


Phylogeny, Expression Profiling, and Coexpression Networks Reveals the Critical Roles of Nucleotide-BindingLeucine-Rich Repeats on Valsa Canker Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Genetic Identification of NLRs of 19 Species
2.2. Phylogenetic and Expansion Rate Analysis
2.3. Gene Duplication Events Analysis
2.4. Expression Analysis of NLR Gene in Response to Vp Metabolites
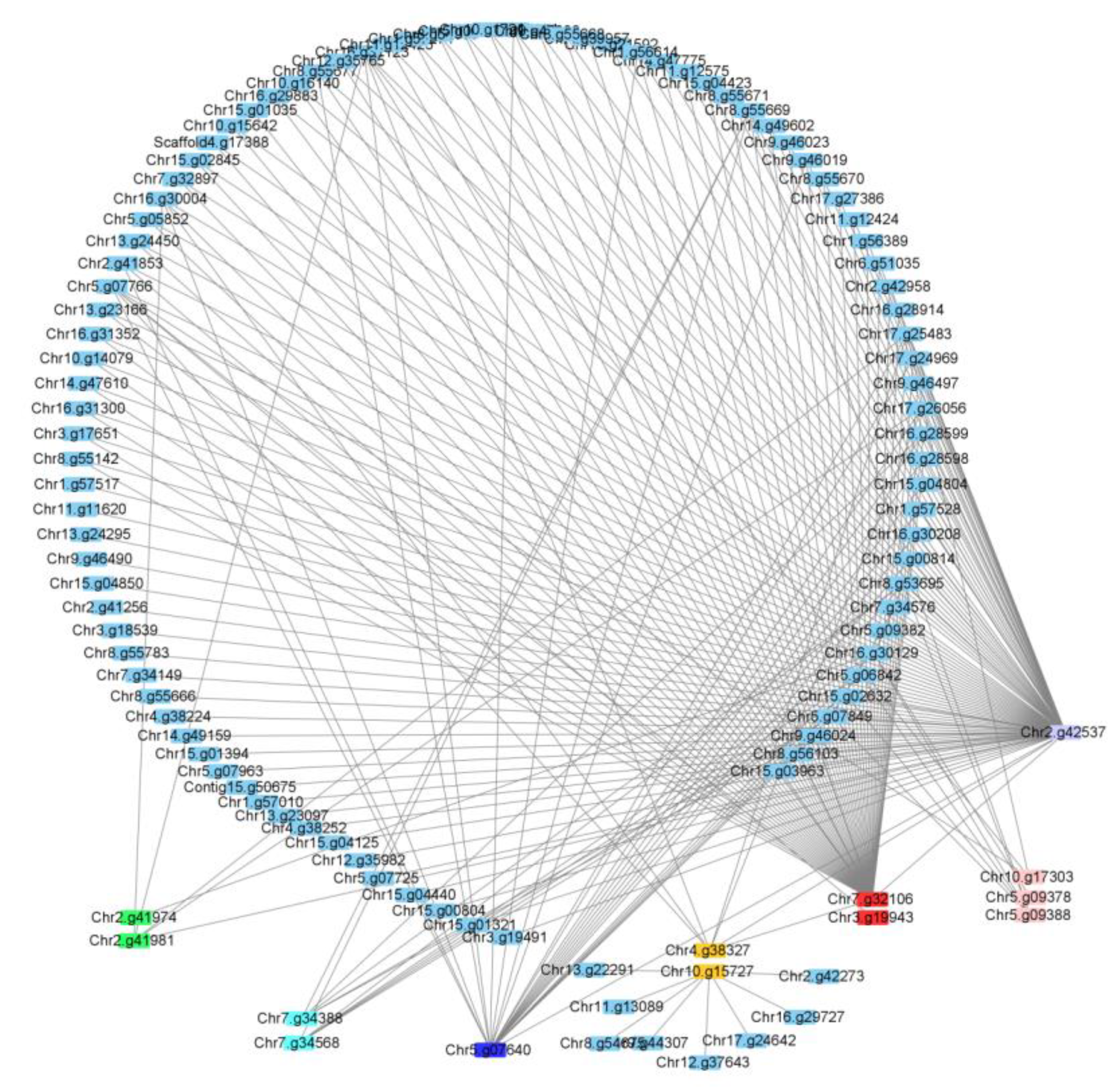
2.5. Co-Expression Network Analysis
2.6. Quantitative Real-Time-PCR Assays
2.7. Statistical Analysis
3. Results
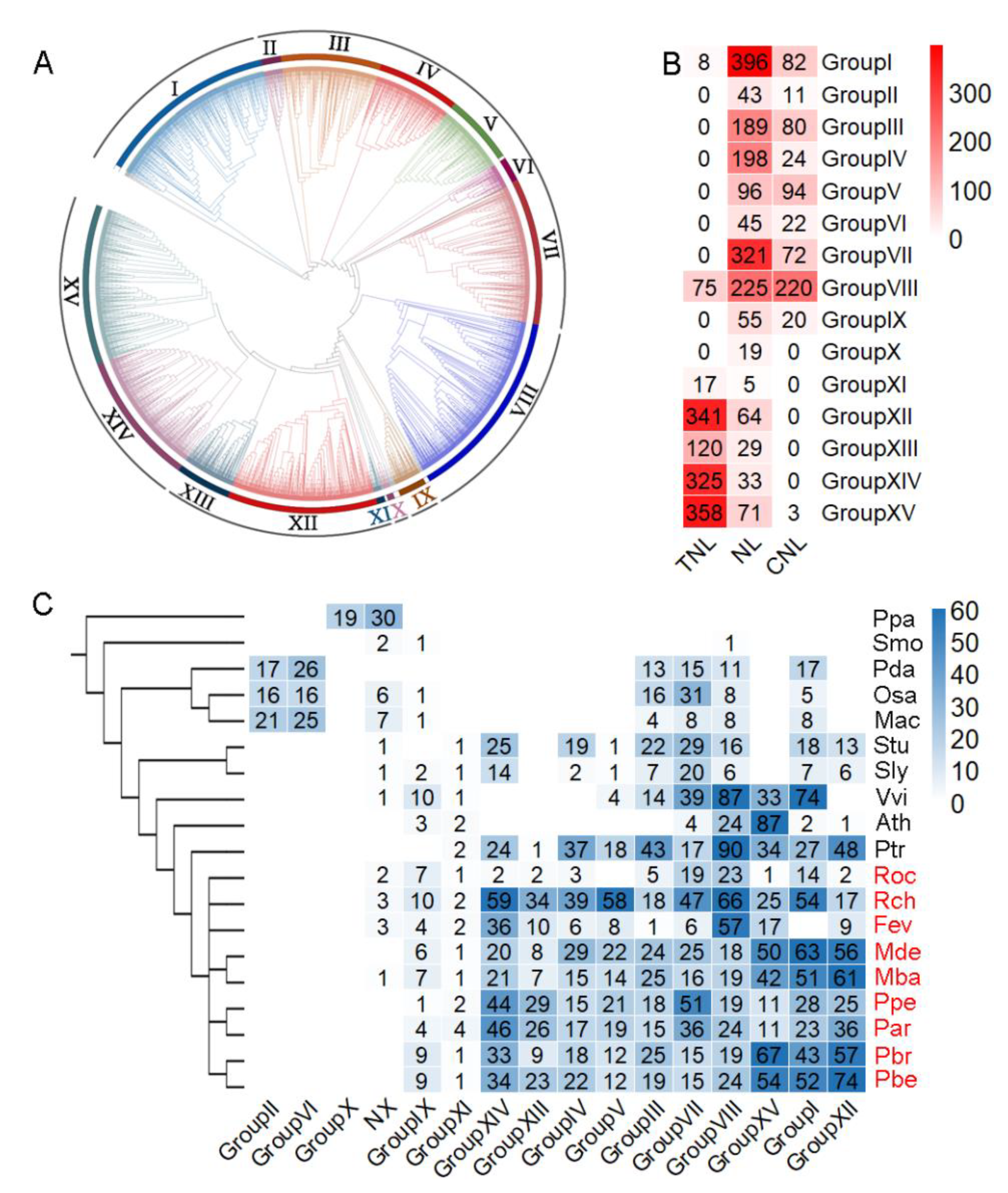
3.1. Genome-Wide Identification and Domain Composition of NBS-Encoding Genes
3.2. Classification of NLRs Based on Phylogenetic Tree Construction
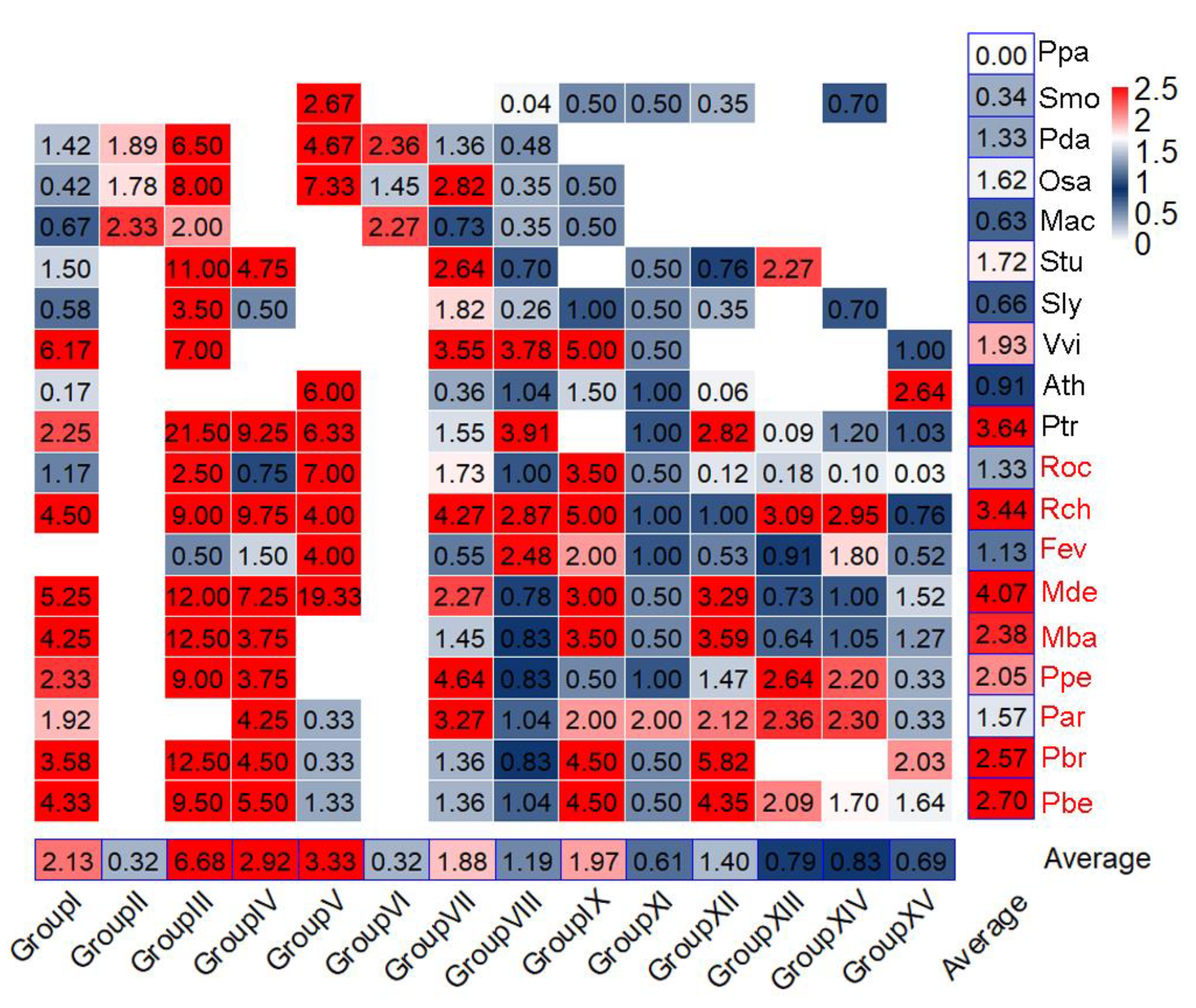
3.3. Expansion of NLRs among Species and Groups
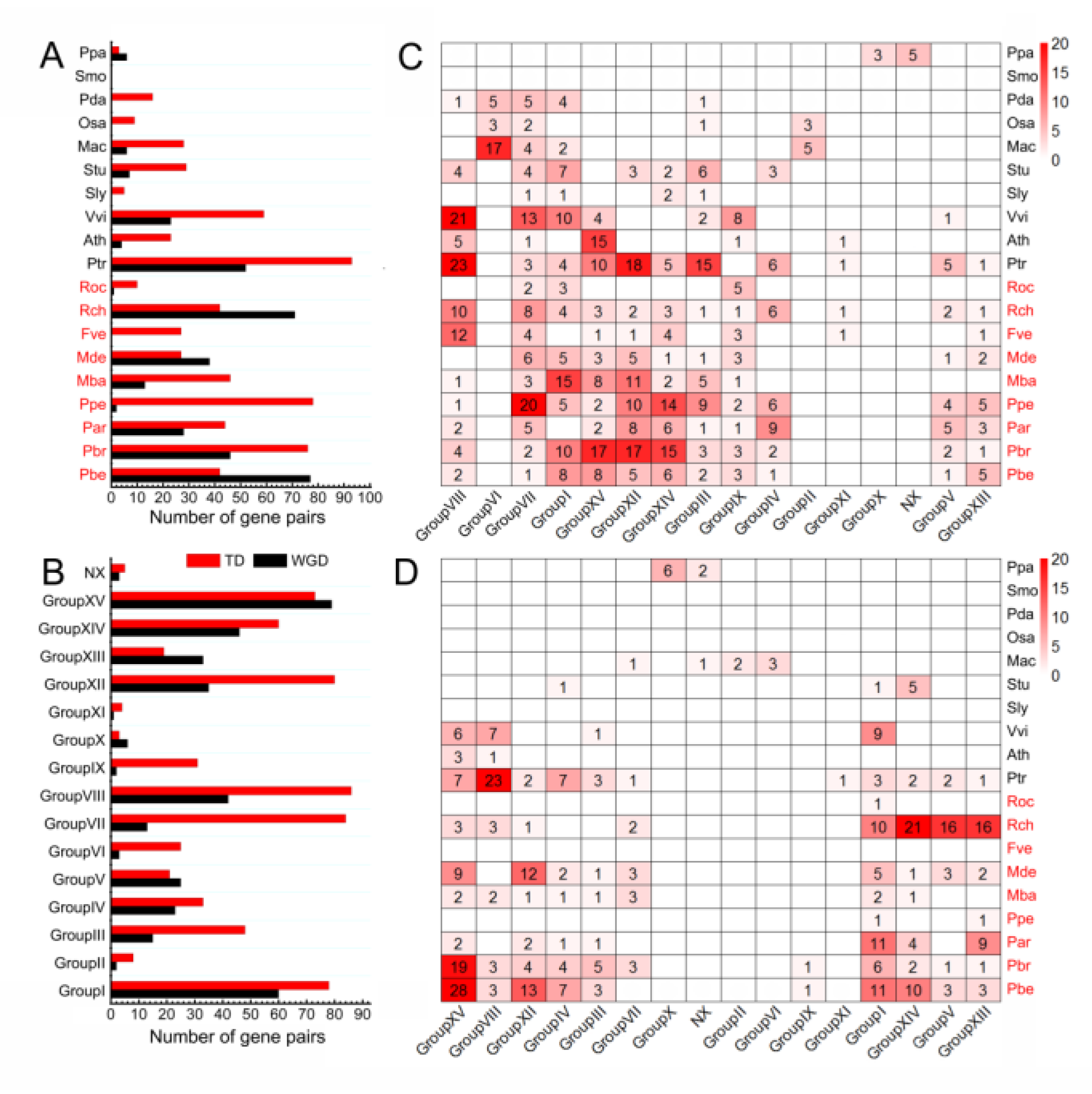
3.4. TD Events Are More Important for the Rapid Expansion of NLRs
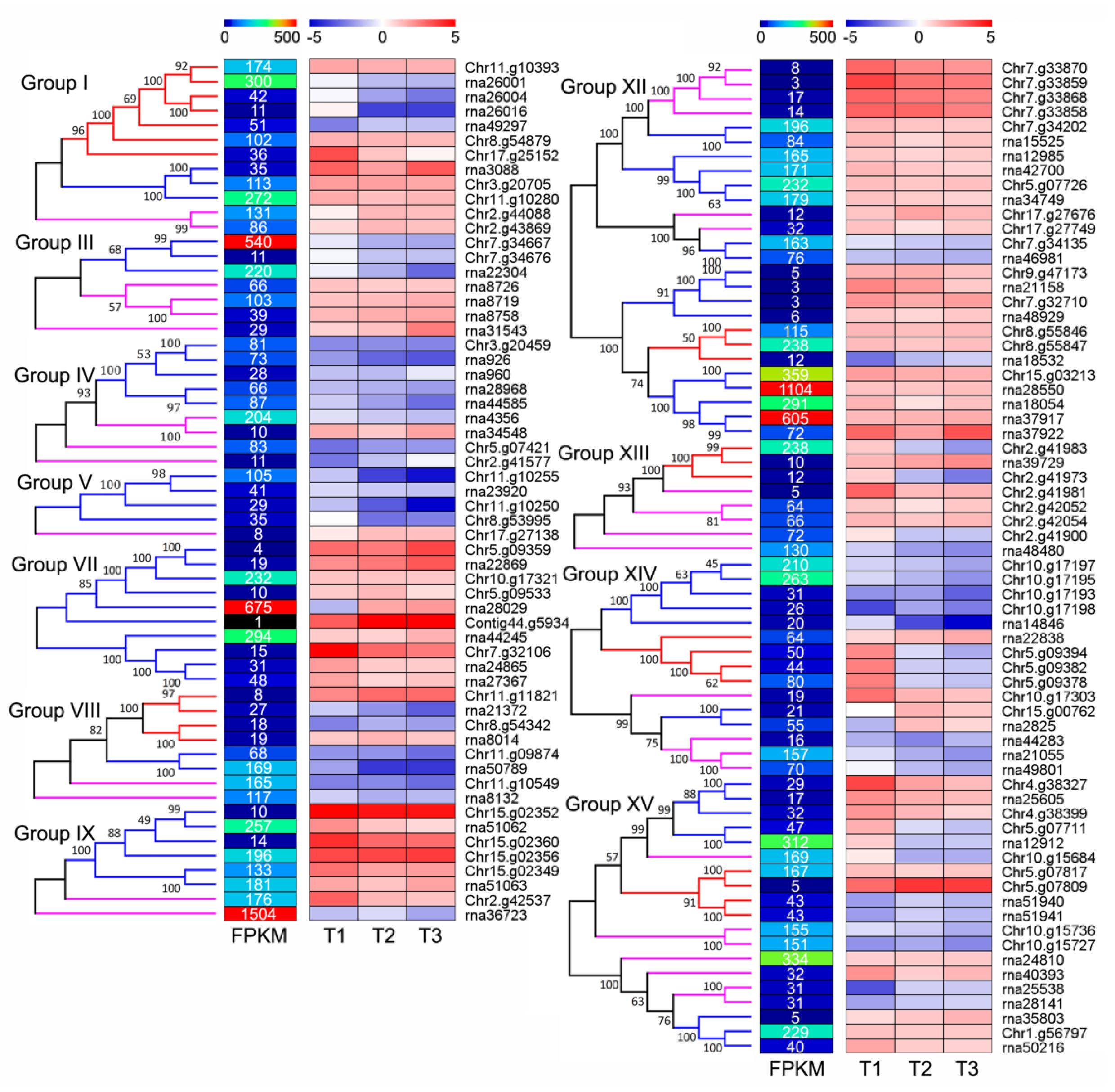
3.5. NLRs Are Low Expressed in Various Tissues under Normal Growth Conditions
3.6. Large Numbers of NLRs Respond to Signals of Valsa Canker
3.7. NLRs Take a Crucial Role in ‘Duli-G03’ Response to Vm Signals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lang, J.; Genot, B.; Bigeard, J.; Colcombet, J. MPK3 and MPK6 control salicylic acid signaling by up-regulating NLR receptors during pattern- and effector-triggered immunity. J. Exp. Bot. 2022, 73, 2190–2205. [Google Scholar] [CrossRef] [PubMed]
