
Reverse Curve Fitting Approach for Quantitative Deconvolution of Closely Overlapping Triplets in Fourier Transform Nuclear Magnetic Resonance Spectroscopy Using Odd-Order Derivatives

Abstract
1. Introduction
- (I)
- Loose overlap as overlapping degrees γ ≥ 2.0;
- (II)
- Moderate overlap as γ from 1.0 to 2.0;
- (III)
- Close overlap as γ from 0.5 to 1.0;
- (IV)
- Tight overlap as 0 < γ < 0.5.
2. Materials and Methods
2.1. Materials and Curve Fitting Strategy
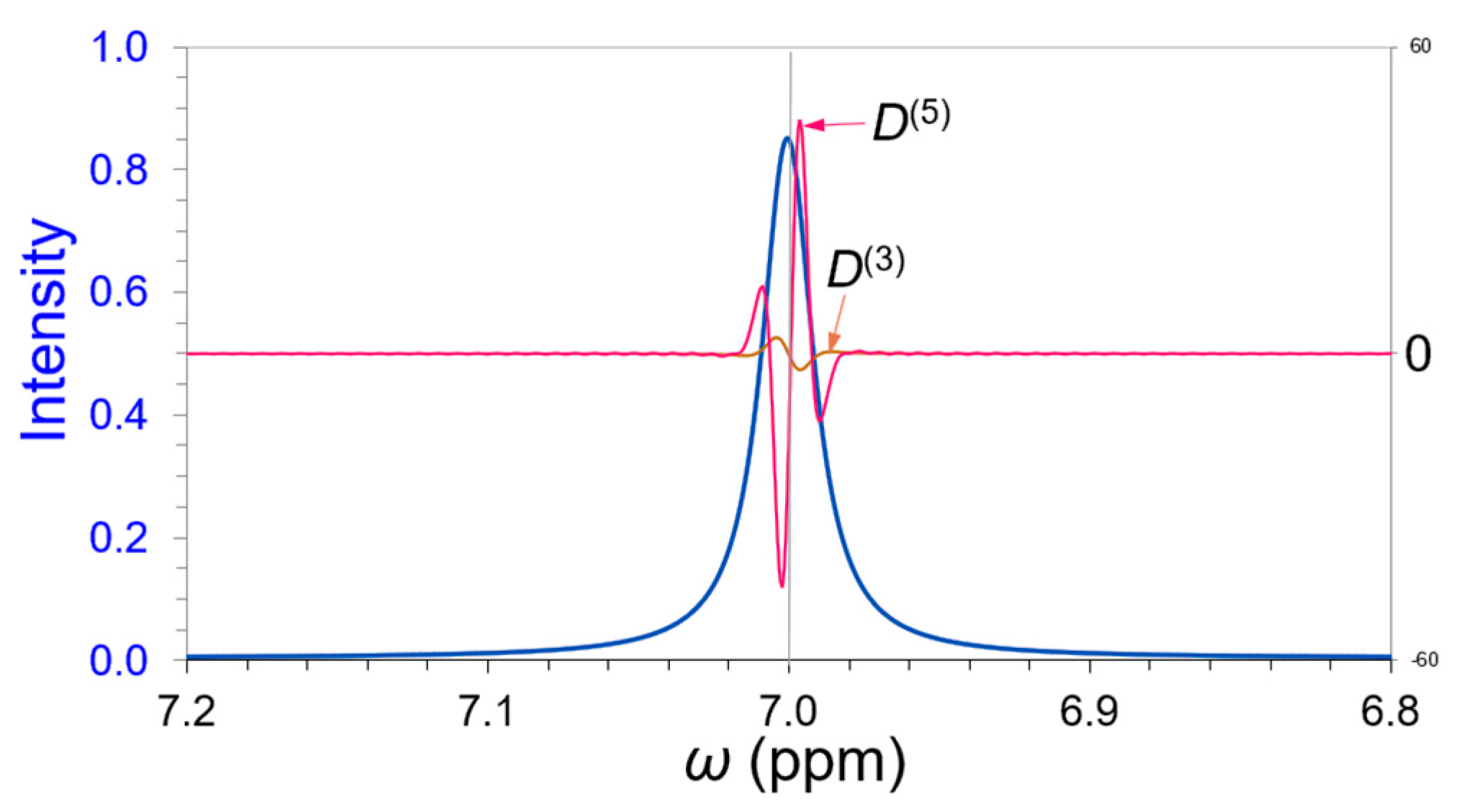
2.2. Peak Position and Derivatives
3. Results
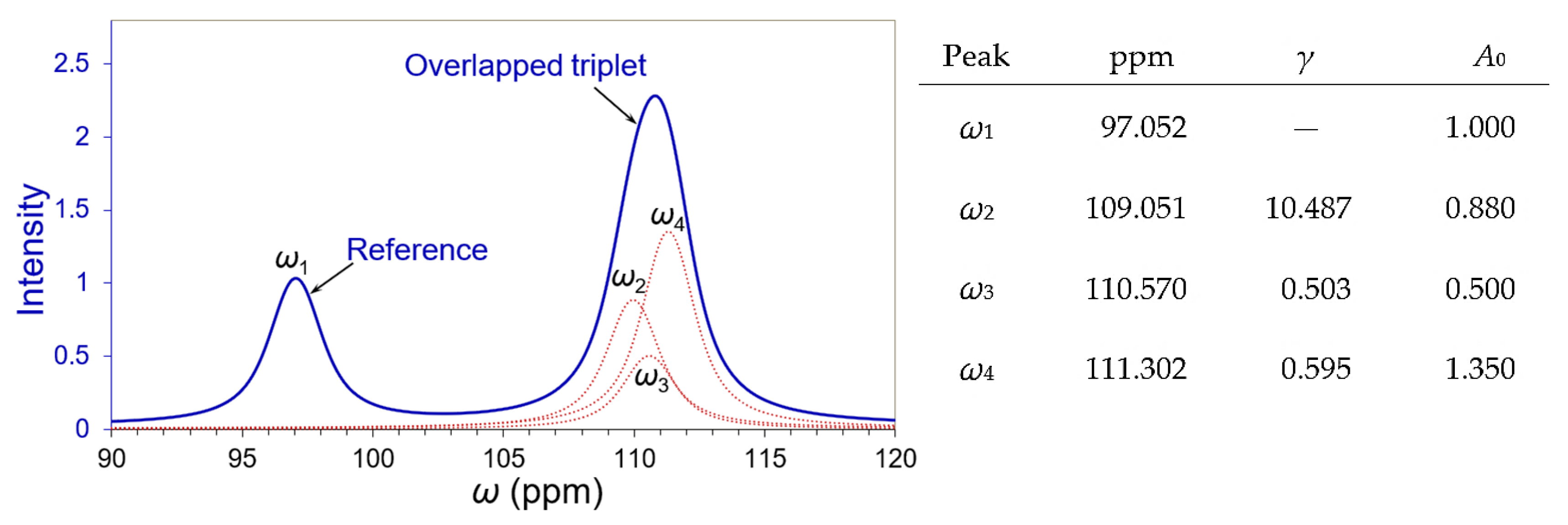
3.1. Deconvolution of a Simulated Overlapping Triplet
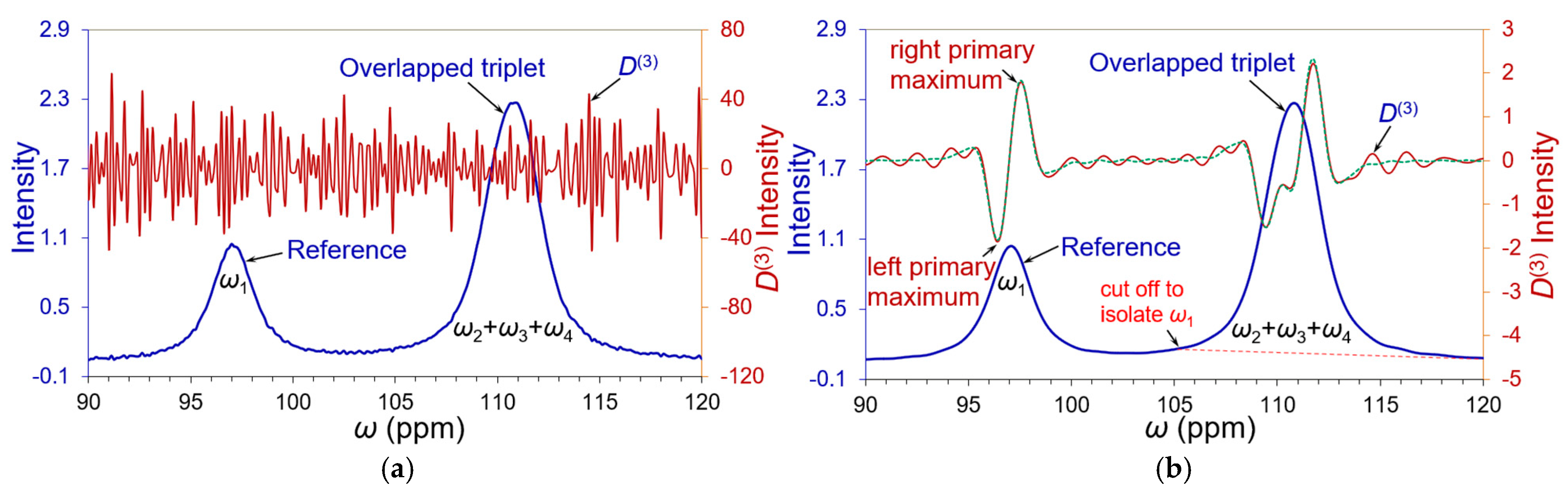
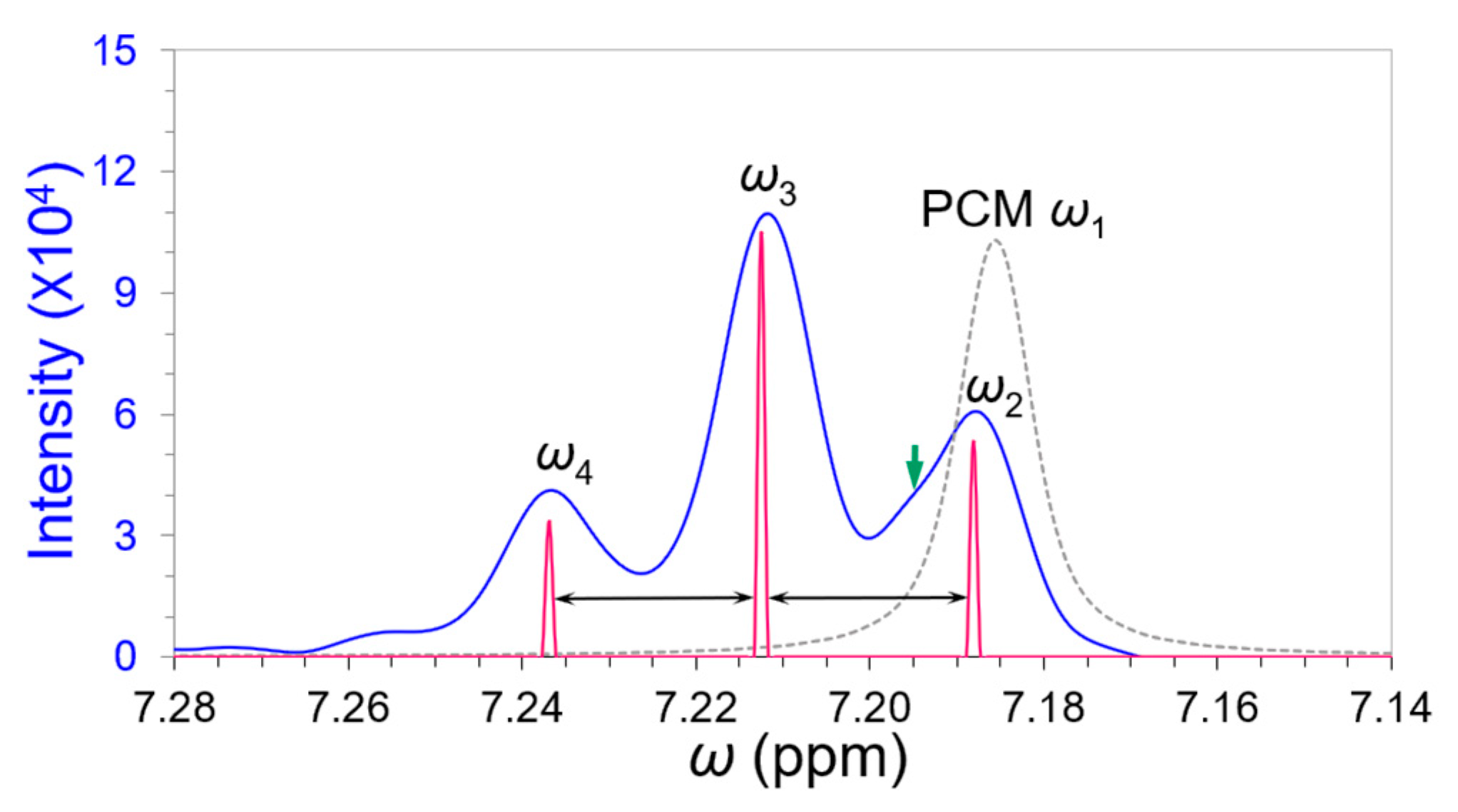
