
Genome-Wide Identification, Characterization, and Expression Pattern of MYB Gene Family in Melastoma candidum
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of MYB Transcription Factors
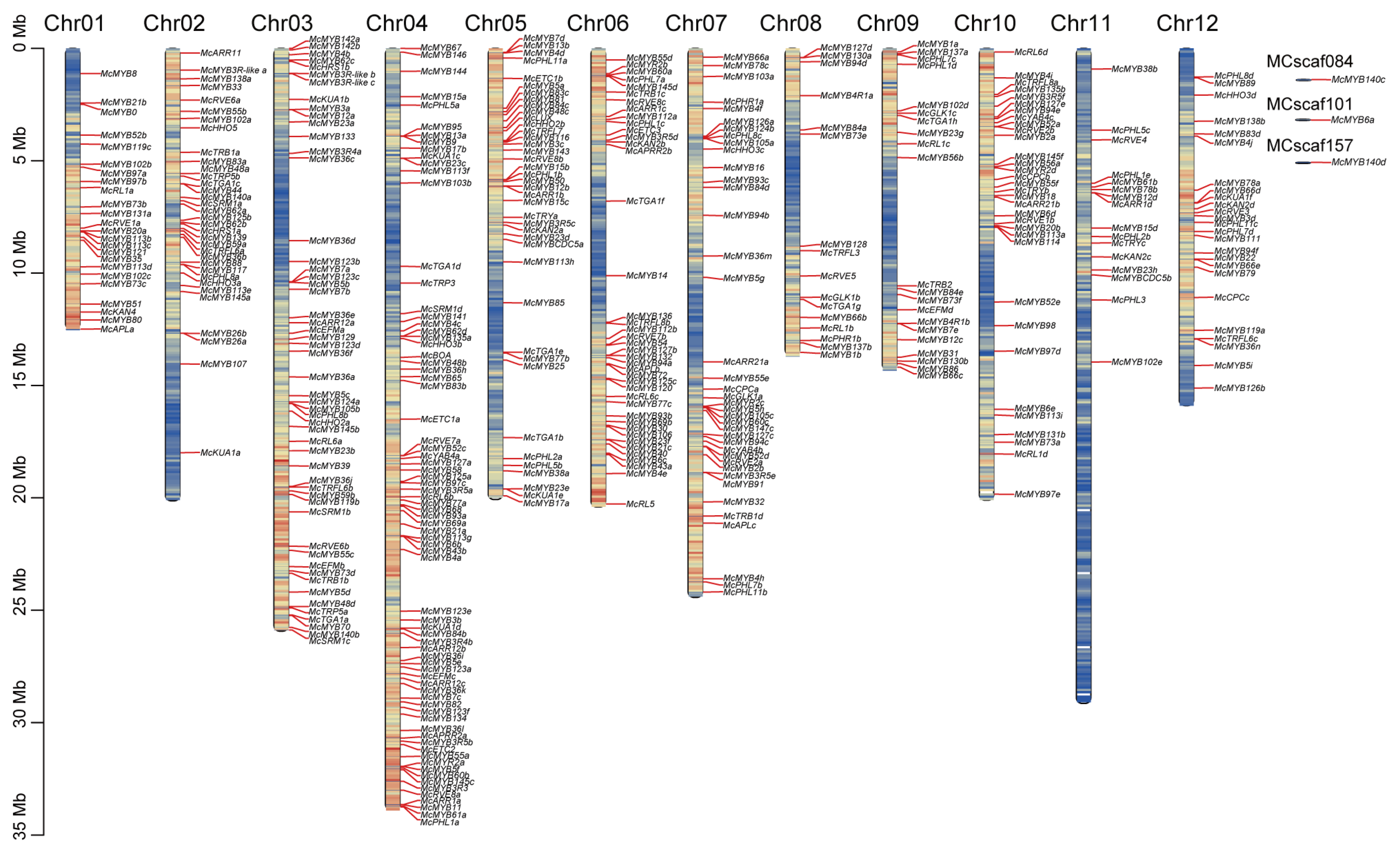
2.2. Phylogenetic Tree Construction and Distribution of MYB Gene Family Members in Chromosome
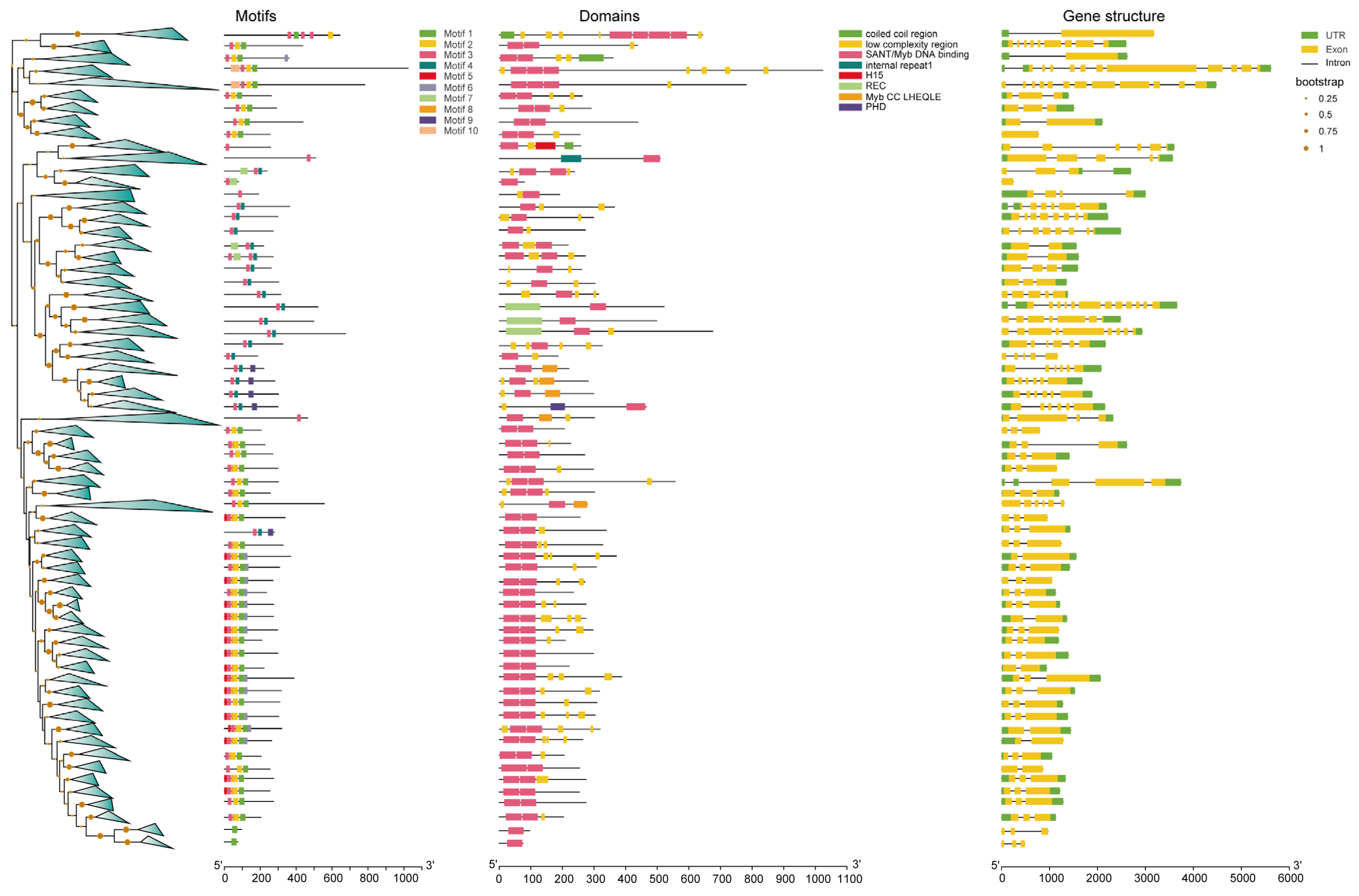
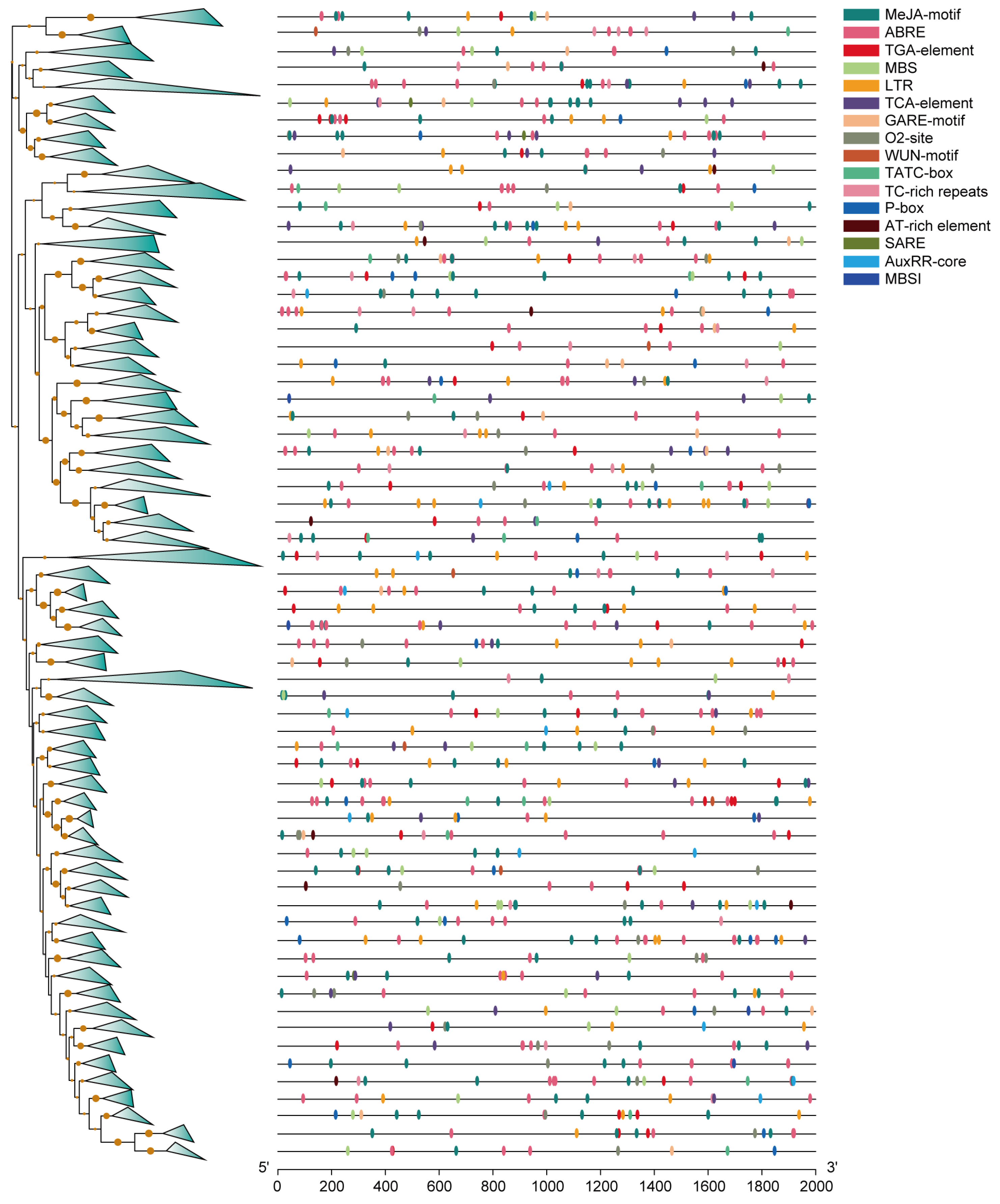
2.3. Visualizing the Motifs, Domains, Gene Structure, and Promoter Regions of MYBs
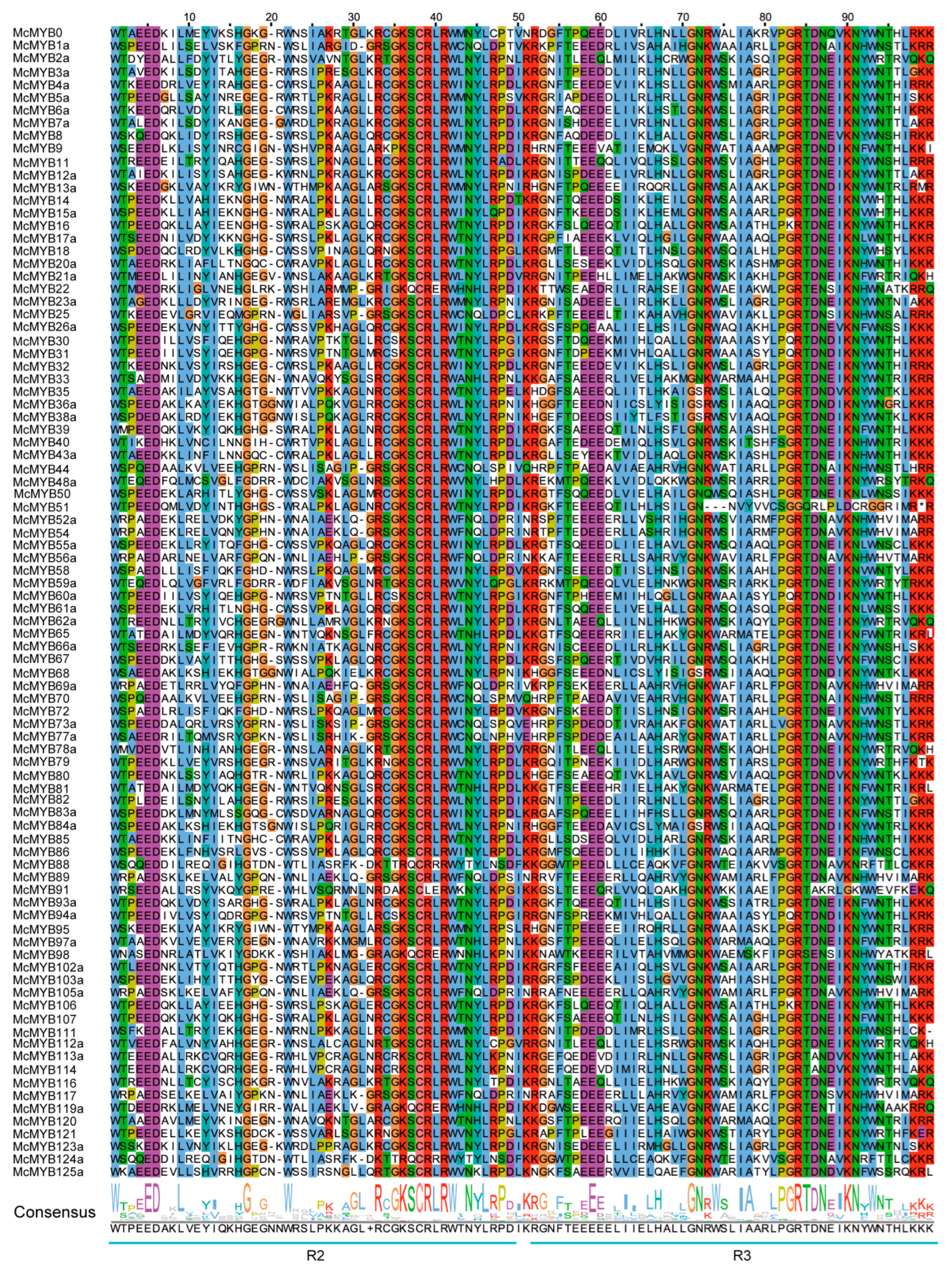
2.4. Conservation Region of the MYBs
2.5. Collinearity Analysis of Identified MYBs and Divergence Times Estimation
2.6. Collinearity and Phylogenetic Analyses for MYB Genes across Three Species
2.7. RNA-seq, Differentially Expressed Gene Analysis (DEGS), WGCNA (Weighted Correlation Network Analysis) Analysis, and Functional Annotation
2.8. Quantitative Real-Time PCR of the Selected MYB Genes in Different Tissues of M. candidum
3. Results
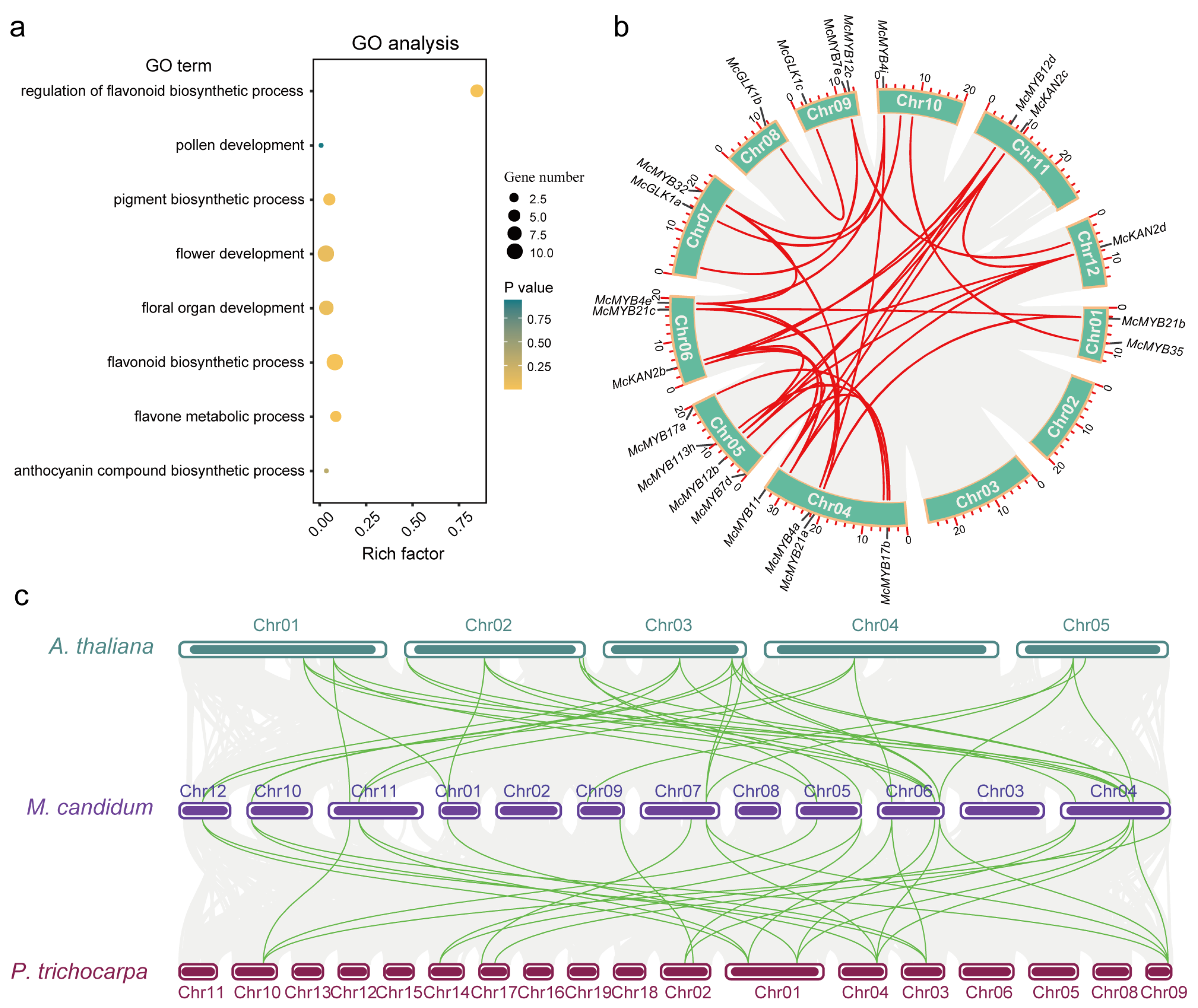
3.1. Evolution Relationship and Chromosome Distribution of MYB Genes
3.2. Motif, Domain, and Gene Structure Analyses of the Identified MYBs
3.3. Promoter Analysis of All the Identified MYBs
3.4. Conservation Analysis of Different Types of MYB TFs
3.5. Duplication Events, Divergence, and Collinearity of Flower Development and Color-Formation-Related MYB Genes
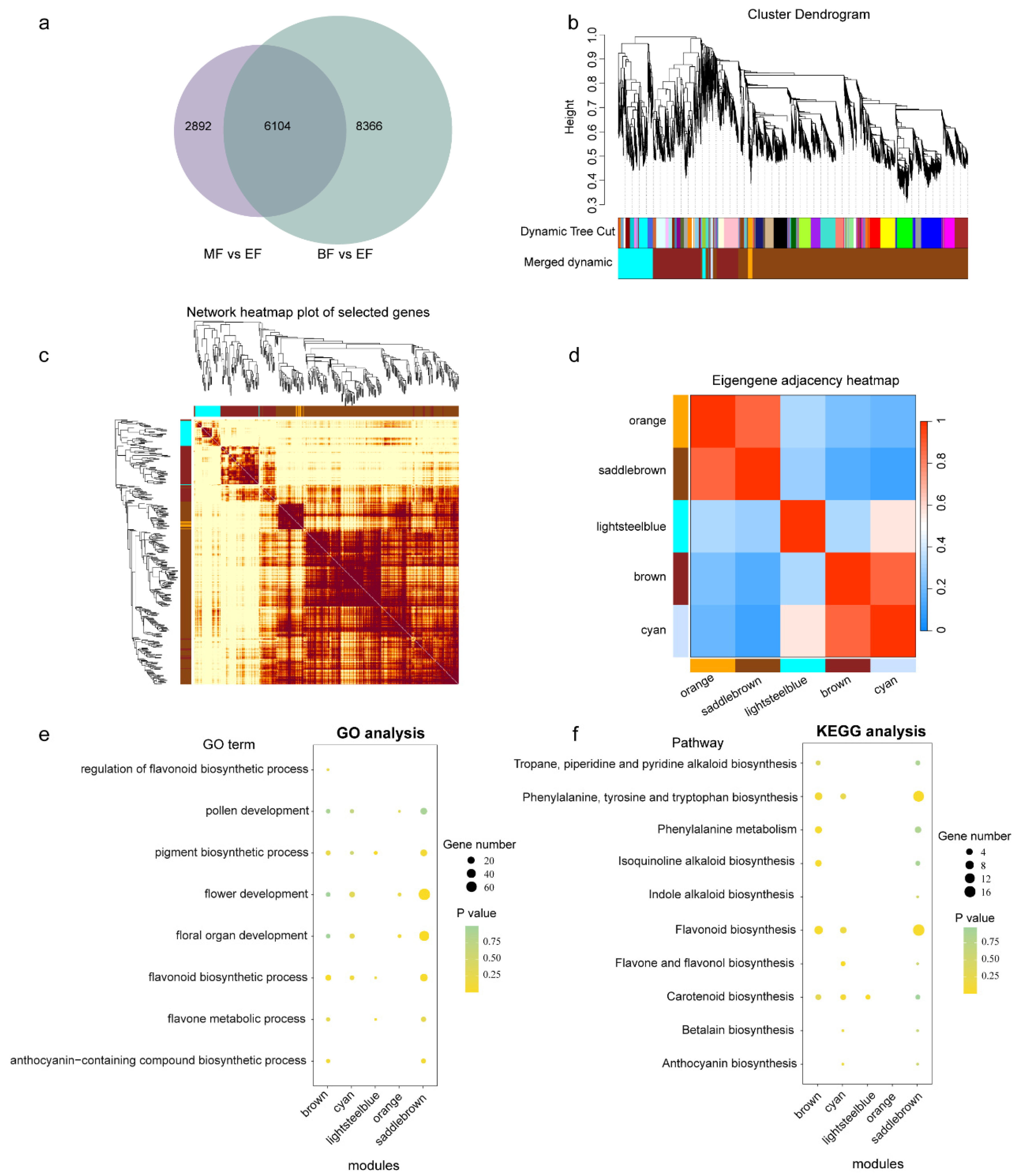
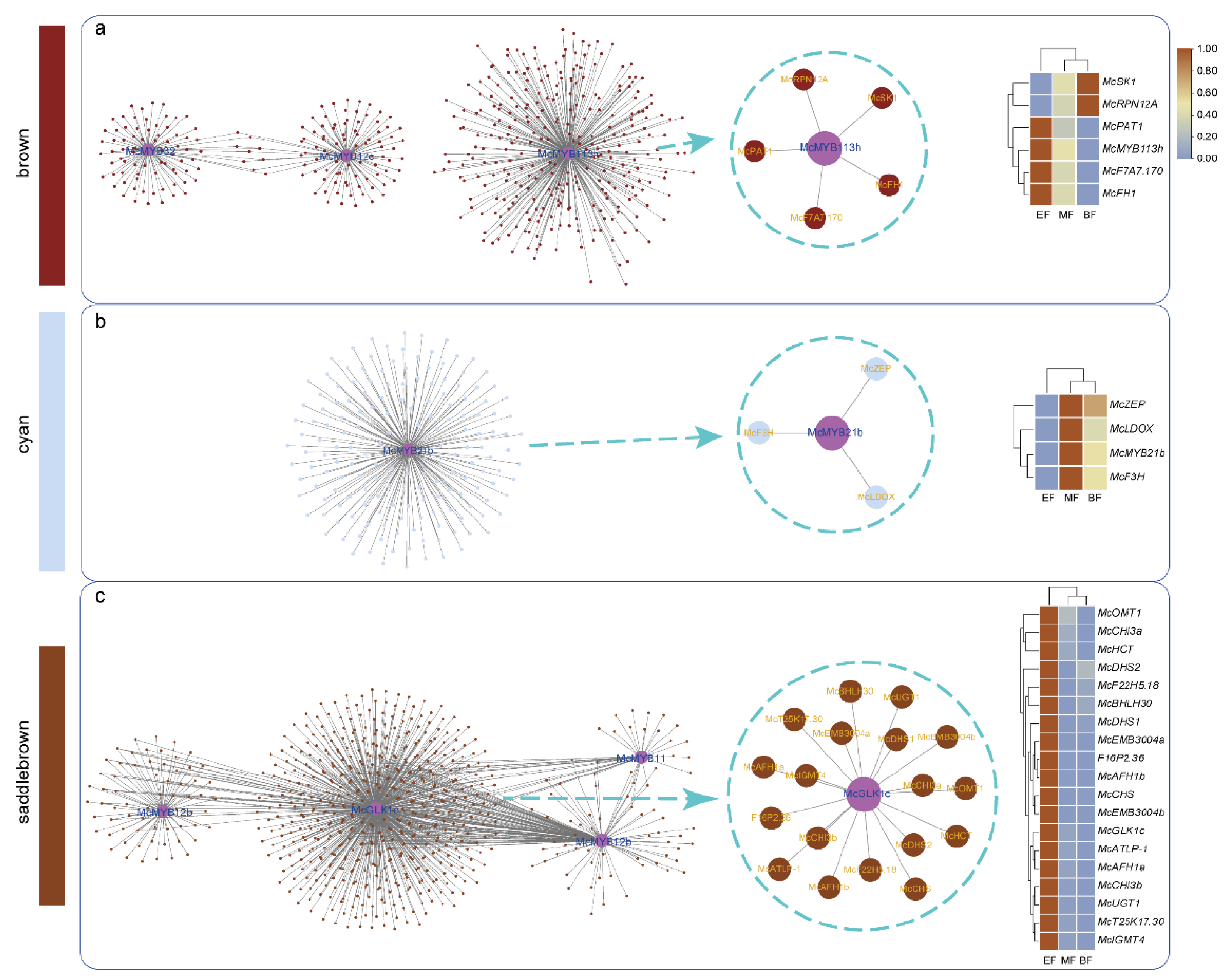
3.6. WGCNA Analysis, Functional Analysis of DEGs, and MYB-Centered Network Building
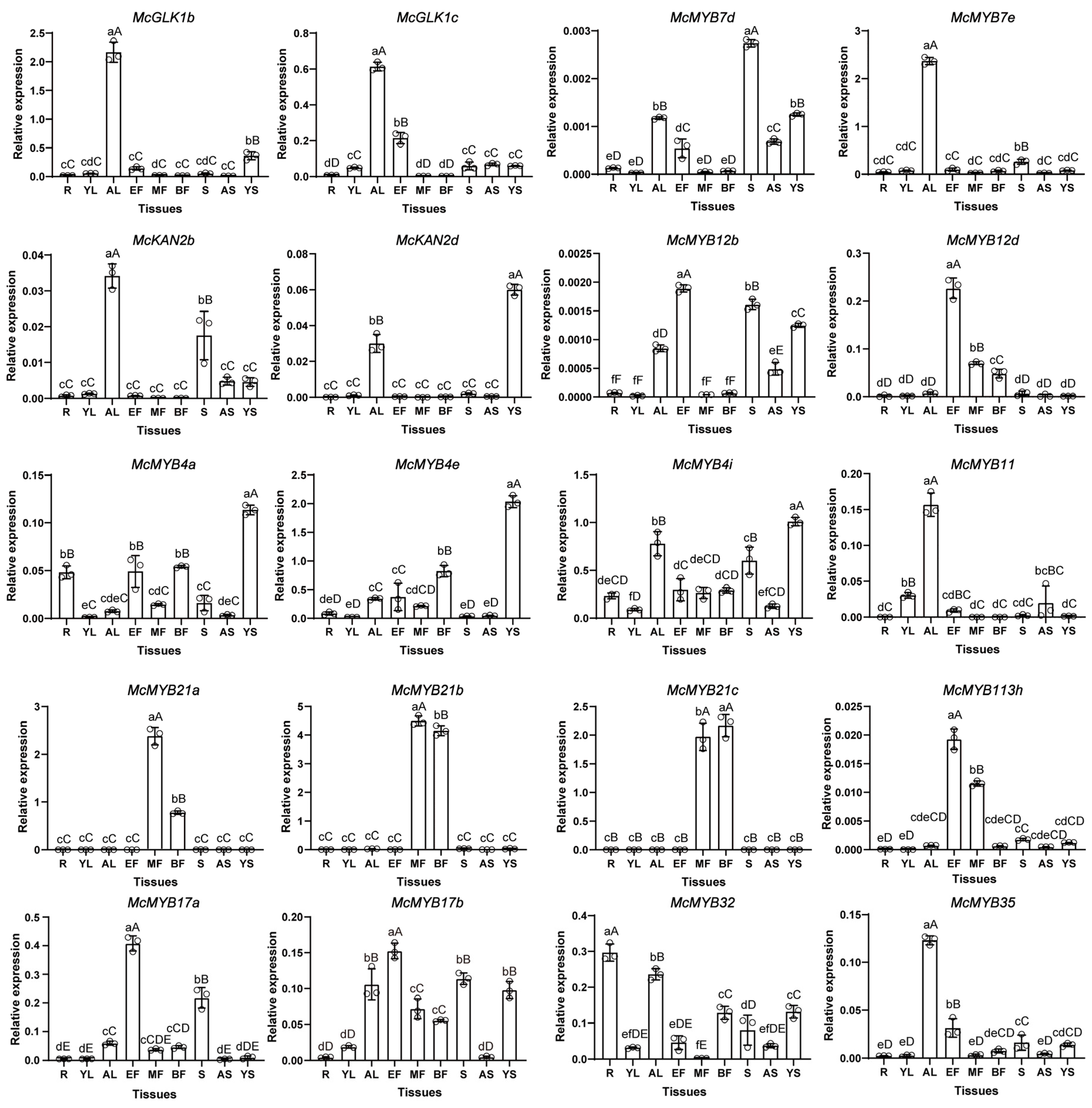
3.7. Expression Pattern of Selected MYBs in Different Tissues of M. candidum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feller, A.; Machemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011, 66, 94–116. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Xiong, J.-S.; Jiang, Y.-T.; Li, W.; Cheng, Z.-M. Evolution of the R2R3-MYB gene family in six Rosaceae species and expression in woodland strawberry. J. Integr. Agric. 2019, 18, 2753–2770. [Google Scholar]
- Klempnauer, K.-H.; Gonda, T.J.; Bishop, J.M. Nucleotide sequence of the retroviral leukemia gene v-myb and its cellular progenitor c-myb: The architecture of a transduced oncogene. Cell 1982, 31, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Paz-Ares, J. MYB transcription factors in plants. Trends Genet. 1997, 13, 67–73. [Google Scholar] [CrossRef]