- Sukarta, O.; Zheng, Q.; Slootweg, E.; Mekken, M.; Mendel, M.; Putker, V.; Bertran, A.; Brand, A.; Overmars, H.; Pomp, R.; et al. GLYCINE-RICH RNA-BINDING PROTEIN 7 potentiates effector-triggered immunity through an RNA recognition motif. Plant Physiol. 2022, 189, 972–987. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Ngou, B.; Ding, P.; Xin, X. PTI-ETI crosstalk: An integrative view of plant immunity. Curr. Opin. Plant Biol. 2021, 62, 102030. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Chen, S.; Wu, Z.; Ao, K.; Yaghmaiean, H.; Sun, T.; Huang, W.; Xu, F.; Zhang, Y.; Wang, S.; et al. Activation of TIR signaling is required for pattern-triggered immunity. Nature 2021, 598, 500–503. [Google Scholar] [CrossRef]
- Yuan, M.; Jiang, Z.; Bi, G.; Nomura, K.; Liu, M.; Wang, Y.; Cai, B.; Zhou, J.; He, S.; Xin, X. Pattern-recognition receptors are required for NLR-mediated plant immunity. Nature 2021, 592, 105–109. [Google Scholar] [CrossRef]
- Schulze, S.; Yu, L.; Ehinger, A.; Kolb, D.; Saile, S.; Stahl, M.; Kemmerling, B. The TIR-NBS-LRR protein CSA1 is required for autoimmune cell death in Arabidopsis pattern recognition co-receptor bak1 and bir3 mutants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Monteiro, F.; Nishimura, M. Structural, Functional, and Genomic Diversity of Plant NLR Proteins: An Evolved Resource for Rational Engineering of Plant Immunity. Annu. Rev. Phytopathol. 2018, 56, 243–267. [Google Scholar] [CrossRef]
- Collier, S.; Hamel, L.; Moffett, P. Cell death mediated by the N-terminal domains of a unique and highly conserved class of NB-LRR protein. Mol. Plant Microbe Interact. 2011, 24, 918–931. [Google Scholar] [CrossRef]
- Dodds, P.; Lawrence, G.; Catanzariti, A.; Teh, T.; Wang, C.; Ayliffe, M.; Kobe, B.; Ellis, J. Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc. Natl. Acad. Sci. USA 2006, 103, 8888–8893. [Google Scholar] [CrossRef]