3.2. Partial Curve Matching Strategy and Reverse Curve Fitting Procedure
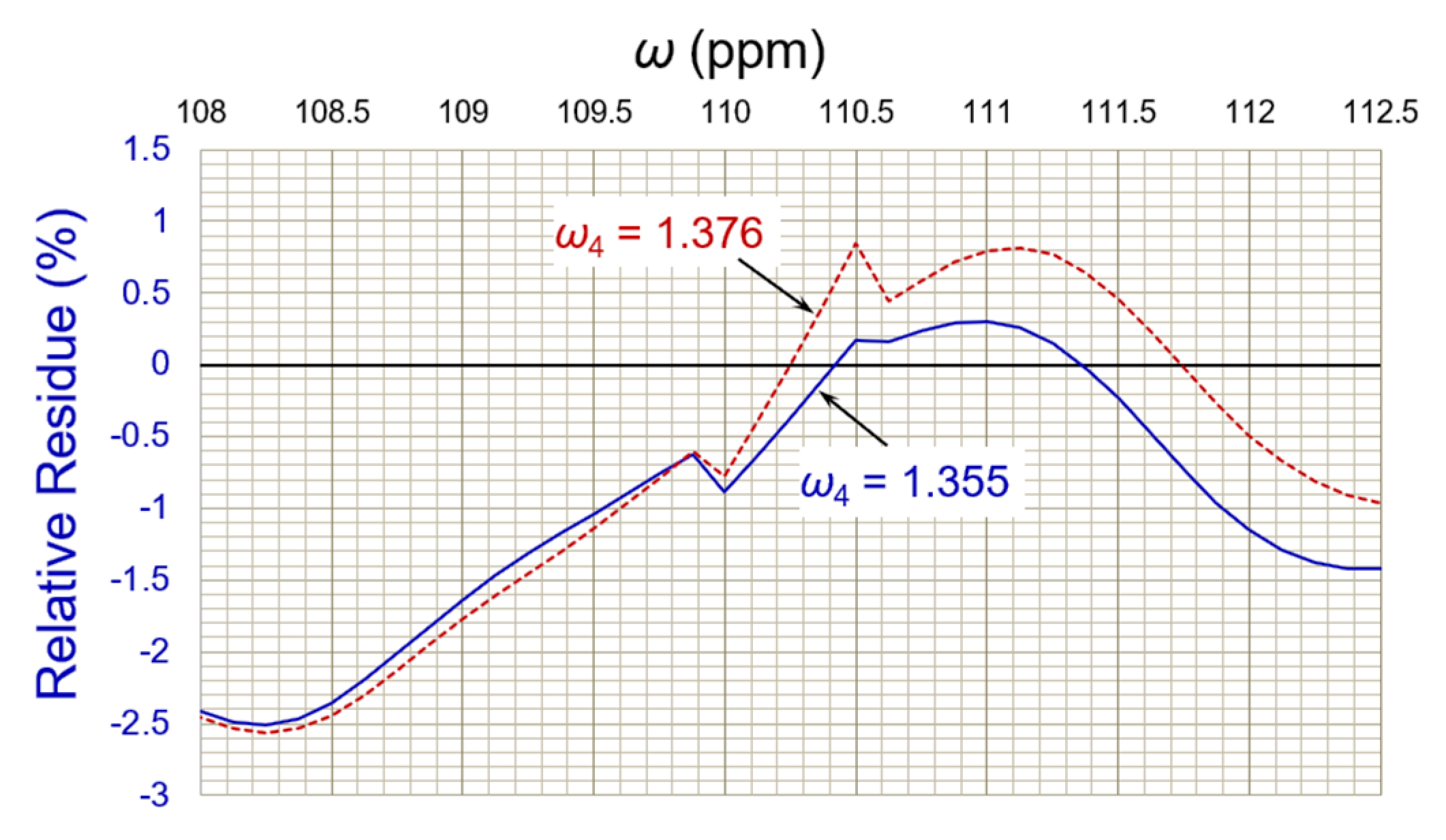
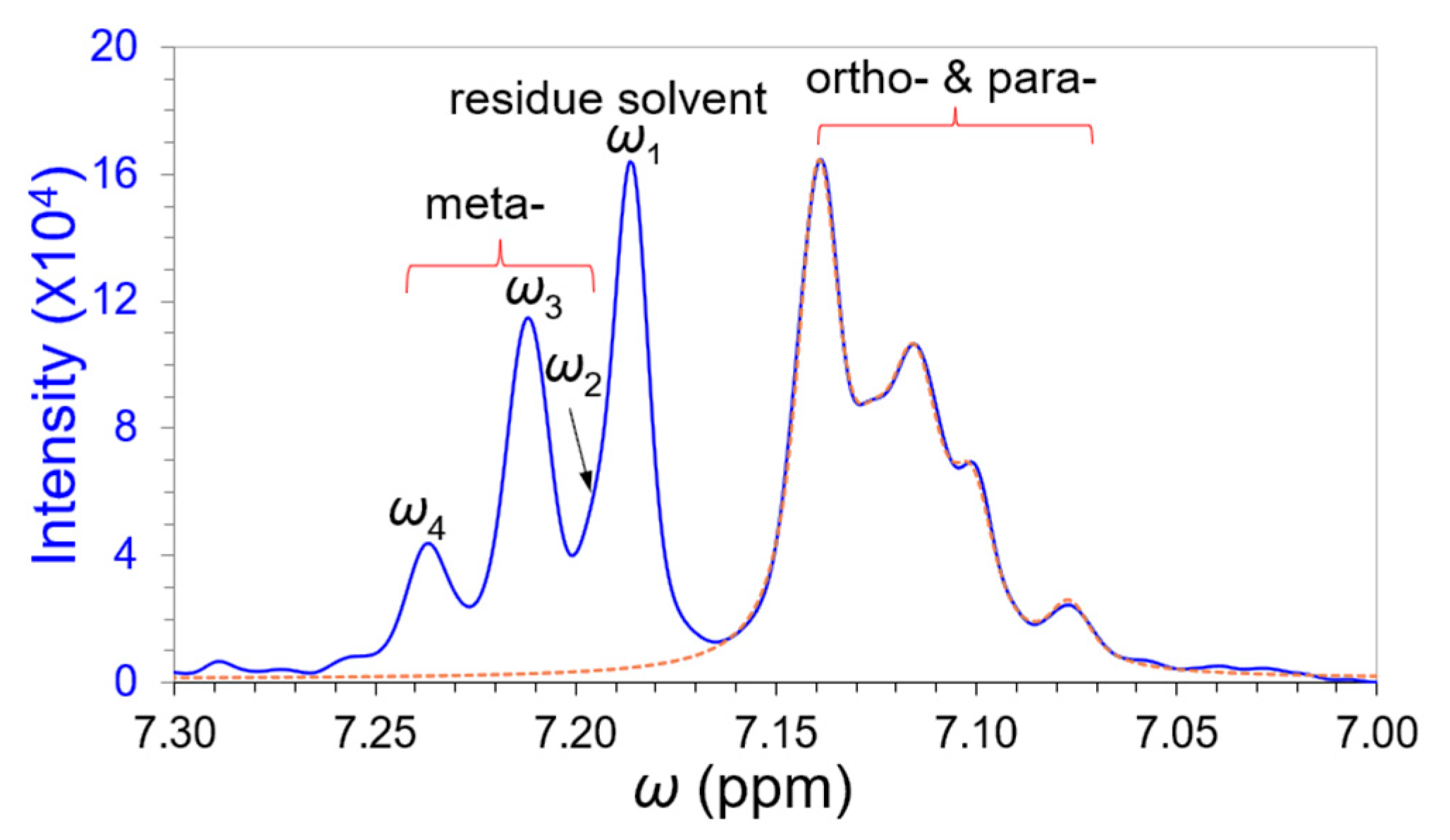
3.3. Deconvolution of the “Closely” Overlapped NMR Peaks
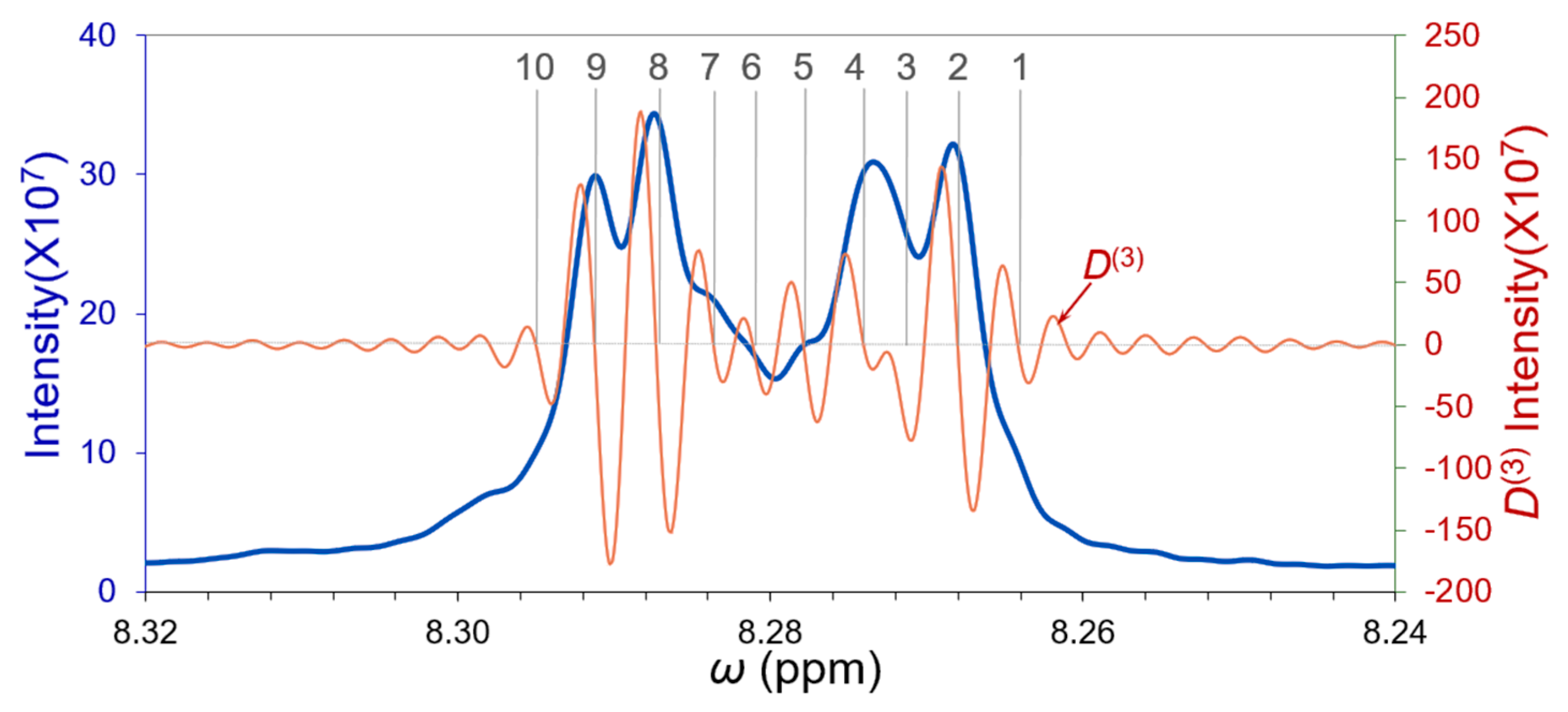
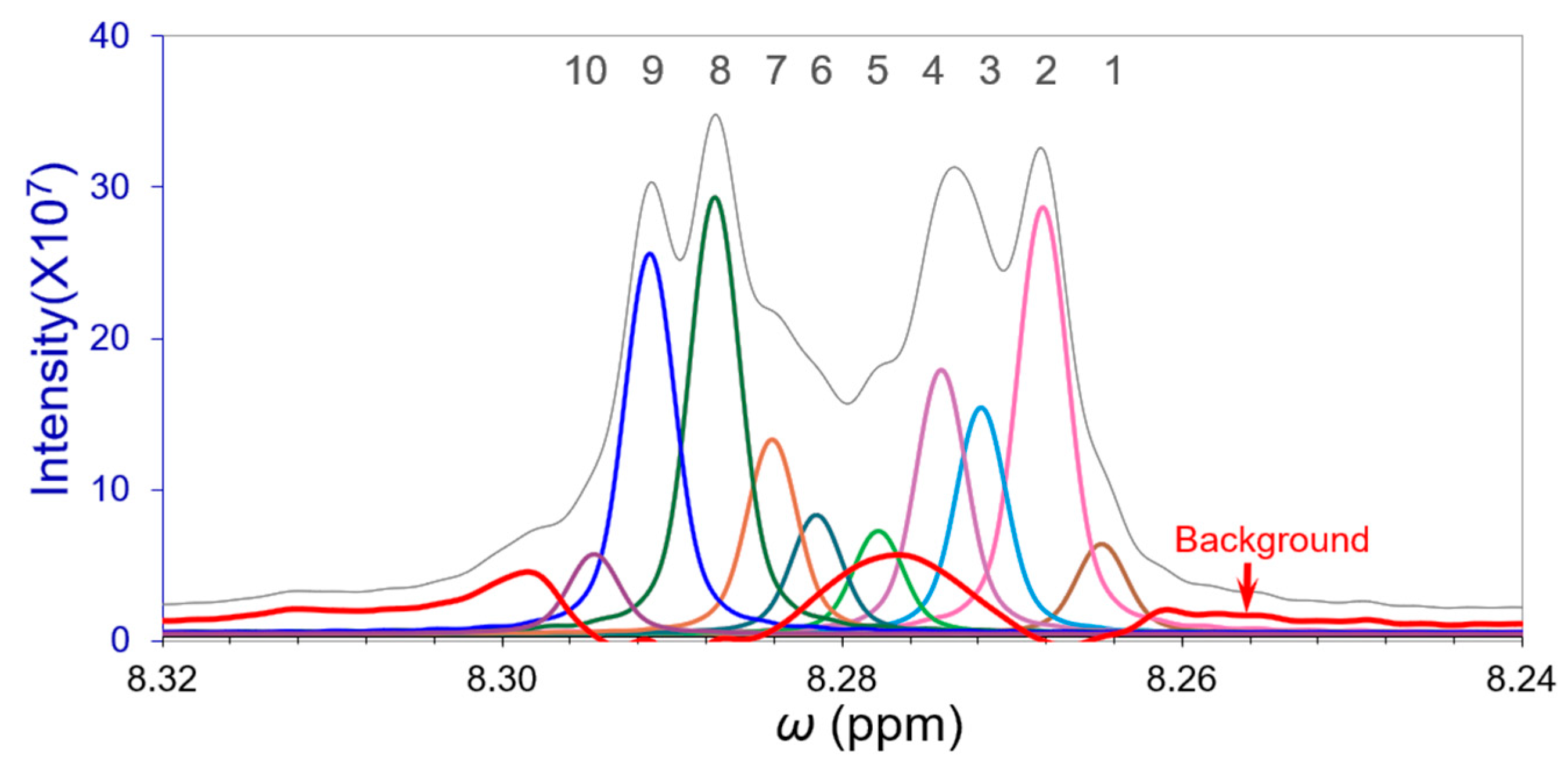
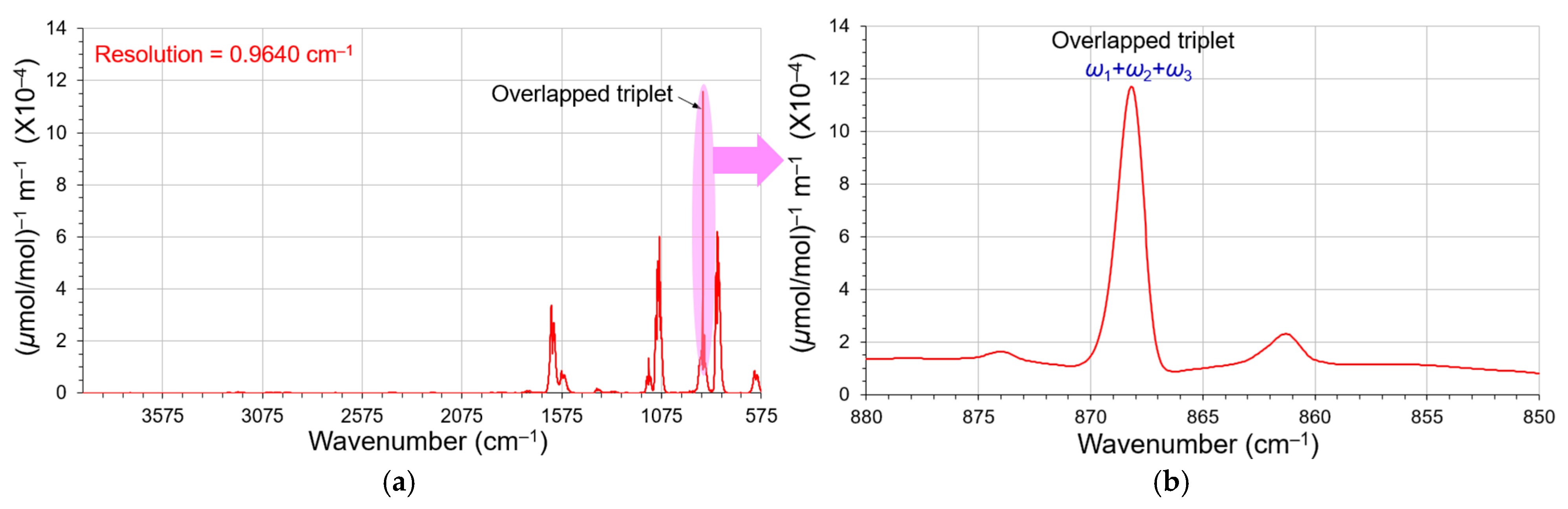
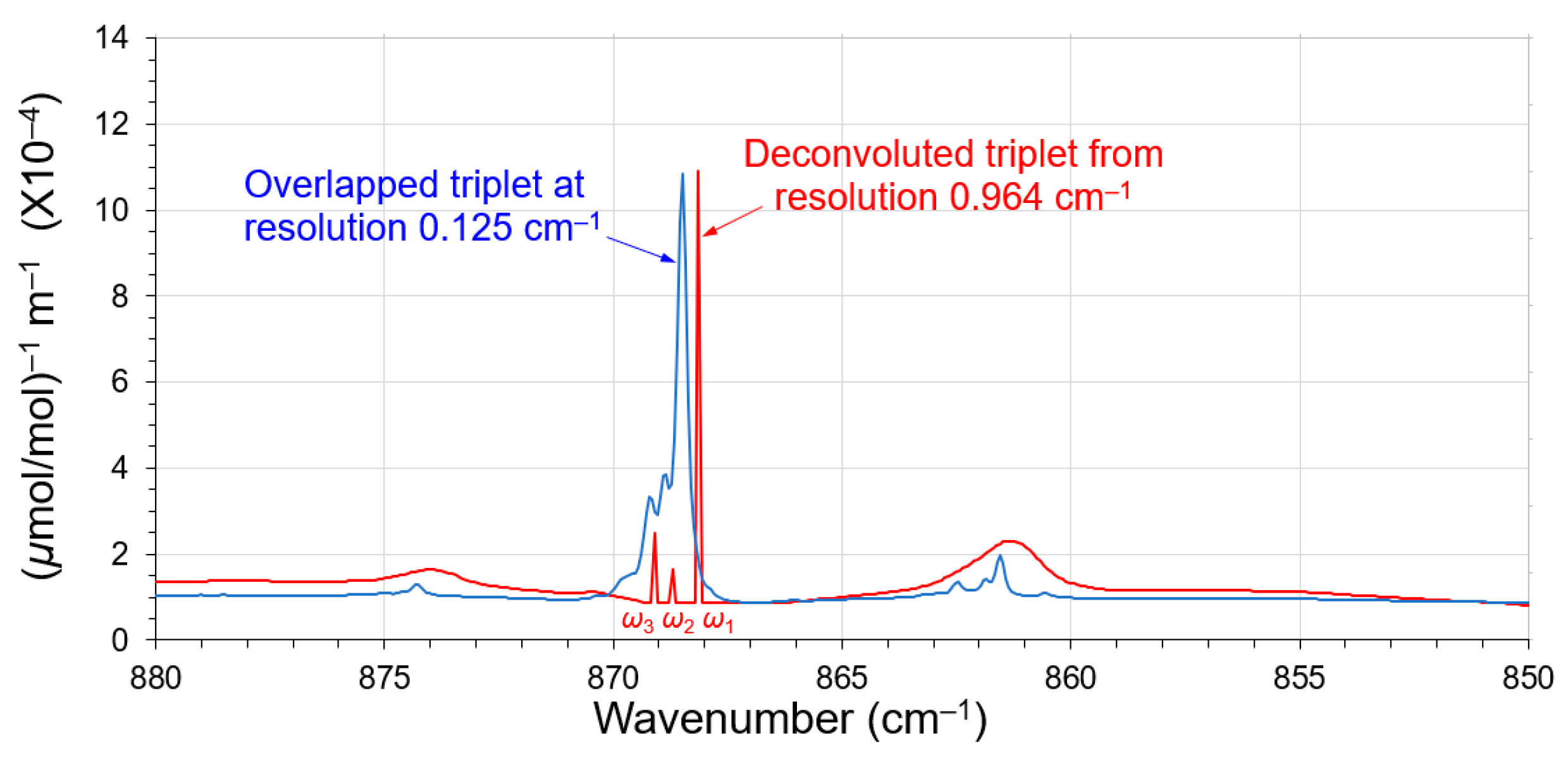
3.4. Deconvolution of the Closely Overlapped FT-IR Peaks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FT | Fourier transform |
| FT-IR | Fourier transform infrared |
| FT-NMR | Fourier transform nuclear magnetic resonance |
| FWHM | Full width at half maximum |
| NMR | Nuclear magnetic resonance |
| PCM | Partial curve matching |
| SNR | Signal-to-noise ratio |
References