- Rosinski, J.A.; Atchley, W.R. Molecular evolution of the Myb family of transcription factors: Evidence for polyphyletic origin. J. Mol. Evol. 1998, 46, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Ito, M. Conservation and diversification of three-repeat Myb transcription factors in plants. J. Plant Res. 2005, 118, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Du, H.; Yang, S.-S.; Liang, Z.; Feng, B.-R.; Liu, L.; Huang, Y.-B.; Tang, Y.-X. Genome-wide analysis of the MYB transcription factor superfamily in soybean. BMC Plant Biol. 2012, 12, 106. [Google Scholar] [CrossRef]
- Du, H.; Feng, B.-R.; Yang, S.-S.; Huang, Y.-B.; Tang, Y.-X. The R2R3-MYB transcription factor gene family in maize. PLoS ONE 2012, 7, e37463. [Google Scholar] [CrossRef]
- Katiyar, A.; Smita, S.; Lenka, S.K.; Rajwanshi, R.; Chinnusamy, V.; Bansal, K.C. Genome-wide classification and expression analysis of MYB transcription factor families in rice and Arabidopsis. BMC Genom. 2012, 13, 544. [Google Scholar] [CrossRef]
- Wilkins, O.; Nahal, H.; Foong, J.; Provart, N.J.; Campbell, M.M. Expansion and diversification of the Populus R2R3-MYB family of transcription factors. Plant Physiol. 2009, 149, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Matus, J.T.; Aquea, F.; Arce-Johnson, P. Analysis of the grape MYB R2R3 subfamily reveals expanded wine quality-related clades and conserved gene structure organization across Vitis and Arabidopsis genomes. BMC Plant Biol. 2008, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, C.; Li, J.; Wang, L.; Ren, Z. Genome-wide identification and characterization of R2R3MYB family in Cucumis sativus. PLoS ONE 2012, 7, e47576. [Google Scholar] [CrossRef]
- Yanhui, C.; Xiaoyuan, Y.; Kun, H.; Meihua, L.; Jigang, L.; Zhaofeng, G.; Zhiqiang, L.; Yunfei, Z.; Xiaoxiao, W.; Xiaoming, Q. The MYB transcription factor superfamily of Arabidopsis: Expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 2006, 60, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Gu, X.; Peterson, T. Identification of conserved gene structures and carboxy-terminal motifs in the Myb gene family of Arabidopsis and Oryza sativa L. ssp. indica. Genome Biol. 2004, 5, R46. [Google Scholar] [CrossRef] [PubMed]