- De la Concepcion, J.; Franceschetti, M.; MacLean, D.; Terauchi, R.; Kamoun, S.; Banfield, M. Protein engineering expands the effector recognition profile of a rice NLR immune receptor. eLife 2019, 8, e47713. [Google Scholar] [CrossRef]
- Axtell, M.; Staskawicz, B. Initiation of RPS2-specified disease resistance in Arabidopsis is coupled to the AvrRpt2-directed elimination of RIN4. Cell 2003, 112, 369–377. [Google Scholar] [CrossRef]
- Castel, B.; Ngou, P.; Cevik, V.; Redkar, A.; Kim, D.; Yang, Y.; Ding, P.; Jones, J. Diverse NLR immune receptors activate defence via the RPW8-NLR NRG1. N. Phytol. 2019, 222, 966–980. [Google Scholar] [CrossRef]
- Jubic, L.; Saile, S.; Furzer, O.; El Kasmi, F.; Dangl, J. Help wanted: Helper NLRs and plant immune responses. Curr. Opin. Plant Biol. 2019, 50, 82–94. [Google Scholar] [CrossRef]
- Lapin, D.; Kovacova, V.; Sun, X.; Dongus, J.; Bhandari, D.; von Born, P.; Bautor, J.; Guarneri, N.; Rzemieniewski, J.; Stuttmann, J.; et al. A Coevolved EDS1-SAG101-NRG1 Module Mediates Cell Death Signaling by TIR-Domain Immune Receptors. Plant Cell 2019, 31, 2430–2455. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Seong, K.; Thomazella, D.; Kim, J.; Pham, J.; Seo, E.; Cho, M.; Schultink, A.; Staskawicz, B. NRG1 functions downstream of EDS1 to regulate TIR-NLR-mediated plant immunity in Nicotiana benthamiana. Proc. Natl. Acad. Sci. USA 2018, 115, E10979–E10987. [Google Scholar] [CrossRef]