- Richards, S.A.; Hollerton, J.C. Essential Practical NMR for Organic Chemistry, 2nd ed.; Wiley: Hoboken, NJ, USA, 2023. [Google Scholar]
- Chu, X.; Huang, Y.; Yun, Y.-H.; Bian, X. Chemometric Methods in Analytical Spectroscopy Technology; Springer: Singapore, 2022. [Google Scholar]
- Dubrovkin, J. Mathematical Processing of Spectral Data in Analytical Chemistry: A Guide to Error Analysis; Cambridge Scholars: Newcastle upon Tyne, UK, 2018. [Google Scholar]
- Martinson, D.G. Quantitative Methods of Data Analysis for the Physical Sciences and Engineering; Cambridge University Press: Cambridge, UK, 2018. [Google Scholar]
- Schmid, N.; Bruderer, S.; Paruzzo, F.; Fischetti, G.; Toscano, G.; Graf, D.; Fey, M.; Henrici, A.; Ziebart, V.; Heitmann, B.; et al. Deconvolution of 1D NMR spectra: A deep learning-based approach. J. Magn. Reson. 2023, 347, 107357. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-W.; Bruschweiler-Li, L.; Hansen, A.L.; Brüschweiler, R. DEEP Picker1D and Voigt Fitter1D: A versatile tool set for the automated quantitative spectral deconvolution of complex 1D-NMR spectra. Magn. Reson. 2023, 4, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.S.; Paluru, N.; Yalavarthy, P.K. Model resolution-based deconvolution for improved quantitative susceptibility mapping. NMR Biomed. 2024, 37, e5055. [Google Scholar] [CrossRef] [PubMed]
- Wenchel, L.; Gampp, O.; Riek, R. Super-resolution NMR spectroscopy. J. Magn. Reson. 2024, 366, 107746. [Google Scholar] [CrossRef] [PubMed]