- Ogata, K.; Morikawa, S.; Nakamura, H.; Sekikawa, A.; Inoue, T.; Kanai, H.; Sarai, A.; Ishii, S.; Nishimura, Y. Solution structure of a specific DNA complex of the Myb DNA-binding domain with cooperative recognition helices. Cell 1994, 79, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.; Fuertes, A.; Benito, M.J.; Malpica, J.M.; Leyva, A.; Paz-Ares, J. More than 80R2R3-MYB regulatory genes in the genome of Arabidopsis thaliana. Plant J. 1998, 14, 273–284. [Google Scholar] [CrossRef]
- Xie, Z.; Lee, E.K.; Lucas, J.R.; Morohashi, K.; Li, D.; Murray, J.; Grotewold, S.E. Regulation of Cell Proliferation in the Stomatal Lineage by the Arabidopsis MYB FOUR LIPS via Direct Targeting of Core Cell Cycle Genes. Plant Cell 2010, 22, 2306–2321. [Google Scholar] [CrossRef]
- Howe, K.; Reakes, C.; Watson, R. Characterization of the sequence-specific interaction of mouse c-myb protein with DNA. EMBO J. 1990, 9, 161–169. [Google Scholar] [CrossRef]
- Tanikawa, J.; Yasukawa, T.; Enari, M.; Ogata, K.; Nishimura, Y.; Ishii, S.; Sarai, A. Recognition of specific DNA sequences by the c-myb protooncogene product: Role of three repeat units in the DNA-binding domain. Proc. Natl. Acad. Sci. USA 1993, 90, 9320–9324. [Google Scholar] [CrossRef]
- Boyer, L.A.; Latek, R.R.; Peterson, C.L. The SANT domain: A unique histone-tail-binding module? Nat. Rev. Mol. Cell Biol. 2004, 5, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Lin-Wang, K.; Bolitho, K.; Grafton, K.; Kortstee, A.; Karunairetnam, S.; McGhie, T.K.; Espley, R.V.; Hellens, R.P.; Allan, A.C. An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol. 2010, 10, 50. [Google Scholar] [CrossRef]
- Gális, I.; Šimek, P.; Narisawa, T.; Sasaki, M.; Horiguchi, T.; Fukuda, H.; Matsuoka, K. A novel R2R3 MYB transcription factor NtMYBJS1 is a methyl jasmonate-dependent regulator of phenylpropanoid-conjugate biosynthesis in tobacco. Plant J. 2006, 46, 573–592. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-K.; Ryu, K.H.; Kang, Y.H.; Song, J.H.; Cho, Y.-H.; Yoo, S.-D.; Schiefelbein, J.; Lee, M.M. Cell fate in the Arabidopsis root epidermis is determined by competition between WEREWOLF and CAPRICE. Plant Physiol. 2011, 157, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Stracke, R.; Ishihara, H.; Huep, G.; Barsch, A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2007, 50, 660–677. [Google Scholar] [CrossRef]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef]