- Shao, Z.; Xue, J.; Wu, P.; Zhang, Y.; Wu, Y.; Hang, Y.; Wang, B.; Chen, J. Large-Scale Analyses of Angiosperm Nucleotide-Binding Site-Leucine-Rich Repeat Genes Reveal Three Anciently Diverged Classes with Distinct Evolutionary Patterns. Plant Physiol. 2016, 170, 2095–2109. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Xue, J.; Wang, Q.; Wang, B.; Chen, J. Revisiting the Origin of Plant NBS-LRR Genes. Trends Plant Sci. 2019, 24, 9–12. [Google Scholar] [CrossRef]
- Nam, S.; Ko, G. Analysis of structural types and design factors for fruit tree greenhouses. Prot. Hortic. Plant Fact. 2013, 22, 27–33. [Google Scholar] [CrossRef]
- Mathews, C.; Brown, M.; Bottrell, D. Leaf extrafloral nectaries enhance of a key economic pest; Grapholita molesta (Lepidoptera: Tortricidae); in peach (Rosales: Rosaceae). Environ. Entomol. 2007, 36, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Liu, H.; Li, Z.; Ke, X.; Dou, D.; Gao, X.; Song, N.; Dai, Q.; Wu, Y.; Xu, J.; et al. Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. N. Phytol. 2015, 208, 1202–1216. [Google Scholar] [CrossRef]
- Wang, S.; Hu, T.; Wang, Y.; Luo, Y.; Michailides, T.; Cao, K. New understanding on infection processes of Valsa canker of apple in China. Eur. J. Plant Pathol. 2016, 146, 531–540. [Google Scholar] [CrossRef]
- Wang, N.; Gao, K.; Han, N.; Tian, R.; Zhang, J.; Yan, X.; Huang, L. ChbB increases antifungal activity of Bacillus amyloliquefaciens against Valsa mali and shows synergistic action with bacterial chitinases. Biol. Control 2019, 142, 104150. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Y.; Han, L.; Zhang, X.; Feng, J. Potential use of cuminic acid as a botanical fungicide against Valsa mali. Microb. Pathog. 2017, 106, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Zuo, C.; Liu, H.; Chen, B. Research progress in pathogen-host interaction mechanism and integrated control of apple Valsa canker. Int. J. Fruit Sci. 2019, 36, 240–249. [Google Scholar] [CrossRef]