- Venetos, M.C.; Elkin, M.; Delaney, C.; Hartwig, J.F.; Persson, K.A. Deconvolution and analysis of the 1H NMR spectra of crude reaction mixtures. J. Chem. Inf. Model. 2024, 64, 3008–3020. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.B.; Andersen, M.R.; Siewertsen, M.-M.; Bro, R.; Strube, M.L.; Gotfredsen, G.H. NMR-Onion—a transparent multi-model based 1D NMR deconvolution algorithm. Heliyon 2024, 10, e36998. [Google Scholar] [CrossRef]
- Chen, S.-P.; Taylor, S.M.; Huang, S.; Zheng, B. Application of odd-order derivatives in Fourier transform nuclear magnetic resonance spectroscopy toward quantitative deconvolution. ACS Omega 2024, 9, 36518–36530. [Google Scholar] [CrossRef] [PubMed]
- Callon, M.; Malär, A.A.; Pfister, S.; Římal, V.; Weber, M.E.; Wiegand, T.; Zehnder, J.; Chávez, M.; Cadalbert, R.; Deb, R.; et al. Biomolecular solid state NMR spectroscopy at 1200 MHz: The gain in resolution. J. Biomol. NMR 2021, 75, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Vandeginste, B.G.M.; De Galan, L. Critical evaluation of curve fitting in infrared spectrometry. Anal. Chem. 1975, 47, 2124–2132. [Google Scholar] [CrossRef]
- Maddams, M.F. The Scope and Limitations of Curve Fitting. Appl. Spectrosc. 1980, 34, 245–267. [Google Scholar] [CrossRef]
- Kauppinen, J.; Partanen, J. Fourier Transforms in Spectroscopy; Wiley-VCH: Berlin, Germany, 2001; pp. 14–16, 172. [Google Scholar]
- Acorn NMR Inc. NUTS NMR Software, Demonstration version 20000813; Acorn NMR Inc.: Livermore, CA, USA, 2000.
