
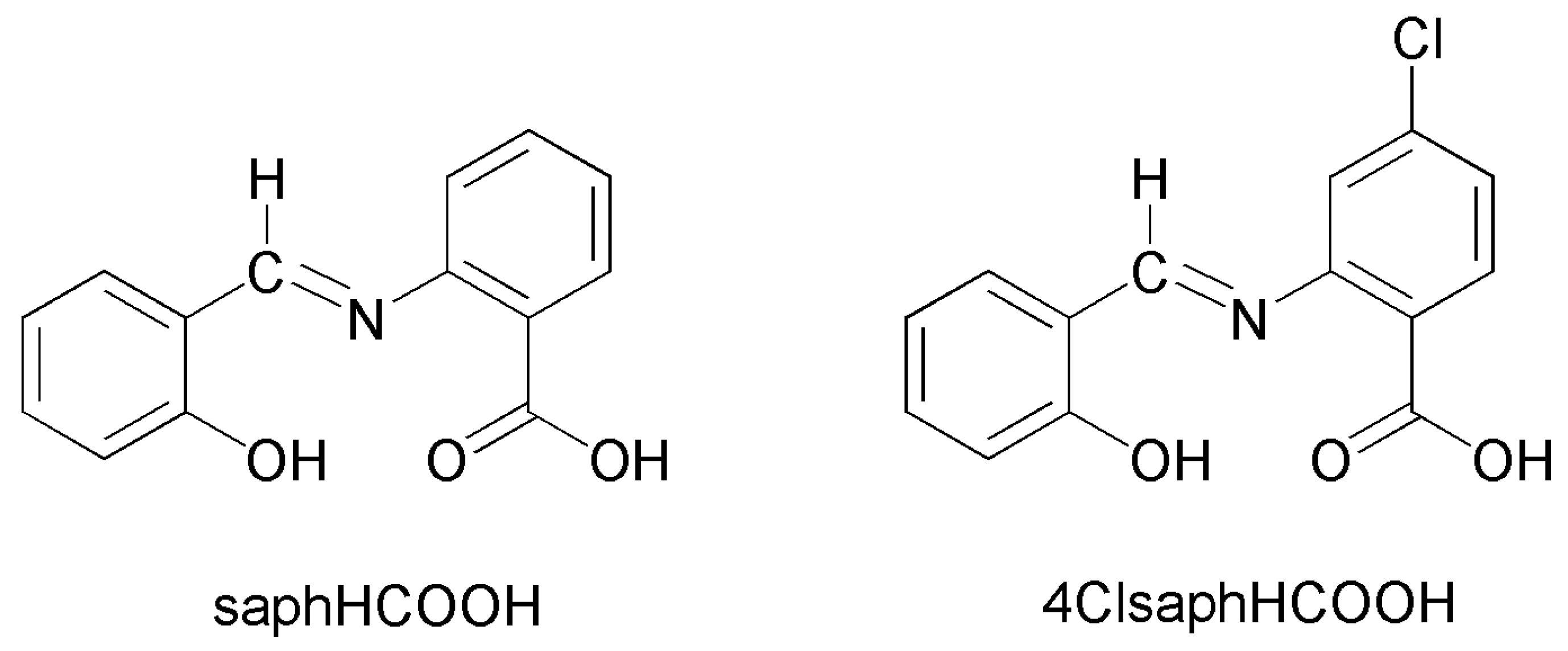
Tetradentate NOO′O″ Schiff-Base Ligands as a Platform for the Synthesis of Heterometallic CdII-FeIII and CdII-CrIII Coordination Clusters †


 ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
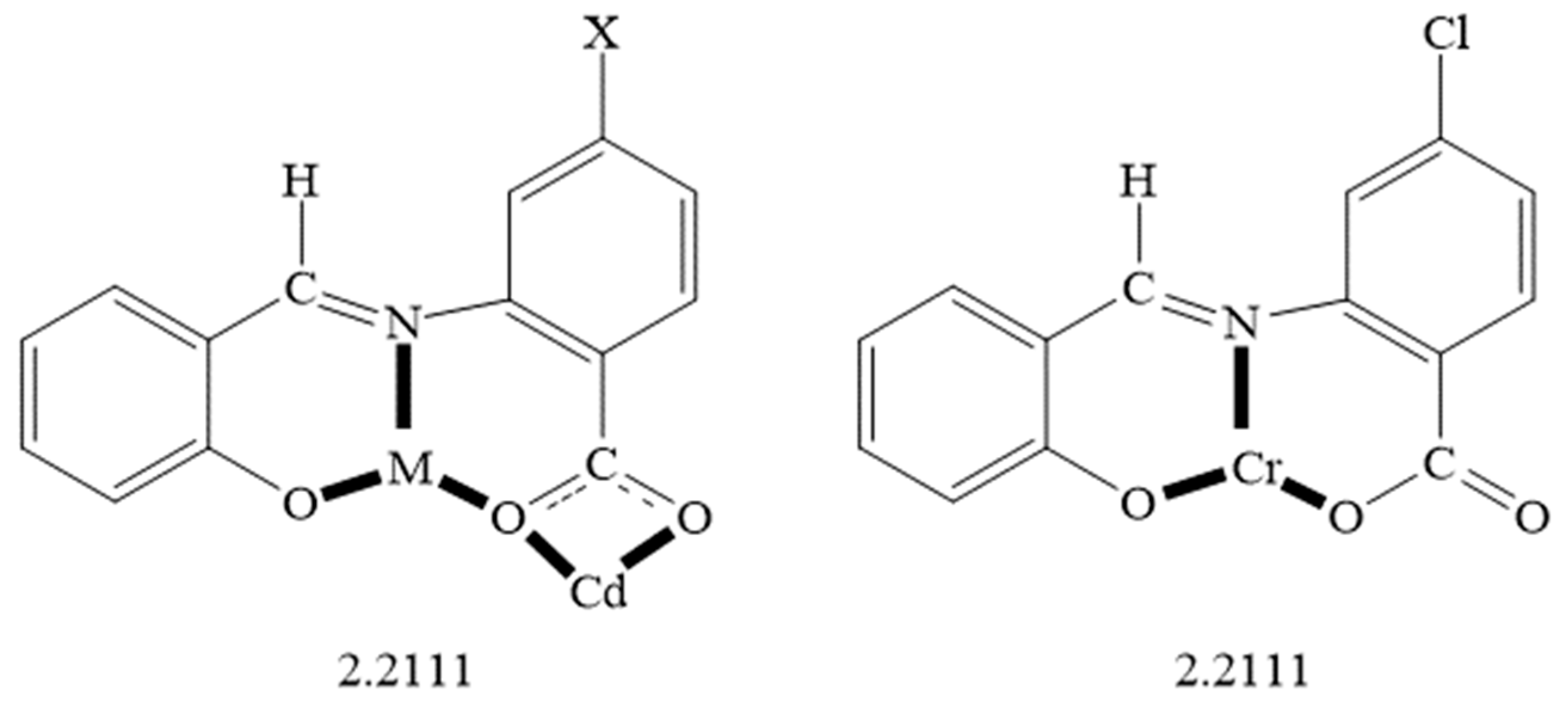
2.1. Synthetic Comments
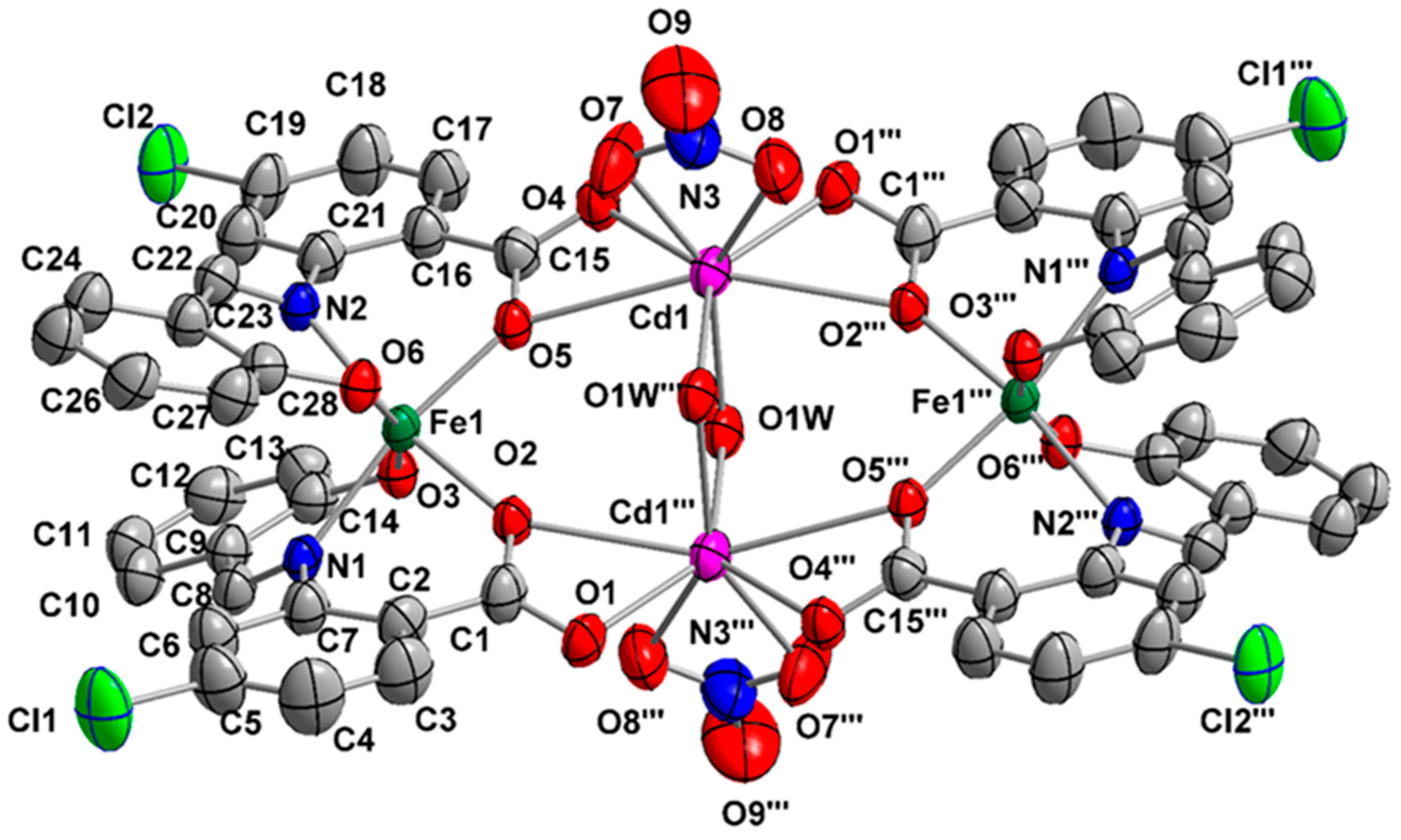
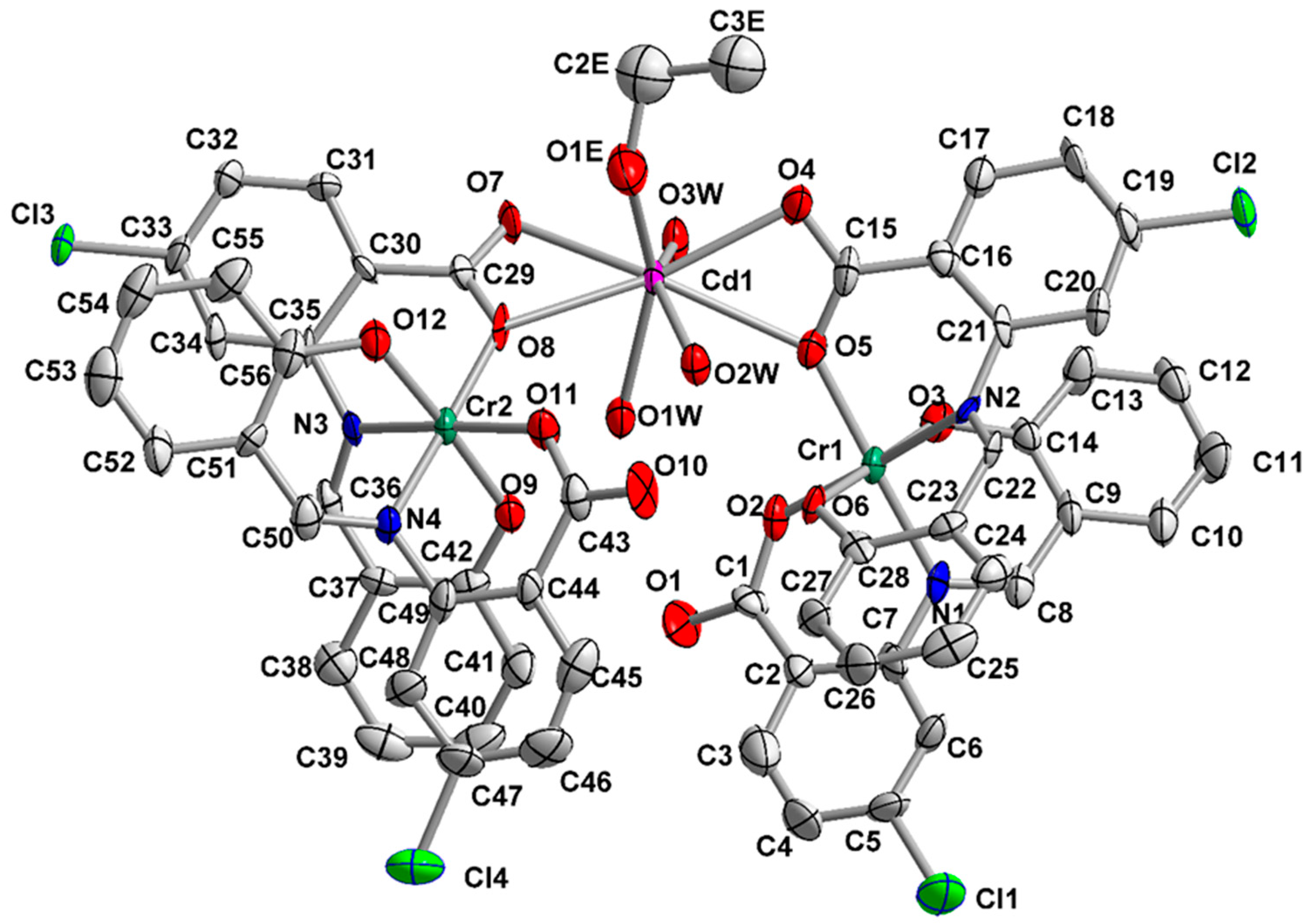
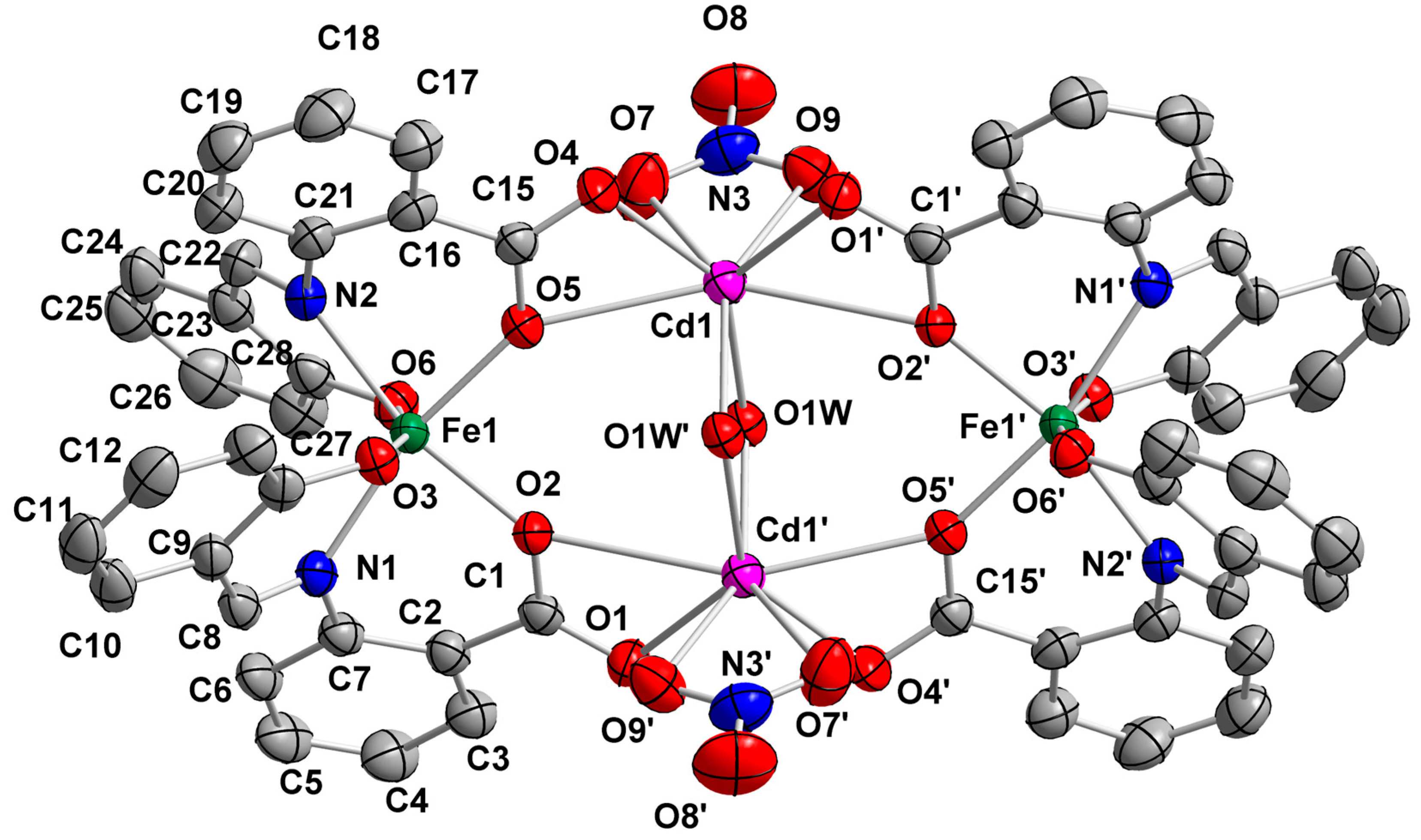
2.2. Description of Structures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interatomic Distances | (Å) | Bond Angles | (°) |
|---|---|---|---|
| Cd1…Cd1′ | 3.659(2) | O1W–Cd1–O1W′ | 76.8(1) |
| Fe1…Fe1′ | 8.122(2) | O1′–Cd1–O4 | 82.3(1) |
| Cd1…Fe1 | 4.371(2) | O1′–Cd1–O1W | 133.5(1) |
| Cd1…Fe1′ | 4.535(2) | O1′–Cd1–O1W′ | 91.2(1) |
| Cd1–O1′ | 2.285(4) | O4–Cd1–O5 | 52.8(1) |
| Cd1–O4 | 2.302(4) | O4–Cd1–O9 | 112.7(2) |
| Cd1–O1W | 2.308(4) | O5–Cd1–O1W′ | 77.1(1) |
| Cd1–O1W′ | 2.362(4) | O5–Cd1–O7 | 75.5(1) |
| Cd1–O5 | 2.593(3) | O5–Cd1–O9 | 127.8(1) |
| Cd1–O7 | 2.449(5) | O7–Cd1–O9 | 52.7(2) |
| Cd1–O9 | 2.379(4) | O2–Fe1–N2 | 166.2(2) |
| Cd1–O2′ | 2.731(4) | O3–Fe1–O6 | 163.1(2) |
| Fe1–O2 | 2.002(3) | O5–Fe1–N1 | 166.0(2) |
| Fe1–O3 | 1.952(4) | O7–N3–O9 | 116.6(5) |
| Fe1–O5 | 2.010(3) | O7–N3–O8 | 123.0(6) |
| Fe1–O6 | 1.950(4) | O8–N3–O9 | 120.4(6) |
| Fe1–N1 | 2.131(4) | Cd1–O1W–Cd1′ | 103.2(1) |
| Fe1–N2 | 2.139(4) |


| Interatomic Distances | (Å) | Bond Angles | (°) |
|---|---|---|---|
| Cd1…Cd1‴ | 3.703(2) | O1W–Cd1–O1W‴ | 75.9(1) |
| Fe1…Fe1‴ | 8.007(1) | O1‴–Cd1–O4 | 83.9(1) |
| Cd1…Fe1 | 4.460(1) | O1‴–Cd1–O1W | 96.0(1) |
| Cd1…Fe1‴ | 4.361(2) | O4–Cd1–O1W | 93.3(1) |
| Cd1–O1‴ | 2.333(3) | O4–Cd1–O7 | 82.8(1) |
| Cd1–O4 | 2.302(2) | O4–Cd1–O8 | 122.6(1) |
| Cd1–O1W | 2.345(3) | O7–Cd1–O8 | 52.0(1) |
| Cd1–O1W‴ | 2.352(3) | O7–Cd1–O1W | 148.7.(1) |
| Cd1–O2‴ | 2.589(2) | O7–Cd1–O1W‴ | 85.1(1) |
| Cd1–O5 | 2.668(3) | O8—Cd1–O1W | 143.9(1) |
| Cd1–O7 | 2.396(4) | O2–Fe1–N2 | 169.0(1) |
| Cd1–O8 | 2.424(3) | O3–Fe1–O6 | 163.1(2) |
| Fe1–O2 | 1.997(2) | O5–Fe1–N1 | 169.6(1) |
| Fe1–O3 | 1.957(2) | O7–N3–O8 | 117.0(4) |
| Fe1–O5 | 2.001(2) | O7–N3–O9 | 120.6(5) |
| Fe1–N1 | 2.117(3) | O8–N3–O9 | 122.2(5) |
| Fe1–N2 | 2.124(3) | Cd1–O1W–Cd1‴ | 104.1(1) |
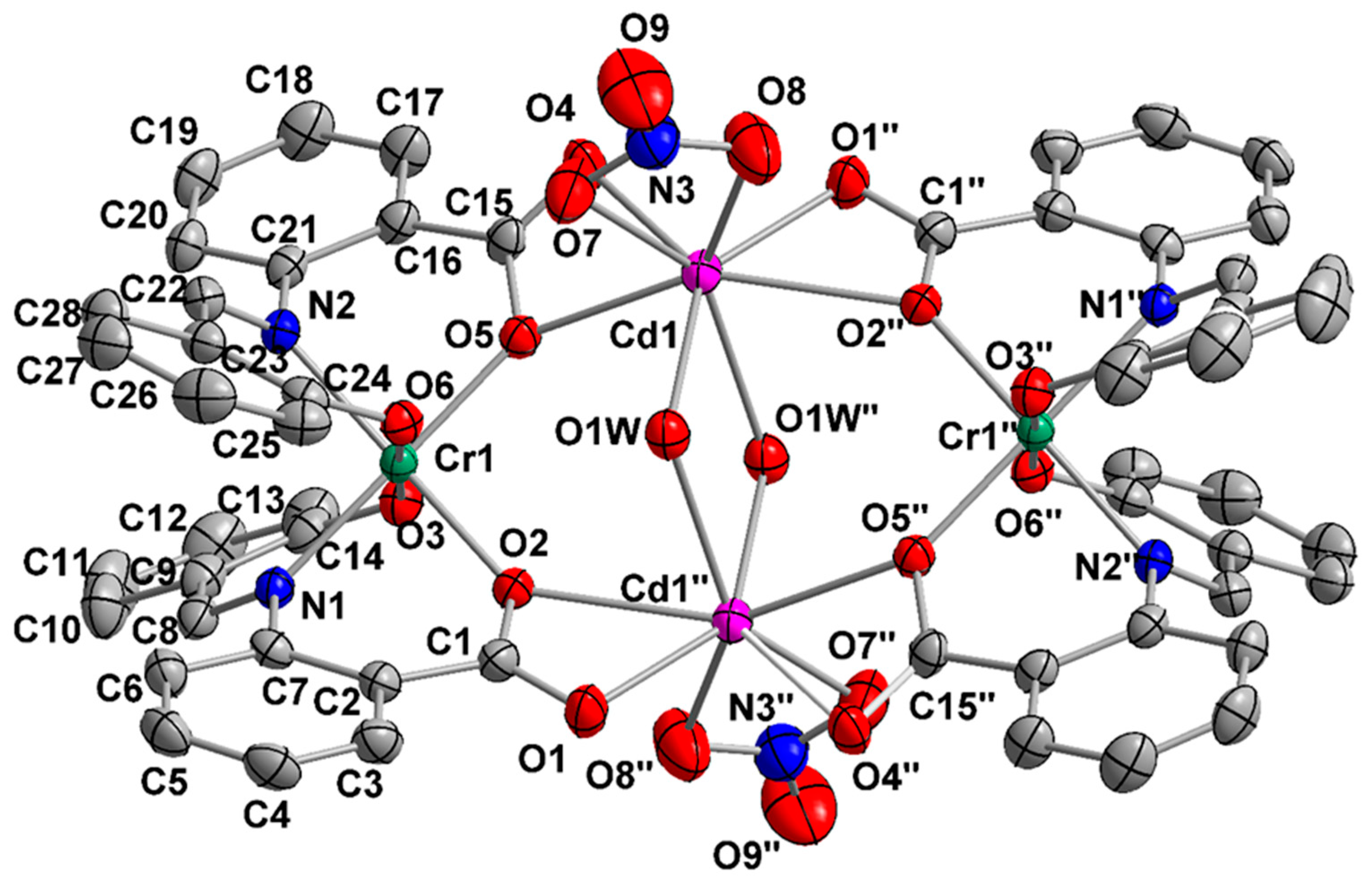
| Interatomic Distances | (Å) | Bond Angles | (°) |
|---|---|---|---|
| Cd1…Cd1″ | 3.946(2) | O1W–Cd1–O1W″ | 72.7(1) |
| Cr1…Cr1″ | 7.321(1) | O1″–Cd1–O4 | 79.8(1) |
| Cd1…Cr1 | 4.080(1) | O1″–Cd1–O1W | 141.6(1) |
| Cd1…Cr1″ | 4.235(2) | O2″–Cd1–O5 | 148.2(1) |
| Cd1–O1″ | 2.270(3) | O4–Cd1–O5 | 54.0(1) |