- Tan, Y.; Lv, S.; Liu, X.; Gao, T.; Li, T.; Wang, Y.; Han, Z. Development of high-density interspecific genetic maps for the identification of QTLs conferring resistance to Valsa ceratosperma in apple. Euphytica 2017, 213, 10–20. [Google Scholar] [CrossRef]
- Steuernagel, B.; Jupe, F.; Witek, K.; Jones, J.; Wulff, B. NLR-parser: Rapid annotation of plant NLR complements. Bioinformatics 2015, 31, 1665–1667. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, A.; Jiang, T.; Keating, A.; Berger, B. Paircoil2: Improved prediction of coiled coils from sequence. Bioinformatics 2006, 22, 356–358. [Google Scholar] [CrossRef]
- Van de Weyer, A.; Monteiro, F.; Furzer, O.; Nishimura, M.; Cevik, V.; Witek, K.; Jones, J.; Dangl, J.; Weigel, D.; Bemm, F. A Species-Wide Inventory of NLR Genes and Alleles in Arabidopsis thaliana. Cell 2019, 178, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Rozewicki, J.; Li, S.; Amada, K.; Standley, D.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Price, M.; Dehal, P.; Arkin, A. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Fischer, I.; Diévart, A.; Droc, G.; Dufayard, J.; Chantret, N. Evolutionary Dynamics of the Leucine-Rich Repeat Receptor-Like Kinase (LRR-RLK) Subfamily in Angiosperms. Plant Physiol. 2016, 170, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Sun, E.; Mao, X.; Chen, Z.; Xu, T.; Zuo, L.; Jiang, D.; Cao, Y.; Zuo, C. Evolutionary and functional analysis reveals the crucial roles of receptor-like proteins in resistance to Valsa canker in Rosaceae. J. Exp. Bot. 2023, 74, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157–170. [Google Scholar] [CrossRef]