- Chu, P.M.; Guenther, F.R.; Rhoderick, G.C.; Lafferty, W.J. Quantitative Infrared Database. Also see: The NIST Quantitative Infrared Database. J. Res. Natl. Inst. Stand. Technol. 1999, 104, 59–81. Available online: https://webbook.nist.gov/chemistry/quant-ir/ (accessed on 19 September 2024). [CrossRef]
- Marshall, A.G.; Verdun, F.R. Fourier Transforms in NMR, Optical and Mass Spectrometry: A User’s Handbook; Elsevier: Amsterdam, The Netherlands, 1990; p. 75. [Google Scholar]
- Huguenin, R.L.; Jones, J.L. Intelligent information extraction from reflectance spectra: Absorption band positions. J. Geophys. Res. Solid Earth 1986, 91, 9585–9598. [Google Scholar] [CrossRef]
- Gorkovskiy, A.; Thurber, K.R.; Tycko, R.; Wickner, R.B. Locating folds of the in-register parallel β-sheet of the Sup35p prion domain infectious amyloid. Proc. Natl. Acad. Sci. USA 2014, 111, E4615–E4622. [Google Scholar] [CrossRef] [PubMed]
- Dubrovkin, J. Derivative Spectroscopy; Cambridge Scholars: Newcastle upon Tyne, UK, 2021; pp. 3–5, 18. [Google Scholar]
- Kito, K. Digital Fourier Analysis: Advance Techniques; Springer: New York, NY, USA, 2015; pp. 105–119. [Google Scholar]
- Wolberg, J. Data Analysis Using the Method of Least Squares: Extracting the Most Information from Experiments; Springer: Berlin/Heidelberg, Germany, 2006; pp. 34–36. [Google Scholar]
- Reich, H.J. Spin-Spin Splitting: J-Coupling. 2017. UW-Madison. Available online: https://organicchemistrydata.org/hansreich/resources/nmr/ (accessed on 7 October 2024).
- Shimizu, T.; Hashi, K.; Goto, A.; Tansyo, M.; Kiyoshi, T.; Matsumoto, S.; Wada, H.; Fujito, T.; Hasegawa, K.-i.; Kirihara, N.; et al. Overview of the development of high-resolution 920 MHz NMR in NIMS. Physica B 2004, 346/347, 528–530. [Google Scholar] [CrossRef]
- O’Haver, T. A Pragmatic Introduction to Signal Processing with Applications in Scientific Measurement. July 2021. Available online: https://terpconnect.umd.edu/~toh/spectrum/CurveFittingC.html (accessed on 27 May 2025).