- Kang, Y.H.; Kirik, V.; Hulskamp, M.; Nam, K.H.; Hagely, K.; Lee, M.M.; Schiefelbein, J. The MYB23 gene provides a positive feedback loop for cell fate specification in the Arabidopsis root epidermis. Plant Cell 2009, 21, 1080–1094. [Google Scholar] [CrossRef]
- Li, L.; Yu, X.; Thompson, A.; Guo, M.; Yoshida, S.; Asami, T.; Chory, J.; Yin, Y. Arabidopsis MYB30 is a direct target of BES1 and cooperates with BES1 to regulate brassinosteroid-induced gene expression. Plant J. 2009, 58, 275–286. [Google Scholar] [CrossRef]
- Cominelli, E.; Galbiati, M.; Vavasseur, A.; Conti, L.; Sala, T.; Vuylsteke, M.; Leonhardt, N.; Dellaporta, S.L.; Tonelli, C. A guard-cell-specific MYB transcription factor regulates stomatal movements and plant drought tolerance. Curr. Biol. 2005, 15, 1196–1200. [Google Scholar] [CrossRef]
- Seo, P.J.; Park, C.M. MYB96-mediated abscisic acid signals induce pathogen resistance response by promoting salicylic acid biosynthesis in Arabidopsis. New Phytol. 2010, 186, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, T.; Wheatley, K.; Hanzawa, Y.; Wright, L.; Mizoguchi, M.; Song, H.-R.; Carré, I.A.; Coupland, G. LHY and CCA1 are partially redundant genes required to maintain circadian rhythms in Arabidopsis. Dev. Cell 2002, 2, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Schellmann, S.; Schnittger, A.; Kirik, V.; Wada, T.; Okada, K.; Beermann, A.; Thumfahrt, J.; Jürgens, G.; Hülskamp, M. TRIPTYCHON and CAPRICE mediate lateral inhibition during trichome and root hair patterning in Arabidopsis. EMBO J. 2002, 21, 5036–5046. [Google Scholar] [CrossRef] [PubMed]
- Kirik, V.; Simon, M.; Huelskamp, M.; Schiefelbein, J. The Enhancer of try and CPC1 gene acts redundantly with Triptycho and Caprice in trichome and root hair cell patterning in Arabidopsis. Dev. Biol. 2004, 268, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Araki, S.; Matsunaga, S.; Itoh, T.; Nishihama, R.; Machida, Y.; Doonan, J.H.; Watanabe, A. G2/M-phase–specific transcription during the plant cell cycle is mediated by c-Myb–like transcription factors. Plant Cell 2001, 13, 1891–1905. [Google Scholar]
- Burns, C.G.; Ohi, R.; Krainer, A.R.; Gould, K.L. Evidence that Myb-related CDC5 proteins are required for pre-mRNA splicing. Proc. Natl. Acad. Sci. USA 1999, 96, 13789–13794. [Google Scholar] [CrossRef]