- Wang, M.; Yue, H.; Feng, K.; Deng, P.; Song, W.; Nie, X. Genome-wide identification; phylogeny and expressional profiles of mitogen activated protein kinase kinase kinase (MAPKKK) gene family in bread wheat (Triticum aestivum L.). BMC Genom. 2016, 17, 668–689. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Yuan, S.; Chen, J.; Chen, Y.; Li, Z.; Lin, W.; Zhang, Y.; Liu, J.; Lin, W. Molecular evolution and lineage-specific expansion of the PP2C family in Zea mays. Planta 2019, 250, 1521–1538. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Qiao, X.; Yin, H.; Li, L.; Wang, R.; Wu, J.; Wu, J.; Zhang, S. Different Modes of Gene Duplication Show Divergent Evolutionary Patterns and Contribute Differently to the Expansion of Gene Families Involved in Important Fruit Traits in Pear (Pyrus bretschneideri). Front. Plant Sci. 2018, 9, 161. [Google Scholar] [CrossRef]
- Zhao, D.; Tian, Y.; Yu, H.; Mao, X.; Wang, C.; Duo, H.; Zuo, C. Establishment of the “Valsa pyri metabolites (VpM)-suspension cell”-based system to study the response of pears to VpM. Physiol. Mol. Plant Pathol. 2022, 120, 101850. [Google Scholar] [CrossRef]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550–570. [Google Scholar] [CrossRef]
- Duo, H.; Yu, H.; Sun, E.; Zhao, D.; Zuo, C. RNA sequencing reveals that cell wall, Ca2+, hypersensitive response and salicylic acid signals are involved in pear suspension cells responses to Valsa pyri infection. Sci. Hortic. 2022, 305, 111422. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 498–504. [Google Scholar] [CrossRef]