- Ye, T.; Zheng, C.; Zhang, S.; Gowda, G.A.N.; Vitek, O.; Raftery, D. “Add to Subtract”: A simple method to remove complex background signals from the 1H nuclear magnetic resonance spectra of mixtures. Anal. Chem. 2012, 84, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Alexandersson, E.; Sandstrom, C.; Lundqvist, L.C.E.; Nestor, G. Band-selective NMR experiments for suppression of unwanted signals in complex mixtures. RSC Adv. 2020, 10, 32511–32515. [Google Scholar] [CrossRef] [PubMed]
- Callon, M.; Luder, D.; Malär, A.A.; Wiegand, T.; Římal, V.; Lecoq, L.; Böckmann, A.; Samoson, A.; Meier, B.H. High and fast: NMR protein–proton side-chain assignments at 160 kHz and 1.2 GHz. Chem. Sci. 2023, 14, 10824–10834. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.P.; Comisarow, M.B. The phase dependence of magnitude spectra. J. Magn. Reson. 1987, 72, 139–142. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1. Segregate an overlapping band and include a well-separated peak nearby (as a reference) if possible. 2. Set an analytical threshold according to noise level (expected SNR). 3. Perform denoising and/or smoothing with a proper denoising technology, such as Kauppinen’s band-pass filtering and smoothing in time domain signals [15] (p. 172). 4. Isolate the reference peak or predominated peak and estimate its peak width by Hilbert transform. Its position and intensity are deduced from the zero-crossing point and primary maximum of 3rd-order derivative in the frequency spectrum by a partial curve matching (PCM) strategy to imitate a harmonic signal in the time domain. Refine the peak position and intensity to reach the appropriate match with the derivative primary maximum of the reference peak via FT of the imitated time signal. 5. Implement the reverse curve fitting procedure by partially curve matching the primary maxima of the edge peaks and progressively reduce the members in the overlapping band to filter out independent peaks individually. 6. Repeat Step 5 until the intensity and peak width of each filtered-out peak are convergent. |
| SNR | Peak | ω2 | ω3 | ω4 | |||
|---|---|---|---|---|---|---|---|
| ppm | A0 | ppm | A0 | ppm | A0 | ||
| Real value | 109.951 | 0.880 | 110.570 | 0.500 | 111.302 | 1.350 | |
| 20:1 | Deconvoluted (deviation) | 109.949 (−0.002%) | 0.882 (+0.227%) | 110.573 (+0.003%) | 0.481 (−3.800%) | 111.297 (−0.004%) | 1.355 (+0.370%) |
| Filtered * | 109.950 | 0.886 | 110.547 | 0.480 | 111.308 | 1.353 | |
| 40:1 | Deconvoluted (deviation) | 109.954 (+0.003%) | 0.886 (+0.682%) | 110.605 (+0.032%) | 0.498 (−0.400%) | 111.315 (+0.012%) | 1.342 (−0.593%) |
| Filtered * | 109.949 | 0.899 | 110.591 | 0.500 | 111.314 | 1.343 | |
| Peak | ω1 | ω2 | Unknown | ω3 | ω4 | |
|---|---|---|---|---|---|---|
| Overlapping γ | – | 0.383 | 3.577 | 3.512 | ||
| Cycle-6 | ppm | 7.186 | 7.188 | 7.213 | 7.237 | |
| A0 | 103098 | 53298 | 104942 | 33428 | ||
| FWHM | 0.01004 | 0.01359 | 0.01406 | 0.01359 | ||
| Filtered * | ppm | 7.186 | 7.187 | 7.195 | 7.213 | 7.237 |
| A0 | 101440 | 53375 | 1958 | 104436 | 33291 |
| Cycles | Peaks | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cycle-1 | ppm A0 (×107) | 8.2648 6.094 | 8.2682 27.565 | 8.2718 9.845 | 8.2741 17.376 | 8.2777 6.558 | 8.2816 5.334 | 8.2842 9.997 | 8.2876 31.055 | 8.2914 27.207 | 8.2945 5.194 |
| Cycle-2 | ppm A0 (×107) | 8.2648 5.975 | 8.2682 27.920 | 8.2718 14.314 | 8.2742 17.580 | 8.2778 6.713 | 8.2816 5.891 | 8.2842 11.694 | 8.2875 28.813 | 8.2913 24.885 | 8.2946 5.326 |
| Cycle-3 | ppm A0 (×107) | 8.2647 5.960 | 8.2682 27.877 | 8.2718 15.038 | 8.2742 17.540 | 8.2779 6.653 | 8.2815 7.336 | 8.2841 12.333 | 8.2875 28.640 | 8.2913 24.897 | 8.2946 5.276 |
| Cycle-4 | ppm A0 (×107) | 8.2647 5.957 | 8.2682 27.918 | 8.2718 14.934 | 8.2742 17.436 | 8.2779 6.720 | 8.2815 7.657 | 8.2841 12.588 | 8.2875 28.613 | 8.2913 24.906 | 8.2946 5.271 |
| Cycle-5 | ppm A0 (×107) | 8.2647 5.952 | 8.2682 27.932 | 8.2718 14.803 | 8.2742 17.327 | 8.2779 6.813 | 8.2815 7.818 | 8.2841 12.766 | 8.2875 28.582 | 8.2913 24.906 | 8.2946 5.260 |
| Filtered * | ppm A0 (×107) | 8.2647 5.952 | 8.2682 27.933 | 8.2718 17.166 | 8.2742 21.799 | 8.2777 12.017 | 8.2814 10.741 | 8.2840 13.312 | 8.2874 28.582 | 8.2913 24.906 | 8.2945 5.320 |
| Overlapping γ | 1.950 | 2.006 | 1.337 | 2.061 | 2.006 | 1.448 | 1.894 | 2.117 | 1.838 | ||
| Peak | ω1 | ω2 | ω3 | |
|---|---|---|---|---|
| Overlapping γ | – | 0.896 | 0.602 | |
| Cycle-0 | cm−1 | 868.150 | – | 869.062 |
| A0 | 9.815 × 104 | – | 1.615 × 104 | |
| Cycle-1 | cm−1 | 868.136 | 868.669 | 869.067 |
| A0 | 10.014 × 104 | 0.714 × 104 | 1.593 × 104 | |
| Cycle-2 | cm−1 | 868.136 | 868.693 | 869.067 |
| A0 | 10.014 × 104 | 0.763 × 104 | 1.593 × 104 | |
| Filtered * | cm−1 | 868.134 | 868.686 | 869.062 |
| A0 | 10.005 × 104 | 0.763 × 104 | 1.593 × 104 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-P.; Taylor, S.M.; Huang, S.; Zheng, B. Reverse Curve Fitting Approach for Quantitative Deconvolution of Closely Overlapping Triplets in Fourier Transform Nuclear Magnetic Resonance Spectroscopy Using Odd-Order Derivatives. Magnetochemistry 2025, 11, 50. https://doi.org/10.3390/magnetochemistry11060050
Chen S-P, Taylor SM, Huang S, Zheng B. Reverse Curve Fitting Approach for Quantitative Deconvolution of Closely Overlapping Triplets in Fourier Transform Nuclear Magnetic Resonance Spectroscopy Using Odd-Order Derivatives. Magnetochemistry. 2025; 11(6):50. https://doi.org/10.3390/magnetochemistry11060050
Chicago/Turabian StyleChen, Shu-Ping, Sandra M. Taylor, Sai Huang, and Baoling Zheng. 2025. "Reverse Curve Fitting Approach for Quantitative Deconvolution of Closely Overlapping Triplets in Fourier Transform Nuclear Magnetic Resonance Spectroscopy Using Odd-Order Derivatives" Magnetochemistry 11, no. 6: 50. https://doi.org/10.3390/magnetochemistry11060050
APA StyleChen, S.-P., Taylor, S. M., Huang, S., & Zheng, B. (2025). Reverse Curve Fitting Approach for Quantitative Deconvolution of Closely Overlapping Triplets in Fourier Transform Nuclear Magnetic Resonance Spectroscopy Using Odd-Order Derivatives. Magnetochemistry, 11(6), 50. https://doi.org/10.3390/magnetochemistry11060050