- Lei, X.-H.; Shen, X.; Xu, X.-Q.; Bernstein, H.S. Human Cdc5, a regulator of mitotic entry, can act as a site-specific DNA binding protein. J. Cell Sci. 2000, 113, 4523–4531. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Wei, B.; Zhang, R.-Z.; Guo, J.-J.; Liu, D.-M.; Li, A.-L.; Fan, R.-C.; Mao, L.; Zhang, X.-Q. Genome-wide analysis of the MADS-box gene family in Brachypodium distachyon. PLoS ONE 2014, 9, e84781. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef]
- Gaut, B.S.; Morton, B.R.; McCaig, B.C.; Clegg, M.T. Substitution rate comparisons between grasses and palms: Synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. USA 1996, 93, 10274–10279. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Li, H.; Huang, X.; Li, W.; Lu, Y.; Dai, X.; Zhou, Z.; Li, Q. MicroRNA comparison between poplar and larch provides insight into the different mechanism of wood formation. Plant Cell Rep. 2020, 39, 1199–1217. [Google Scholar] [CrossRef] [PubMed]
- Sazegari, S.; Niazi, A.; Ahmadi, F.S. A study on the regulatory network with promoter analysis for Arabidopsis DREB-genes. Bioinformation 2015, 11, 101. [Google Scholar] [CrossRef]
- Wu, Y.; Ke, Y.; Wen, J.; Guo, P.; Ran, F.; Wang, M.; Liu, M.; Li, P.; Li, J.; Du, H. Evolution and expression analyses of the MADS-box gene family in Brassica napus. PLoS ONE 2018, 13, e0200762. [Google Scholar] [CrossRef]
- Brown, A.; Dunn, M.; Goddard, N.; Hughes, M. Identification of a novel low-temperature-response element in the promoter of the barley (Hordeum vulgare L.) gene blt101.1. Planta 2001, 213, 770–780. [Google Scholar] [CrossRef]
- Jiang, C.-K.; Rao, G.-Y. Insights into the diversification and evolution of R2R3-MYB transcription factors in plants. Plant Physiol. 2020, 183, 637–655. [Google Scholar] [CrossRef]
- Wu, Y.; Wen, J.; Xia, Y.; Zhang, L.; Du, H. Evolution and functional diversification of R2R3-MYB transcription factors in plants. Hortic. Res. 2022, 9, uhac058. [Google Scholar] [CrossRef] [PubMed]
- Airoldi, C.A.; Davies, B. Gene duplication and the evolution of plant MADS-box transcription factors. J. Genet. Genom. 2012, 39, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-L.; Irish, V.F. Gene duplication and loss in a MADS box gene transcription factor circuit. Mol. Biol. Evol. 2011, 28, 3367–3380. [Google Scholar] [CrossRef] [PubMed]
- Gaut, B.S. Evolutionary dynamics of grass genomes. New Phytol. 2002, 154, 15–28. [Google Scholar] [CrossRef]