- Arocho, A.; Chen, B.; Ladanyi, M.; Pan, Q. Validation of the 2−ΔΔCt calculation as an alternate method of data analysis for quantitative PCR of BCR-ABL P210 transcripts. Diagn. Mol. Pathol. 2006, 15, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.; Kozik, A.; Griego, A.; Kuang, H.; Michelmore, R. Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 2003, 15, 809–834. [Google Scholar] [CrossRef]
- Seong, K.; Seo, E.; Witek, K.; Li, M.; Staskawicz, B. Evolution of NLR resistance genes with noncanonical N-terminal domains in wild tomato species. N. Phytol. 2020, 227, 1530–1543. [Google Scholar] [CrossRef]
- Celton, J.; Gaillard, S.; Bruneau, M.; Pelletier, S.; Aubourg, S.; Martin-Magniette, M.; Renou, J. Widespread anti-sense transcription in apple is correlated with si RNA production and indicates a large potential for transcriptional and/or post-transcriptional control. N. Phytol. 2014, 203, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, J.; Keller, B. NLR immune receptors and diverse types of non-NLR proteins control race-specific resistance in Triticeae. Curr. Opin. Plant Biol. 2021, 62, 102053. [Google Scholar] [CrossRef]
- Guo, Y.; Fitz, J.; Schneeberger, K.; Ossowski, S.; Cao, J.; Weigel, D. Genome-wide comparison of nucleotide-binding site-leucine-rich repeat-encoding genes in Arabidopsis. Plant Physiol. 2011, 157, 757–769. [Google Scholar] [CrossRef]
- Huang, Z.; Qiao, F.; Yang, B.; Liu, J.; Liu, Y.; Wulff, B.; Hu, P.; Lv, Z.; Zhang, R.; Chen, P.; et al. Genome-wide identification of the NLR gene family in Haynaldia villosa by SMRT-RenSeq. BMC Genom. 2022, 23, 118–140. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, X.; Shao, Z. Genome-Wide Analysis of NLR Disease Resistance Genes in an Updated Reference Genome of Barley. Front. Genet. 2021, 12, 694682. [Google Scholar] [CrossRef]