| Cd1–O4 | 2.417(2) | O4–Cd1–O8 | 110.5(1) |
| Cd1–O1W | 2.343(3) | O5–Cd1–O7 | 83.3(1) |
| Cd1–O1W″ | 2.554(3) | O5–Cd1–O8 | 134.5.(1) |
| Cd1–O5 | 2.379(2) | O7–Cd1–O8 | 51.7(1) |
| Cd1–O7 | 2.459(3) | O8–Cd1–O1W | 91.8(1) |
| Cd1–O8 | 2.377(4) | O3–Cr1–O6 | 178.0(1) |
| Cd1–O2″ | 2.514(2) | O2–Cr1–N2 | 173.3(1) |
| Cr1–O2 | 1.979(2) | O5–Cr1–N1 | 176.8(1) |
| Cr1–O3 | 1.946(2) | O3–Cr1–O5 | 88.6(2) |
| Cr1–O5 | 2.001(2) | O7–N3–O8 | 115.6(4) |
| Cr1–O6 | 1.931(2) | O7–N3–O9 | 121.7(4) |
| Cr1–N1 | 2.007(3) | O8–N3–O9 | 122.7(4) |
| Cr1–N2 | 2.005(5) | Cd1–O1W–Cd1″ | 102.3(1) |
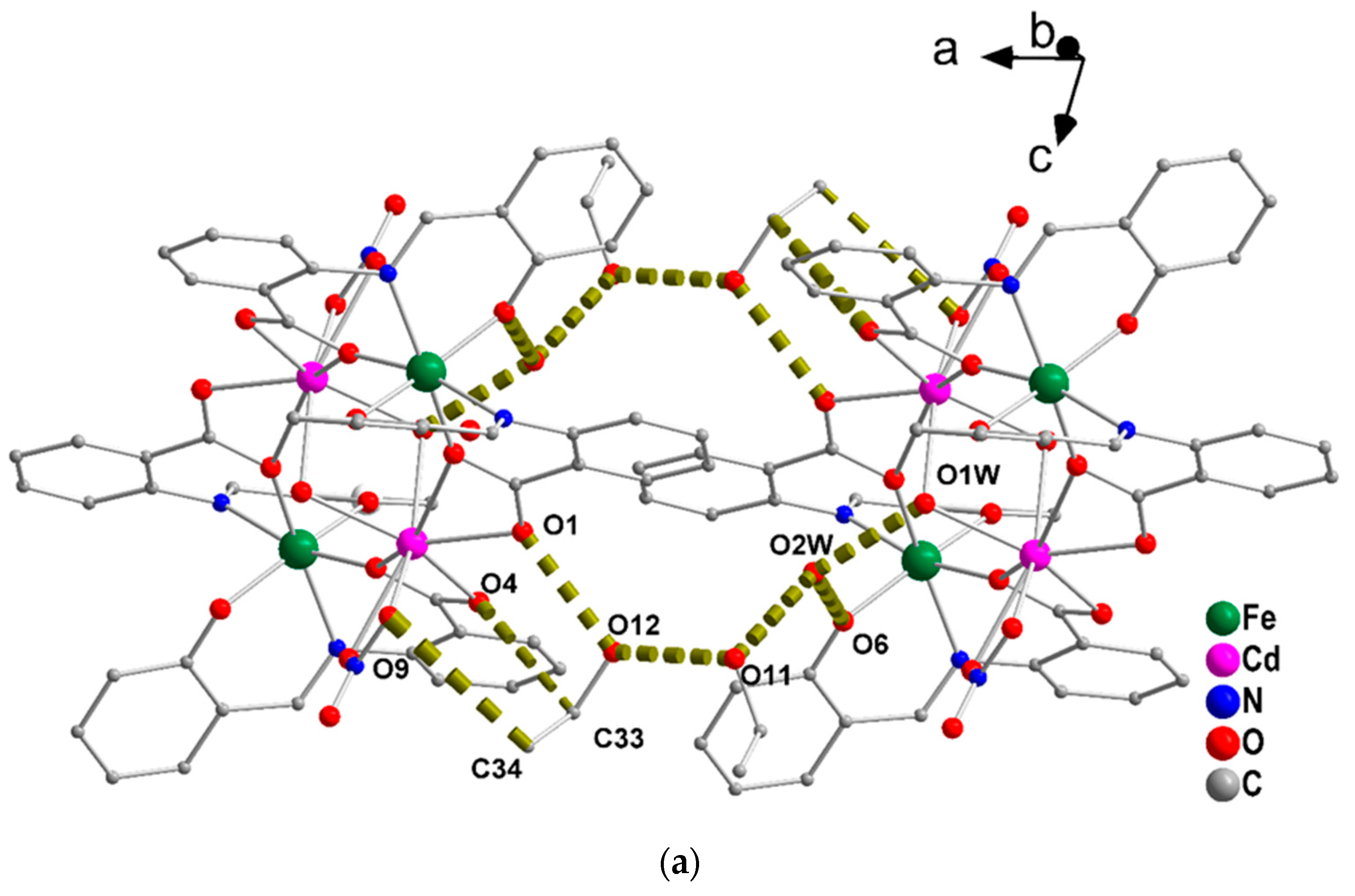
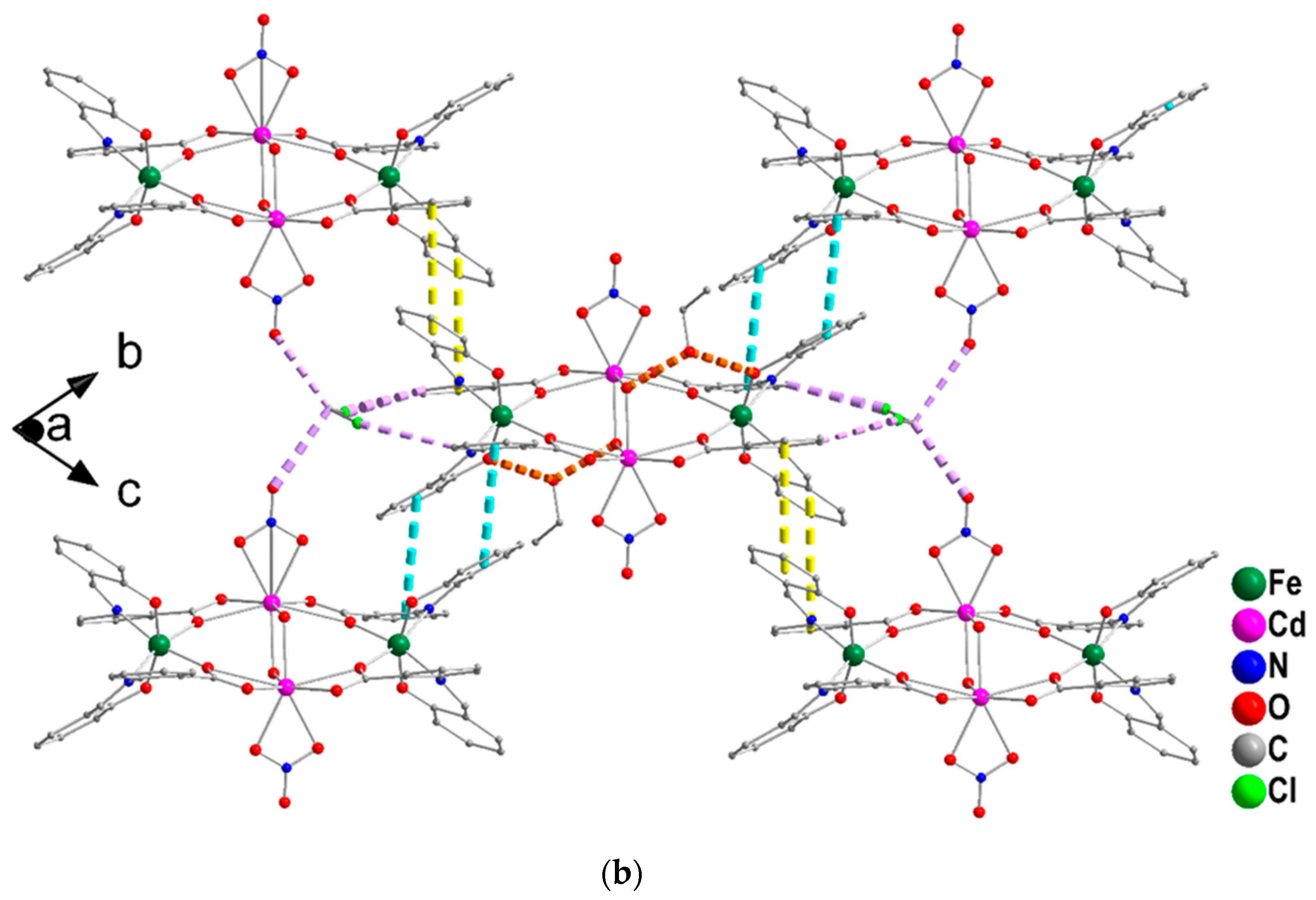
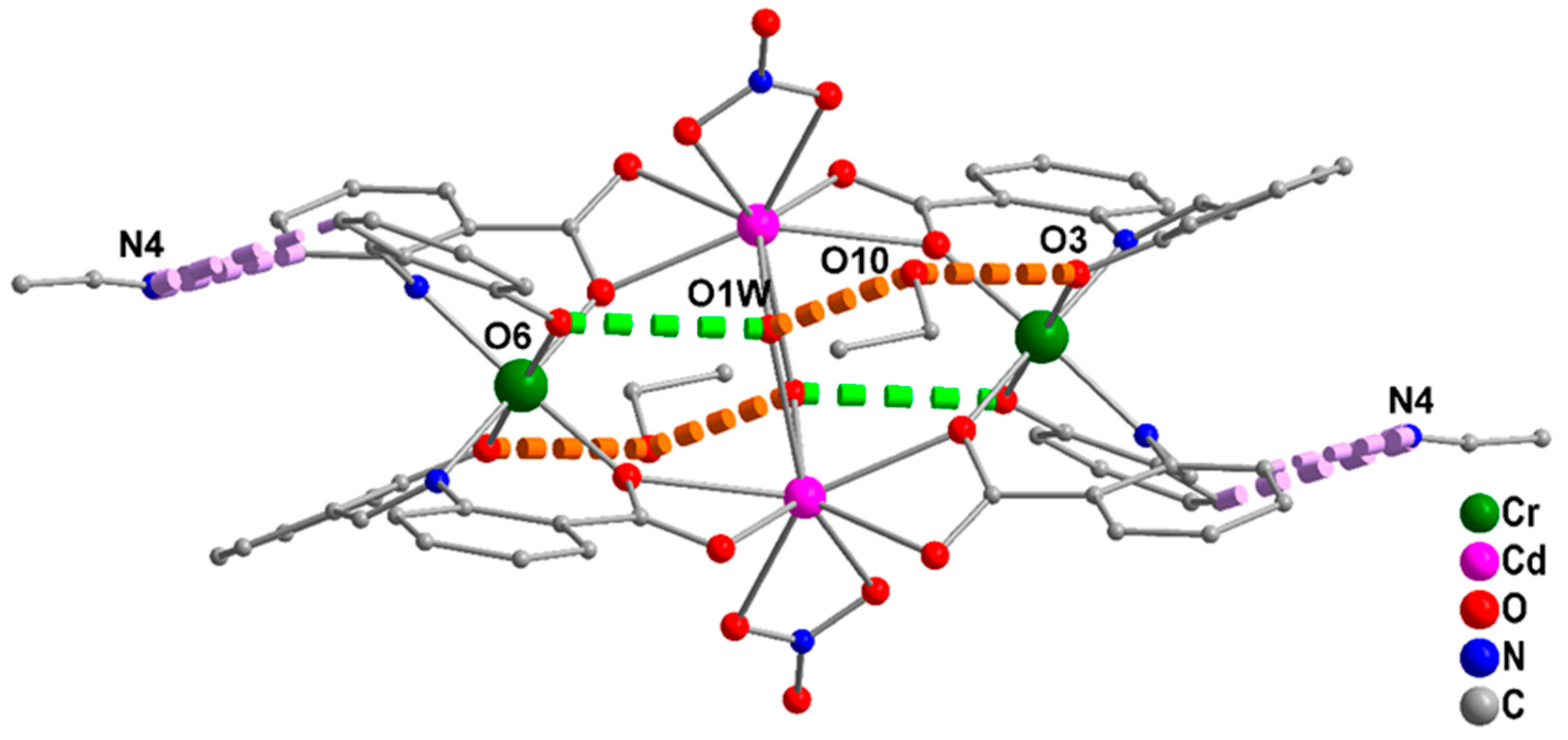
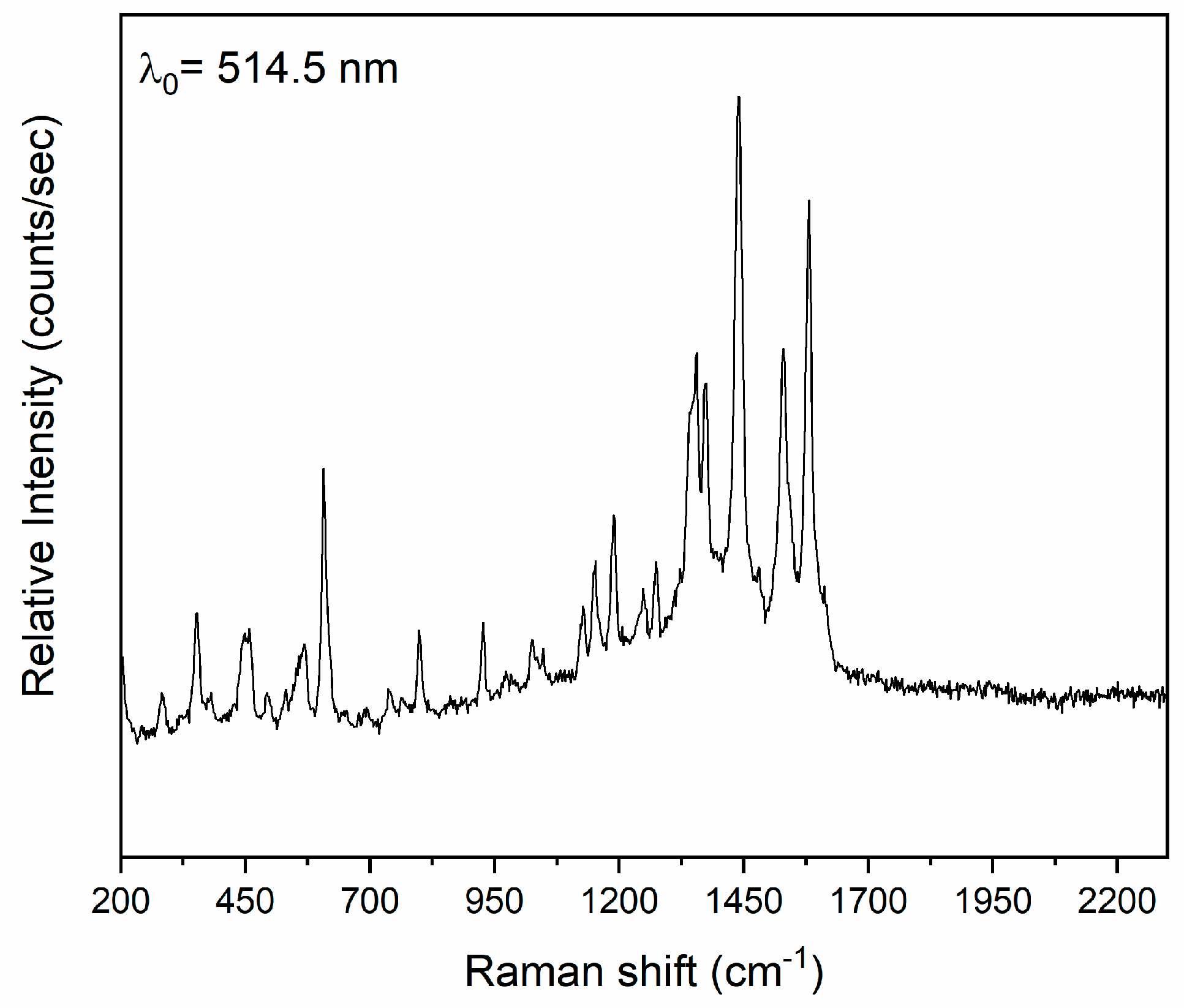
2.3. Conventional Spectroscopic Characterization in Brief
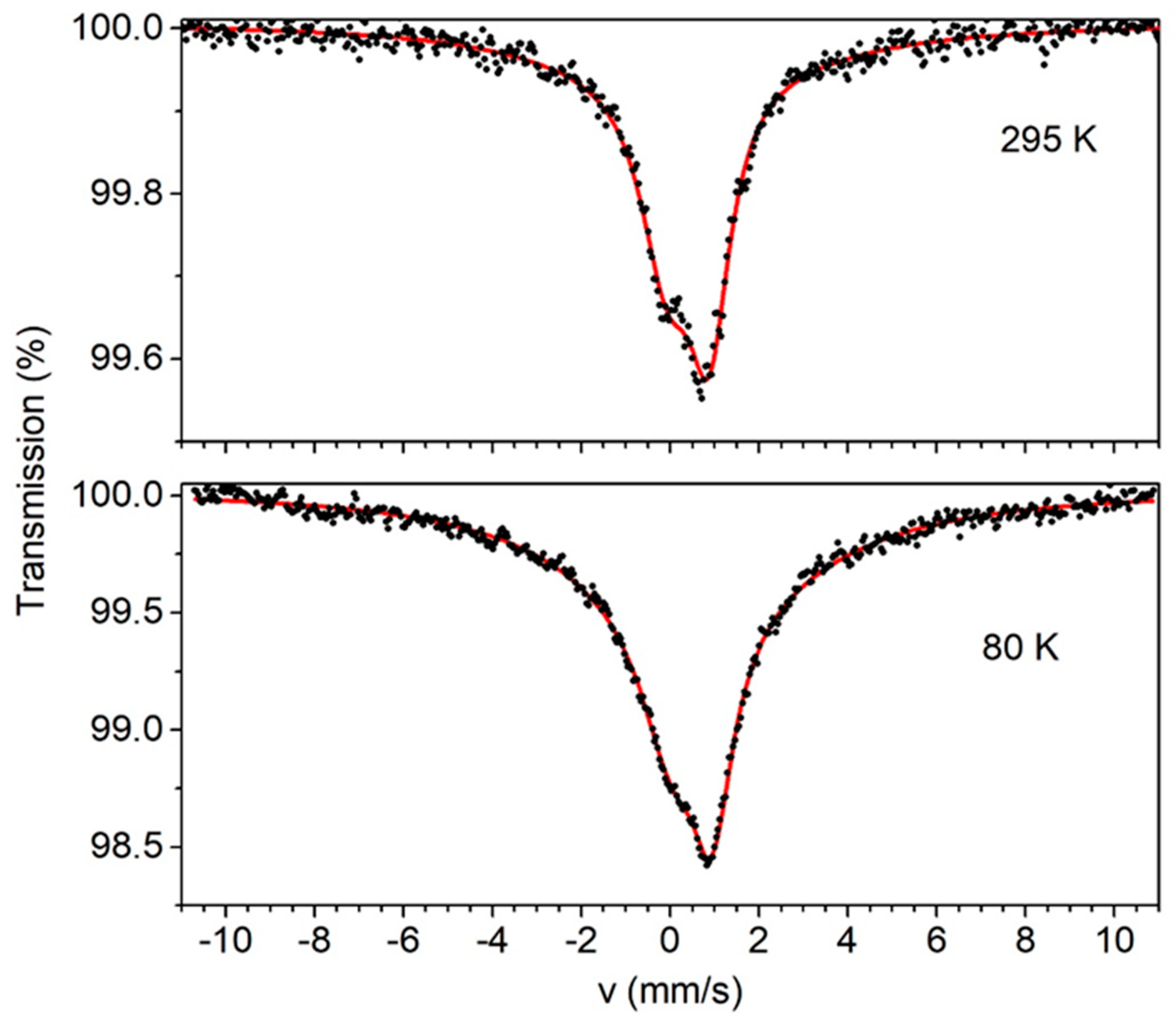
2.4. Mössbauer Spectra
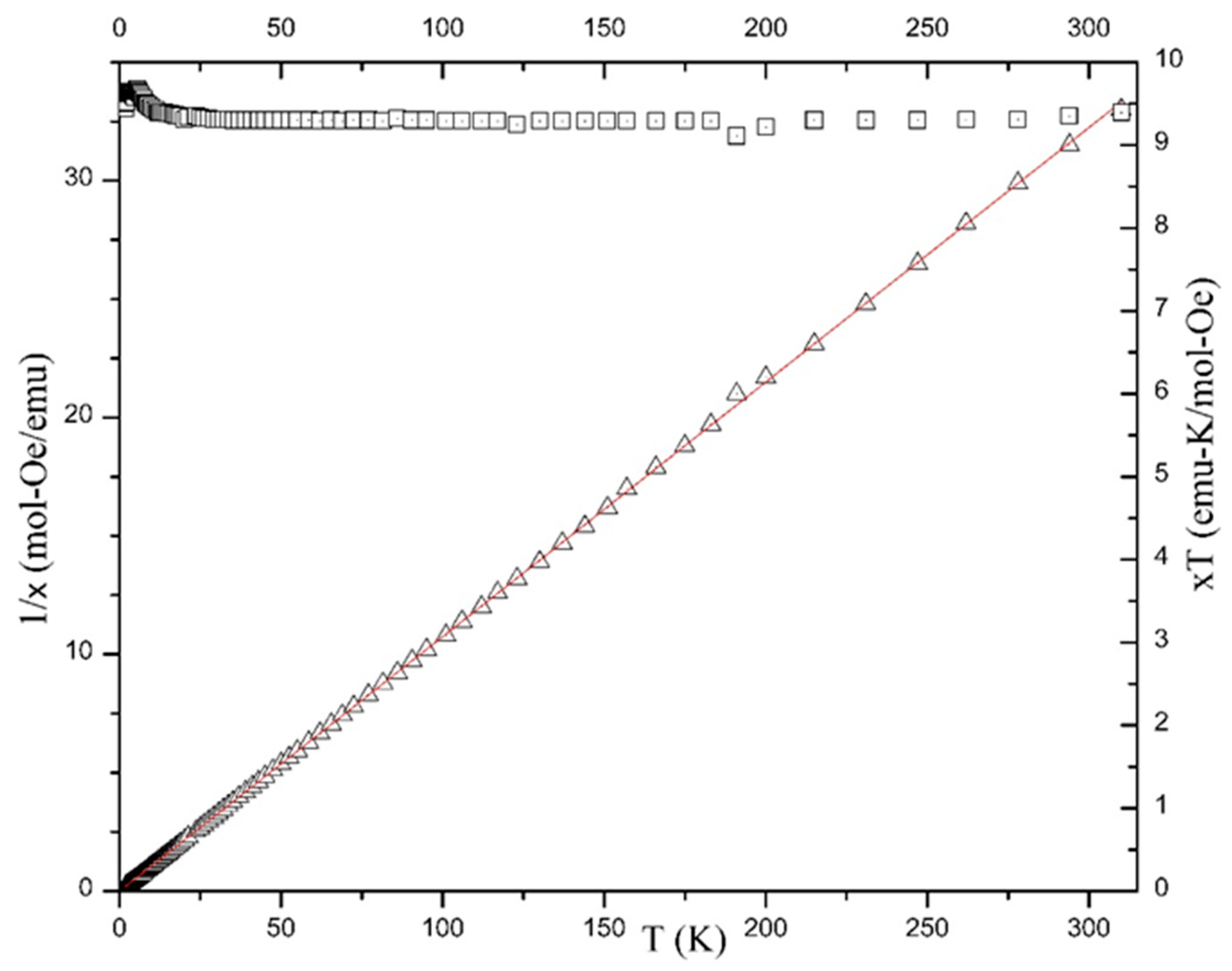
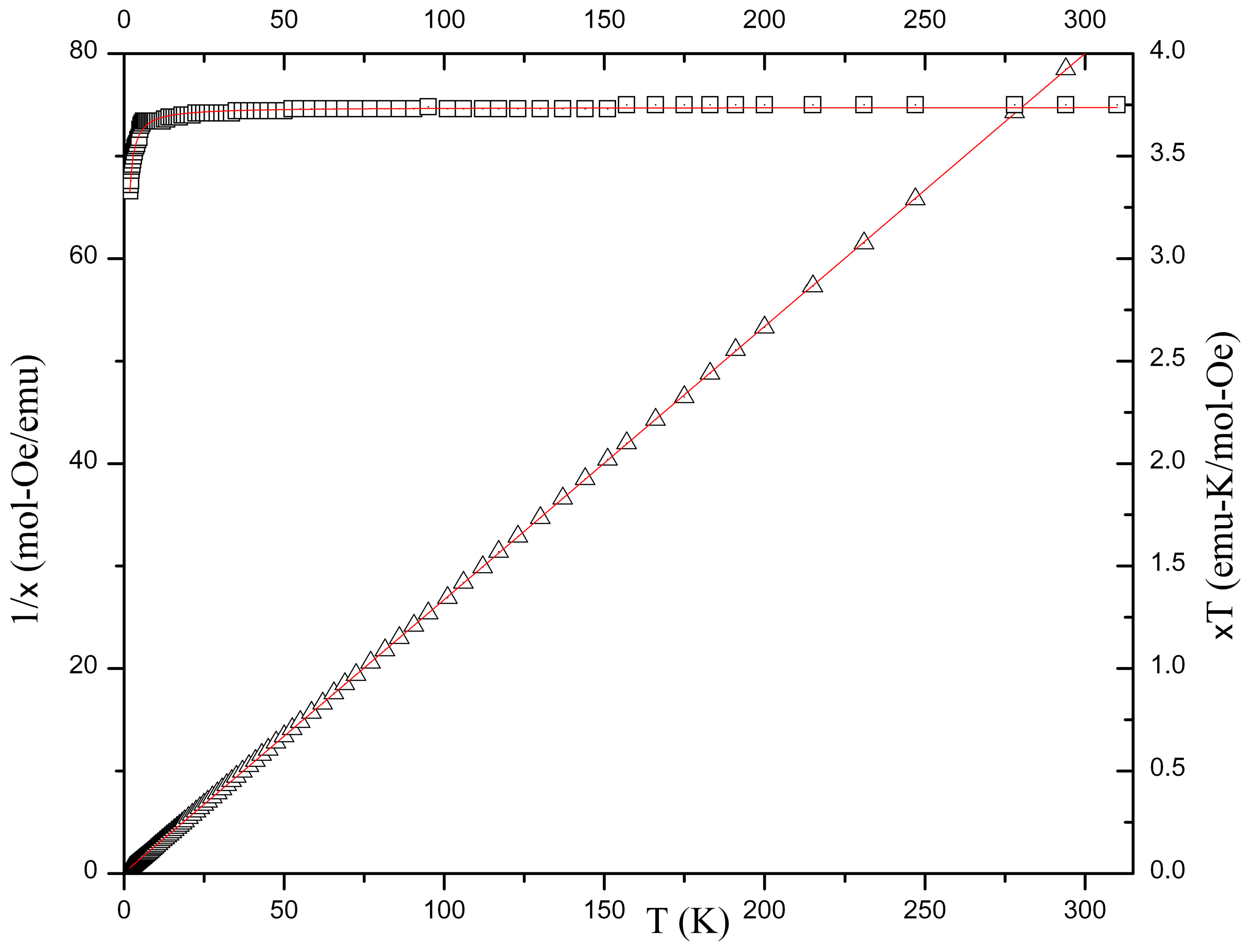
2.5. Magnetochemistry
3. Experimental Section
3.1. Materials and Instrumentation
3.2. Preparation of the Complexes
3.3. Single-Crystal X-ray Crystallography
4. Concluding Comments and Prognosis for the Future
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, N.; Homann, C.; Morfin, S.; Kesanakurti, M.S.; Calvert, N.D.; Shuhendler, A.J.; Al, T.; Hemmer, E. Core-multi-shell design: Unlocking multimodal capabilities in lanthanide-based nanoparticles as upconverting, T2-weighted MRI and CT probes. Nanoscale 2013, 15, 19546–19556. [Google Scholar] [CrossRef] [PubMed]
- Cheong, S.-W.; Mostovoy, M. Multiferroics: A magnetic twist for ferroelectricity. Nat. Mater. 2007, 6, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kanatzidis, M.G.; Pöttgen, R.; Jeitschko, W. The Metal Flux: A Preparative Tool for the Exploration of Intermetallic Compounds. Angew. Chem. Int. Ed. 2005, 44, 6996–7023. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, H.; Julve, M.; Yamashita, M.; Clérac, R. Slow Dynamics of the Magnetization in One-Dimensional Coordination Polymers: Single-Chain Magnets. Inorg. Chem. 2009, 48, 3420–3437. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar] [CrossRef] [PubMed]
- O’ Keeffe, M.; Yaghi, O.M. Deconstructing the Crystal Structures of Metal-Organic Frameworks and Related Materials into Their Underlying Nets. Chem. Rev. 2012, 112, 675–702. [Google Scholar] [CrossRef]