- Initiative, T. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar]
- D’hont, A.; Denoeud, F.; Aury, J.-M.; Baurens, F.-C.; Carreel, F.; Garsmeur, O.; Noel, B.; Bocs, S.; Droc, G.; Rouard, M. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012, 488, 213–217. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Jia, C.; Miao, H.; Zhang, J.; Liu, J.; Xu, B.; Jin, Z. Genome-wide identification and transcript analysis of TCP gene family in Banana (Musa acuminata L.). Biochem. Genet. 2022, 60, 204–222. [Google Scholar] [CrossRef]
- Ma, K.; Xiao, J.; Li, X.; Zhang, Q.; Lian, X. Sequence and expression analysis of the C3HC4-type RING finger gene family in rice. Gene 2009, 444, 33–45. [Google Scholar] [CrossRef]
- Bai, J.; Sun, F.; Wang, M.; Su, L.; Li, R.; Caetano-Anollés, G. Genome-wide analysis of the MYB-CC gene family of maize. Genetica 2019, 147, 1–9. [Google Scholar] [CrossRef]
- Ehsan, H.; Reichheld, J.-P.; Durfee, T.; Roe, J.L. TOUSLED kinase activity oscillates during the cell cycle and interacts with chromatin regulators. Plant Physiol. 2004, 134, 1488–1499. [Google Scholar] [CrossRef]
- Rawat, R.; Schwartz, J.; Jones, M.A.; Sairanen, I.; Cheng, Y.; Andersson, C.R.; Zhao, Y.; Ljung, K.; Harmer, S.L. REVEILLE1, a Myb-like transcription factor, integrates the circadian clock and auxin pathways. Proc. Natl. Acad. Sci. USA 2009, 106, 16883–16888. [Google Scholar] [CrossRef] [PubMed]
- Gatz, C. From pioneers to team players: TGA transcription factors provide a molecular link between different stress pathways. Mol. Plant-Microbe Interact. 2013, 26, 151–159. [Google Scholar] [CrossRef]
- Yan, Y.; Shen, L.; Chen, Y.; Bao, S.; Thong, Z.; Yu, H. A MYB-domain protein EFM mediates flowering responses to environmental cues in Arabidopsis. Dev. Cell 2014, 30, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, K.; Sheng, S.; Wang, M.; Hua, P.; Wang, Y.; Chen, P.; Wang, K.; Zhao, M.; Wang, Y. Transcriptome analysis of MYB transcription factors family and PgMYB genes involved in salt stress resistance in Panax ginseng. BMC Plant Biol. 2022, 22, 479. [Google Scholar] [CrossRef] [PubMed]
