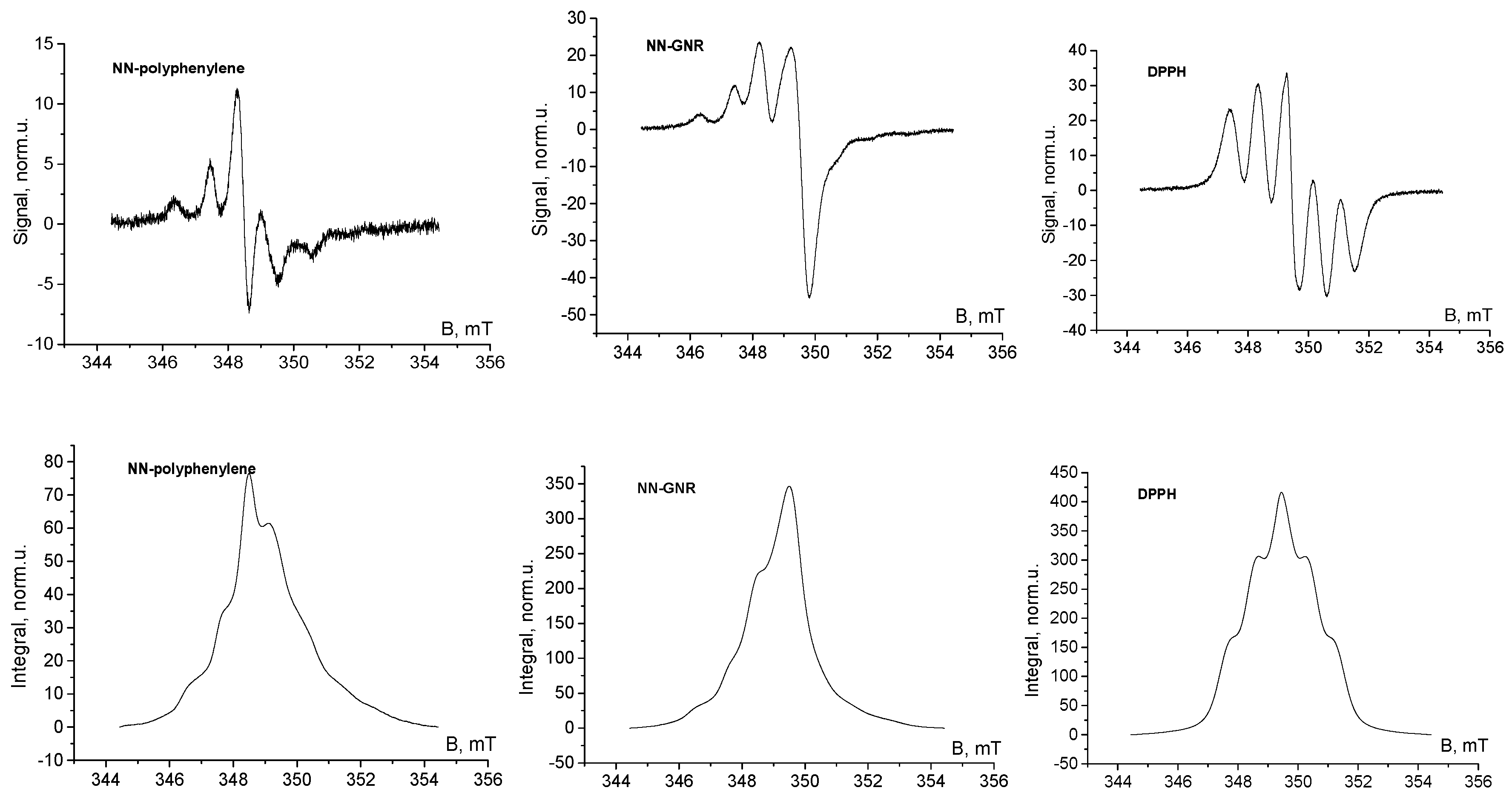
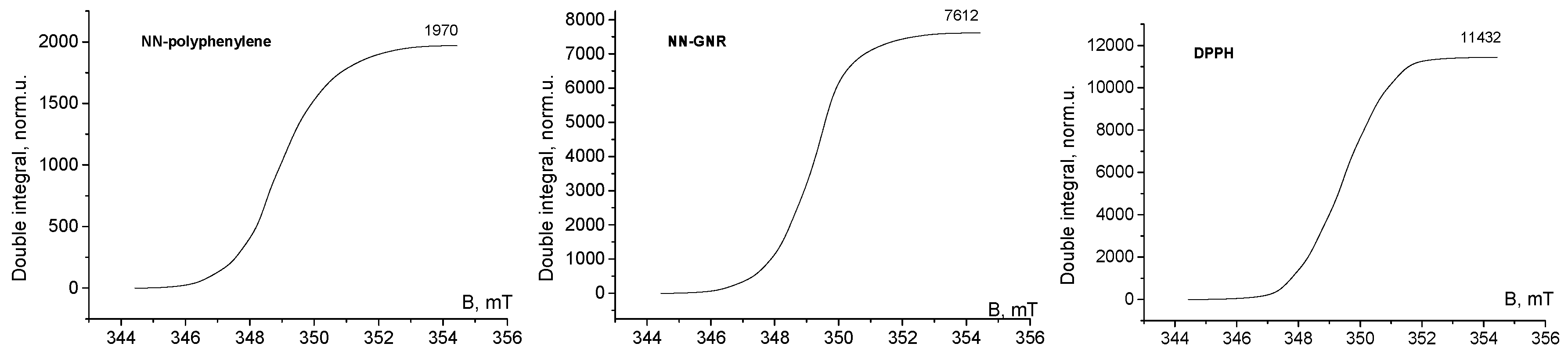
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Recher, P.; Trauzettel, B. Quantum dots and spin qubits in graphene. Nanotechnology 2010, 21, 302001. [Google Scholar] [CrossRef] [PubMed]
- Meunier, V.; Souza Filho, A.G.; Barros, E.B.; Dresselhaus, M.S. Physical properties of low-dimensional sp2-based carbon nanostructures. Rev. Mod. Phys. 2016, 88, 025005. [Google Scholar] [CrossRef]
- Pesin, D.; MacDonald, A.H. Spintronics and pseudospintronics in graphene and topological insulators. Nat. Mater. 2012, 11, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Trauzettel, B.; Bulaev, D.V.; Loss, D.; Burkard, G. Spin qubits in graphene quantum dots. Nat. Phys. 2007, 3, 192–196. [Google Scholar] [CrossRef]
- Kunstmann, J.; Özdoğan, C.; Quandt, A.; Fehske, H. Stability of edge states and edge magnetism in graphene nanoribbons. Phys. Rev. B 2011, 83, 045414. [Google Scholar] [CrossRef]
- Barone, V.; Hod, O.; Scuseria, G.E. Electronic structure and stability of semiconducting graphene nanoribbons. NANO Lett. 2006, 6, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Slota, M.; Keerthi, A.; Myers, W.K.; Tretyakov, E.; Baumgarten, M.; Ardavan, A.; Sadeghi, H.; Lambert, C.J.; Narita, A.; Müllen, K.; et al. Magnetic edge states and coherent manipulation of graphene nanoribbons. Nature 2018, 557, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Narita, A.; Wang, X.-Y.; Feng, X.; Müllen, K. New advances in nanographene chemistry. Chem. Soc. Rev. 2015, 44, 6616–6643. [Google Scholar] [CrossRef] [PubMed]
- Eaton, G.R.; Eaton, S.S.; Barr, D.P.; Weber, R.T. Quantitative EPR; Springer Science & Business Media: New York, NY, USA, 2010; 185p. [Google Scholar]