- Aguilà, D.; Barrios, L.; Belasco, V.; Roubeau, O.; Repollés, A.; Alonso, P.J.; Sesé, J.; Teat, S.J.; Luis, F.; Aromi, G. Heterodimetallic [LnLn’] Lanthanide Complexes: Toward a Chemical Design of Two-Qubit Molecular Spin Quantum Gates. J. Am. Chem. Soc. 2014, 136, 14215–14222. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadis, N.C.; Polyzou, C.D.; Kostakis, G.E.; Bekiari, V.; Lan, Y.; Perlepes, S.P.; Konidaris, K.F.; Powell, A.K. Dinuclear lanthanide(III)/zinc(II) complexes with methyl 2-pyridyl Ketone oxime. Dalton Trans. 2015, 44, 19791–19795. [Google Scholar] [CrossRef]
- Aguilà, D.; Prado, Y.; Koumousi, E.S.; Mathonière, C.; Clérac, R. Switchable Fe/Co Prussian blue networks and molecular analogues. Chem. Soc. Rev. 2016, 45, 203–224. [Google Scholar] [CrossRef]
- Lada, Z.G.; Polyzou, C.D.; Nika, V.; Stamatatos, T.C.; Konidaris, K.F.; Perlepes, S.P. Adventures in the coordination chemistry of 2-pyridyl oximes: On the way to 3d/4f-metal coordination clusters. Inorg. Chim. Acta 2022, 539, 120954. [Google Scholar] [CrossRef]
- Liu, K.; Shi, W.; Cheng, P. Toward heterometallic single-molecule magnets: Synthetic strategy, structures and properties of 3d–4f discrete complexes. Coord. Chem. Rev. 2015, 289–290, 74–122. [Google Scholar] [CrossRef]
- Sessoli, R.; Powell, A.K. Strategies towards single-molecule magnets based on lanthanide ions. Coord. Chem. Rev. 2009, 253, 2328–2341. [Google Scholar] [CrossRef]
- Li, D.; Clérac, R.; Roubeau, O.; Harté, E.; Mathonière, C.; Le Bris, R.; Holmes, S.M. Magnetic and Optical Bistability Driven by Thermally and Photoinduced Intramolecular Electron Transfer in a Molecular Cobalt-Iron Prussian Blue Analogue. J. Am. Chem. Soc. 2008, 130, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Rosado Piquer, L.; Sañudo, E.C. Heterometallic 3d–4f single-molecule magnets. Dalton Trans. 2015, 44, 8771–8780. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-S.; Zhang, K.; Song, Y.; Pan, Z.-Q. Recent advances in 3d–4f magnetic complexes with several types of non-carboxylate organic ligands. Inorg. Chim. Acta 2021, 521, 120318. [Google Scholar] [CrossRef]
- Dey, A.; Acharya, J.; Chandrasekhar, V. Heterometallic 3d–4f Complexes as Single-Molecule Magnets. Chem. Asian J. 2019, 14, 4433–4453. [Google Scholar] [CrossRef]
- Long, J. Luminescent Schiff-Base Lanthanide Single-Molecule Magnets: The Association between Optical and Magnetic Properties. Front. Chem. 2019, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, I.C.; Funes, A.V.; Carrella, L.; Sorace, L.; Rentschler, E.; Alborés, P. Magnetic Study of a Pentanuclear {CoIII2CoII3} Cluster with a Bent {CoII3} Motif. Eur. J. Inorg. Chem. 2014, 2561–2568. [Google Scholar] [CrossRef]
- Ke, X.; Xu, G.-F.; Guo, Y.-N.; Gamez, P.; Beavers, C.M.; Teat, S.J.; Tang, J. A linear tetranuclear dysprosium(III) compound showing single-molecule magnet behavior. Chem. Commun. 2010, 46, 6057–6059. [Google Scholar] [CrossRef]