- Tombuloglu, H.; Kekec, G.; Sakcali, M.S.; Unver, T. Transcriptome-wide identification of R2R3-MYB transcription factors in barley with their boron responsive expression analysis. Mol. Genet. Genom. 2013, 288, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cai, Z.; Chen, S.; Wang, D.; Wu, Z. Transcriptome-wide identification and characterization of the MYB gene family in sickle seagrass (Thalassia hemprichii). Ecol. Genet. Genom. 2021, 20, 100093. [Google Scholar] [CrossRef]
- Bowman, J.L.; Kohchi, T.; Yamato, K.T.; Jenkins, J.; Shu, S.; Ishizaki, K.; Yamaoka, S.; Nishihama, R.; Nakamura, Y.; Berger, F. Insights into land plant evolution garnered from the Marchantia polymorpha genome. Cell 2017, 171, 287–304.e15. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Li, P.; Wen, J.; Chen, P.; Guo, P.; Ke, Y.; Wang, M.; Liu, M.; Tran, L.-S.P.; Li, J.; Du, H. MYB superfamily in Brassica napus: Evidence for hormone-mediated expression profiles, large expansion, and functions in root hair development. Biomolecules 2020, 10, 875. [Google Scholar] [CrossRef]
- Yin, Y.; Guo, C.; Shi, H.; Zhao, J.; Ma, F.; An, W.; He, X.; Luo, Q.; Cao, Y.; Zhan, X. Genome-wide comparative analysis of the R2R3-MYB gene family in five Solanaceae species and identification of members regulating carotenoid biosynthesis in Wolfberry. Int. J. Mol. Sci. 2022, 23, 2259. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujita, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. ABA-mediated transcriptional regulation in response to osmotic stress in plants. J. Plant Res. 2011, 124, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.-J.; Do Choi, Y. Methyl jasmonate as a vital substance in plants. Trends Genet. 2003, 19, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Czechowski, T.; Bari, R.P.; Stitt, M.; Scheible, W.R.; Udvardi, M.K. Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: Unprecedented sensitivity reveals novel root-and shoot-specific genes. Plant J. 2004, 38, 366–379. [Google Scholar] [CrossRef]
- Cheung, A.Y.; Wu, H.-m. Overexpression of an Arabidopsis formin stimulates supernumerary actin cable formation from pollen tube cell membrane. Plant Cell 2004, 16, 257–269. [Google Scholar] [CrossRef]
- Sparvoli, F.; Martin, C.; Scienza, A.; Gavazzi, G.; Tonelli, C. Cloning and molecular analysis of structural genes involved in flavonoid and stilbene biosynthesis in grape (Vitis vinifera L.). Plant Mol. Biol. 1994, 24, 743–755. [Google Scholar] [CrossRef]