- Potapov, A.I.; Stass, D.V.; Fursova, E.Y.; Lukzen, N.N.; Romanenko, G.V.; Ovcharenko, V.I.; Molin, Y.N. Searching for the Exchange Shift: A Set of Test Systems. Appl. Magn. Reson. 2008, 35, 43–55. [Google Scholar] [CrossRef]
- Yordanov, N.D. Quantitative EPR spectrometry—“State of the art”. Appl. Magn. Reson. 1994, 6, 241–257. [Google Scholar] [CrossRef]
- Yordanov, N.D.; Ivanova, M. The present state of quantitative EPR spectrometry: The results from an international experiment. Appl. Magn. Reson. 1994, 6, 333–340. [Google Scholar] [CrossRef]
- Yordanov, N.D.; Christova, A. DPPH as a primary standard for quantitative EPR spectrometry. Appl. Magn. Reson. 1994, 6, 341–345. [Google Scholar] [CrossRef]
- Kooser, R.G.; Kirchmann, E.; Matkov, T. Measurements of spin concentration in electron paramagnetic resonance spectroscopy preparation of standard solutions from optical absorption. Concept. Magn. Reson. 1992, 4, 145–152. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stass, D.; Tretyakov, E. Estimation of Absolute Spin Counts in Nitronyl Nitroxide-Bearing Graphene Nanoribbons. Magnetochemistry 2019, 5, 32. https://doi.org/10.3390/magnetochemistry5020032
Stass D, Tretyakov E. Estimation of Absolute Spin Counts in Nitronyl Nitroxide-Bearing Graphene Nanoribbons. Magnetochemistry. 2019; 5(2):32. https://doi.org/10.3390/magnetochemistry5020032
Chicago/Turabian StyleStass, Dmitri, and Evgeny Tretyakov. 2019. "Estimation of Absolute Spin Counts in Nitronyl Nitroxide-Bearing Graphene Nanoribbons" Magnetochemistry 5, no. 2: 32. https://doi.org/10.3390/magnetochemistry5020032
APA StyleStass, D., & Tretyakov, E. (2019). Estimation of Absolute Spin Counts in Nitronyl Nitroxide-Bearing Graphene Nanoribbons. Magnetochemistry, 5(2), 32. https://doi.org/10.3390/magnetochemistry5020032