- Lazzarini, I.C.; Carrella, L.; Rentschler, E.; Alborés, P. One dimensional Mn(III) Schiff-base complex organization through very strong symmetrical H-bond interaction. Inorg. Chim. Acta 2016, 453, 692–696. [Google Scholar] [CrossRef]
- Anastasiadis, N.C.; Granadeiro, C.M.; Mayans, J.; Raptopoulou, C.P.; Bekiari, V.; Cunha-Silva, L.; Psycharis, V.; Escuer, A.; Balula, S.S.; Konidaris, K.F.; et al. Multifunctionality in Two Families of Dinuclear Lanthanide(III) Complexes with a Tridentate Schiff-Base Ligand. Inorg. Chem. 2019, 58, 9581–9585. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.-Z.; Shi, L.; Chen, K.; Xue, J.-Y. catena-Poly[[zinc(II)-μ-[2-(2-carboxylatophenyl-iminomethyl)phenolato-1k3O,N,O’:1’kO”] methanol solvate. Acta Crystallogr. E 2005, 61, m1427–m1428. [Google Scholar] [CrossRef]
- Wang, J.; Bei, F.L.; Yang, X.J.; Lu, L.D.; Wang, X. Crystal structure of a carboxylate-bridged one-dimensional linear chain polymer N-salicylidene-2-amino-phenylacidato copper(II) complex. J. Mol. Struct. 2002, 643, 129–133. [Google Scholar] [CrossRef]
- Tonde, S.S.; Kumbhar, A.S.; Padhye, S.B.; Butcher, R.J. Self-activating nuclease activity of copper(II) complexes of hydroxyl-rich ligands. J. Inorg. Biochem. 2006, 100, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Laye, R.H.; Sañudo, E.C. Synthesis of Fe(II) and Cu(II) building blocks for metal-organic frameworks. Inorg. Chim. Acta 2009, 362, 2205–2212. [Google Scholar] [CrossRef]
- Li, X.; Bi, C.-F.; Fan, Y.-H.; Zhang, X.; Wei, X.-D.; Meng, X.-M. Synthesis, characterization, DNA binding and cleavage properties of a ternary copper(II) Schiff base complex. Transit. Met. Chem. 2014, 39, 577–584. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Liu, Z.; Xu, Y.; Wang, D. Synthesis, characterization of a ternary Cu(II) Schiff base complex with degradation activity of organophosphorus pesticides. Inorg. Chim. Acta 2018, 471, 280–289. [Google Scholar] [CrossRef]
- Pantelis, K.N.; Skiadas, S.G.; Lada, Z.G.; Clérac, R.; Sanakis, Y.; Dechambenoit, P.; Perlepes, S.P. Hexanuclear {ZnII4FeIII2} and {ZnII4CrIII2} complexes from the use of potentially tetradentate NOO’O” Schiff-bae ligands. New J. Chem. 2024, 48, 11221–11232. [Google Scholar] [CrossRef]
- Biswas, P.; Ghosh, M.; Dutta, S.K.; Flörke, U.; Nag, K. Synthesis, Reactivities and Magnetostructural Properties of FeIII, FeIII-O-FeIII, and ZnIIFeIII-O-FeIIIZnII Complexes of a Tetraiminodiphenolate Macrocycle. Inorg. Chem. 2006, 45, 4830–4844. [Google Scholar] [CrossRef]