- Abrahams, S.; Lee, E.; Walker, A.R.; Tanner, G.J.; Larkin, P.J.; Ashton, A.R. The Arabidopsis TDS4 gene encodes leucoanthocyanidin dioxygenase (LDOX) and is essential for proanthocyanidin synthesis and vacuole development. Plant J. 2003, 35, 624–636. [Google Scholar] [CrossRef]
- Gauthier, A.; Gulick, P.J.; Ibrahim, R.K. Characterization of two cDNA clones which encode O-methyltransferases for the methylation of both flavonoid and phenylpropanoid compounds. Arch. Biochem. Biophys. 1998, 351, 243–249. [Google Scholar] [CrossRef]
- Shimada, N.; Aoki, T.; Sato, S.; Nakamura, Y.; Tabata, S.; Ayabe, S.-i. A cluster of genes encodes the two types of chalcone isomerase involved in the biosynthesis of general flavonoids and legume-specific 5-deoxy (iso) flavonoids in Lotus japonicus. Plant Physiol. 2003, 131, 941–951. [Google Scholar] [CrossRef]
- Jiang, L.; Yue, M.; Liu, Y.; Ye, Y.; Zhang, Y.; Lin, Y.; Wang, X.; Chen, Q.; Tang, H. Alterations of phenylpropanoid biosynthesis lead to the natural formation of pinkish-skinned and white-fleshed strawberry (Fragaria× ananassa). Int. J. Mol. Sci. 2022, 23, 7375. [Google Scholar] [CrossRef]
- Xie, L.; Li, F.; Zhang, S.; Zhang, H.; Qian, W.; Li, P.; Zhang, S.; Sun, R. Mining for candidate genes in an introgression line by using RNA sequencing: The anthocyanin overaccumulation phenotype in Brassica. Front. Plant Sci. 2016, 7, 1245. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Wen, X.; Wei, M.; Huang, X.; Dai, S.; Ruan, L.; Yu, Y. Genome-Wide Identification, Characterization, and Expression Pattern of MYB Gene Family in Melastoma candidum. Horticulturae 2023, 9, 708. https://doi.org/10.3390/horticulturae9060708
Li H, Wen X, Wei M, Huang X, Dai S, Ruan L, Yu Y. Genome-Wide Identification, Characterization, and Expression Pattern of MYB Gene Family in Melastoma candidum. Horticulturae. 2023; 9(6):708. https://doi.org/10.3390/horticulturae9060708
Chicago/Turabian StyleLi, Hui, Xiaoxia Wen, Mingke Wei, Xiong Huang, Seping Dai, Lin Ruan, and Yixun Yu. 2023. "Genome-Wide Identification, Characterization, and Expression Pattern of MYB Gene Family in Melastoma candidum" Horticulturae 9, no. 6: 708. https://doi.org/10.3390/horticulturae9060708
APA StyleLi, H., Wen, X., Wei, M., Huang, X., Dai, S., Ruan, L., & Yu, Y. (2023). Genome-Wide Identification, Characterization, and Expression Pattern of MYB Gene Family in Melastoma candidum. Horticulturae, 9(6), 708. https://doi.org/10.3390/horticulturae9060708