- Vreshch, V.D.; Lysenko, A.B.; Chernega, A.N.; Howard, J.A.K.; Krautscheid, H.; Sieler, J.; Domasevitch, K.V. Extended coordination frameworks incorporating heterometallic squares. Dalton Trans. 2004, 2899–2903. [Google Scholar] [CrossRef]
- Sayin, E.; Kürkcüoğlu, G.S.; Yesilel, O.Z.; Sahin, O. Unprecedented deca-, penta- and poly-heteronuclear complexes with hexacyanometallates amd 2-(hydroxylmethyl)pyridine. Polyhedron 2021, 210, 115535. [Google Scholar] [CrossRef]
- Zhang, H.; Cai, J.; Feng, X.-L.; Sang, H.-Y.; Liu, J.-Z.; Li, X.-Y.; Ji, L.-N. Assembly chemistry of a cadmium(II) complex with cyanometallate anions [Fe(CN)5NO]2−, [Pd(CN)4]2− and [Pt(CN)6]2−. Polyhedron 2002, 21, 721–728. [Google Scholar] [CrossRef]
- Laha, S.; Sathapathi, D.; Das, M.; Das, M.; Ray, P.P.; Bag, A.; Samanta, B.C.; Das, U.K.; Maity, T. Synthesis, characterization and multi-dimensional application approach for two distinctive tetranuclear, first time reported, Fe3+/Hg2+ and Fe3+/Cd2+ clusters from a new Fe3+ containing metalloligand. New J. Chem. 2022, 46, 16730–16742. [Google Scholar] [CrossRef]
- Ouyang, Z.-J.; Mo, X.-Y.; Yang, M.; Zhong, L.; Chen, W.-B.; Gao, S.; Dong, W. High temperature Fe(III) spin crossover behaviors in three unpredicted FeIII-MII-FeIII (M = Fe, Cd) linear trinuclear complexes. Inorg. Chem. Front. 2020, 7, 1526–1531. [Google Scholar] [CrossRef]
- Laye, R.H.; Larsen, F.K.; Overgaard, J.; Muryn, C.A.; McInnes, E.J.L.; Rentschler, E.; Sanchez, V.; Teat, S.J.; Güdel, H.U.; Waldmann, O.; et al. A family of heterometallic wheels containing potentially fourteen hundred siblings. Chem. Commun. 2005, 1125–1127. [Google Scholar] [CrossRef] [PubMed]
- Timco, G.A.; Batsanov, A.S.; Larsen, F.K.; Muryn, C.A.; Overgaard, J.; Teat, S.J.; Winpenny, R.E.P. Influencing the nuclearity and constitution of heterometallic rings via templates. Chem. Commun. 2005, 3649–3651. [Google Scholar] [CrossRef]
- Larsen, F.K.; McInnes, E.J.L.; El Mkami, H.; Overgaard, J.; Piligkos, S.; Rajaraman, G.; Rentschler, E.; Smith, A.A.; Smith, G.M.; Boote, V.; et al. Synthesis and Characterization of Heterometallic {Cr7M} Wheels. Angew. Chem. Int. Ed. 2003, 42, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.K.; Overgaard, J.; Christensen, M.; McIntyre, G.J.; Timco, G.; Winpenny, R.E.P. Metal distribution and disorder in the crystal structure of [NH2Et2][Cr7MF8(tBuCO2)16] wheel molecules for M=Mn, Fe, Co, Ni, Cu, Zn and Cd. Acta Crystallogr. B 2014, 70, 932–941. [Google Scholar] [CrossRef]
- Ballesteros, B.; Faust, T.B.; Lee, C.-F.; Leigh, D.A.; Muryn, C.A.; Pritchard, R.G.; Schultz, D.; Teat, S.J.; Timco, G.A.; Winpenny, R.E.P. Synthesis, Structure, and Dynamic Properties of Hybrid Organic-Inorganic Rotaxanes. J. Am. Chem. Soc. 2010, 132, 15435–15444. [Google Scholar] [CrossRef]
- Schoedel, A.; Wojtas, L.; Kelley, S.P.; Rogers, R.D.; Eddaoudi, M.; Zaworotko, M.J. Network Diversity through Decoration of Trigonal-Prismatic Nodes: Two-Step Crystal Engineering of Cationic Metal-Organic Materials. Angew. Chem. Int. Ed. 2011, 50, 11421–11424. [Google Scholar] [CrossRef]
- An, H.; Xu, T.; Jia, C.; Zheng, H.; Mu, W. Two new architectures based on Anderson-type polyoxoanions and cadmium fragments. J. Mol. Struct. 2009, 933, 86–91. [Google Scholar] [CrossRef]
- Coxall, R.A.; Harris, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; Winpenny, R.E.P. Inter-ligand reactions: In situ formation of new polydentate ligands. J. Chem. Soc. Dalton Trans. 2000, 2349–2356. [Google Scholar] [CrossRef]
- Katsoulakou, E.; Konidaris, K.F.; Terzis, A.; Raptopoulou, C.P.; Perlepes, S.P.; Manessi-Zoupa, E.; Kostakis, G.E. One-dimensional cadmium(II)/bicinate(-1) complexes: The role of the alkali metal ion used in the reaction medium. Polyhedron 2011, 30, 397–404. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Laidig, K.E. Barriers to Rotation to Double Bonds. 3. The C-O Barrier in Formic Acid, Methyl Formate, Acetic Acid and Methyl Acetate. The Origin of Ester and Amide “Resonance”. J. Am. Chem. Soc. 1987, 109, 5935–5943. [Google Scholar] [CrossRef]
- Peterson, M.R.; Csizmadia, I.G. Determination and Analysis of the Formic Acid Conformational Hypersurface. J. Am. Chem. Soc. 1979, 101, 1076–1079. [Google Scholar] [CrossRef]
- Rardin, R.L.; Tolman, W.B.; Lippard, S.J. Monodentate Carboxylate Complexes and the Carboxylate Shift: Implications for Polymetalloprotein Structure and Function. New J. Chem. 1991, 15, 417–430. [Google Scholar]
- Li, S.-L.; Mak, T.C.W. Synthesis and structural characterization of discrete mono-, bi-, tri- and tetranuclear complexes of cadmium(II) with triphenylphosphoniopropionate. Inorg. Chim. Acta 1997, 258, 11–24. [Google Scholar] [CrossRef]
- Llunell, M.; Casanova, D.; Girera, J.; Alemany, P.; Alvarez, S. SHAPE, Continuous Shape Measures Calculation, Version 2.0; Universitat de Barcelona: Barcelona, Spain, 2010. [Google Scholar]
- Choudhury, R.R.; Chitra, R. Stacking interaction between homostacks of simple aromatics and the factors influencing these interactions. CrystEngComm 2010, 12, 2113–2121. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, J.; Gu, H.; Wei, D.; Xu, Y.-C.; Fu, W.; Yu, Z. Conformational Preferences of π-π Stacking between Ligand and Protein, Analysis Derived from Crystal Structure Data Geometric Preference of π-π Interaction. Interdisip. Sci. Comput. Life Sci. 2015, 7, 211–220. [Google Scholar] [CrossRef]
- Murugesapandian, B.; Roesky, P.W. Synthesis and Structures of Zinc(II) and Cadmium(II) Complexes with η6-Benzenecarboxylato Chromium Tricarbonyl and Pyrazole Derivatives. Z. Anorg. Allg. Chem. 2011, 637, 1818–1823. [Google Scholar] [CrossRef]
- Sarkar, B.; Wen, S.-Y.; Wang, J.-H.; Chiou, L.-S.; Liao, P.-K.; Santra, B.K.; Wang, J.-C.; Liu, C.W. Conjugate Base of a Secondary Phosphine Selenide [P(Se)(OiPr)2]− as the Bridging Unit for the Construction of Heterometallic Fe(II)-Hg(II)/Cd(II) Complexes. Inorg. Chem. 2009, 48, 5129–5140. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th ed.; Wiley: New York, NY, USA, 1986; pp. 231–233, 254–257. [Google Scholar]
- Rao, C.N.R. Ultra-Violet and Visible Spectroscopy, 2nd ed.; Butterworths: London, UK, 1967; pp. 20–33, 58–73. [Google Scholar]
- Lever, A.B.P. Inorganic Electronic Spectroscopy, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1984; pp. 329–332, 417–420, 450, 452, 453. [Google Scholar]
- Savva, M.; Alexandropoulos, D.I.; Pissas, M.; Perlepes, S.P.; Papatriantafyllopoulou, C.; Sanakis, Y.; Tasiopoulos, A.J. Heterometallic clusters based on an uncommon asymmetric “V-shaped” [Fe3+(μ-OR)Ln3+(μ-OR)2Fe3+]6+ (Ln=Gd, Tb, Dy, Ho) structural core and the investigation of the slow relaxation of the magnetization behaviour of the [Fe2Dy} analogue. Dalton Trans. 2023, 52, 6997–7008. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, N.N.; Gibbs, T.C. Mössbauer Spectroscopy; Chapman and Hall: London, UK, 1971; pp. 1–660. [Google Scholar]
- Burdinsky, D.; Bill, E.; Birkelbach, F.; Wieghardt, K.; Chaudhri, P. Long-Range Exchange Interactions and Integer-Spin ST = 2 EPR Spectra of a CrIIIZnIICrIII Species with Multiplet Mixing. Inorg. Chem. 2001, 40, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Carlin, R.L. Magnetochemistry; Springer: Berlin, Germany, 1986. [Google Scholar]
- CrystalClear; Rigaku/MSC Inc.: The Woodlands, TX, USA.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Diamond, Crystal and Molecular Structure Visualization, Version 3.2.; Crystal Impact: Bonn, Germany, 2018.
- Spek, A.L. PLATON SQUEEZE: A tool for calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef]
| Parameter | 1·6EtOH·2CH2Cl2·2H2O | 2·2EtOH·2MeCN | 3·4.5CH2Cl2·4H2O |
|---|---|---|---|
| Formula | C70H84N6O28Cl4Cd2Fe2 | C64H58N8O22Cd2Cr2 | C60.5H53N6O24 Cl13 Cd2Fe2 |
| Formula weight | 1935.73 | 1619.98 | 2045.44 |
| Crystal system | triclinic | monoclinic | triclinic |
| Space group | P | P21/n | P |
| Radiation (wavelength/Å) | Mo Kα (0.71073) | Mo Kα (0.71073) | Mo Kα (0.71073) |
| T/°C | −83 | −83 | −113 |
| a/Å | 11.6608(10) | 12.6281(8) | 13.3233(7) |
| b/Å | 13.9884(11) | 14.1793(9) | 13.8231(7) |
| c/Å | 14.3258(12) | 18.3556(12) | 13.8893(7) |
| α/° | 67.611(2) | 90.00 | 116.159(1) |
| β/° | 73.211(2) | 97.033(2) | 96.288(1) |
| γ/° | 70.602(2) | 90.00 | 104.868(1) |
| V/Å3 | 2002.4(3) | 3262.0(4) | 2144.8(2) |
| Z | 1 | 2 | 1 |
| Dcalcd/g cm−3 | 1.605 | 1.649 | 1.584 |
| μ/mm−1 | 1.10 | 1.05 | 1.30 |
| 2θmax/° | 54 | 54 | 54 |
| Reflections collected | 45,107 | 46,117 | 38,911 |
| Reflections unique (Rint) | 8717(0.045) | 7105(0.083) | 9322(0.0744) |
| Reflections with I > 2σ(I) | 7777 | 5887 | 6887 |
| No. of parameters | 591 | 520 | 412 |
| R1a [I > 2σ(I)] | 0.0643 | 0.0476 | 0.0528 |
| wR2b [I > 2σ(I)] | 0.1741 | 0.1204 | 0.1444 |
| GOF(F2) | 1.07 | 1.05 | 1.03 |
| Δρmax/Δρmin (e Å−3) | 1.60/−1.46 | 1.22/−0.82 | 1.44/−0.61 |
| CCDC | 2,374,217 | 2,374,218 | 2,374,219 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pantelis, K.N.; Skiadas, S.G.; Lada, Z.G.; Raptopoulou, C.P.; Psycharis, V.; Sanakis, Y.; Turnbull, M.M.; Perlepes, S.P. Tetradentate NOO′O″ Schiff-Base Ligands as a Platform for the Synthesis of Heterometallic CdII-FeIII and CdII-CrIII Coordination Clusters. Magnetochemistry 2024, 10, 69. https://doi.org/10.3390/magnetochemistry10100069
Pantelis KN, Skiadas SG, Lada ZG, Raptopoulou CP, Psycharis V, Sanakis Y, Turnbull MM, Perlepes SP. Tetradentate NOO′O″ Schiff-Base Ligands as a Platform for the Synthesis of Heterometallic CdII-FeIII and CdII-CrIII Coordination Clusters. Magnetochemistry. 2024; 10(10):69. https://doi.org/10.3390/magnetochemistry10100069
Chicago/Turabian StylePantelis, Konstantinos N., Sotiris G. Skiadas, Zoi G. Lada, Catherine P. Raptopoulou, Vassilis Psycharis, Yiannis Sanakis, Mark M. Turnbull, and Spyros P. Perlepes. 2024. "Tetradentate NOO′O″ Schiff-Base Ligands as a Platform for the Synthesis of Heterometallic CdII-FeIII and CdII-CrIII Coordination Clusters" Magnetochemistry 10, no. 10: 69. https://doi.org/10.3390/magnetochemistry10100069
APA StylePantelis, K. N., Skiadas, S. G., Lada, Z. G., Raptopoulou, C. P., Psycharis, V., Sanakis, Y., Turnbull, M. M., & Perlepes, S. P. (2024). Tetradentate NOO′O″ Schiff-Base Ligands as a Platform for the Synthesis of Heterometallic CdII-FeIII and CdII-CrIII Coordination Clusters. Magnetochemistry, 10(10), 69. https://doi.org/10.3390/magnetochemistry10100069