- Seo, E.; Kim, S.; Yeom, S.I.; Choi, D. Genome-Wide Comparative Analyses Reveal the Dynamic Evolution of Nucleotide-Binding Leucine-Rich Repeat Gene Family among Solanaceae Plants. Front. Plant Sci. 2016, 7, 1205. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Zhang, Y.; Hang, Y.; Xue, J.; Zhou, G.; Wu, P.; Wu, X.; Wu, X.; Wang, Q.; Wang, B.; et al. Long-term evolution of nucleotide-binding site-leucine-rich repeat genes: Understanding gained from and beyond the legume family. Plant Physiol. 2014, 166, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Xu, X.; Kong, W.; Xia, X.; Zhang, S.; Liu, L.; Zou, L. Genome-wide identification and expression analysis of rice NLR genes responsive to the infections of Xanthomonas oryzae pv. oryzae and Magnaporthe oryzae. Physiol. Mol. Plant Pathol. 2020, 111, 101488. [Google Scholar] [CrossRef]
- Wang, G.; He, Y.; Strauch, R.; Olukolu, B.; Nielsen, D.; Li, X.; Balint-Kurti, P. Maize Homologs of Hydroxycinnamoyltransferase; a Key Enzyme in Lignin Biosynthesis; Bind the Nucleotide Binding Leucine-Rich Repeat Rp1 Proteins to Modulate the Defense Response. Plant Physiol. 2015, 169, 2230–2243. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Chae, E. Variation Patterns of NLR Clusters in Arabidopsis thaliana Genomes. Plant Commun. 2020, 1, 100089. [Google Scholar] [CrossRef]
- Arya, P.; Kumar, G.; Acharya, V.; Singh, A. Genome-wide identification and expression analysis of NBS-encoding genes in Malus × domestica and expansion of NBS genes family in Rosaceae. PLoS ONE 2014, 9, e107987. [Google Scholar] [CrossRef]
- Jia, Y.; Yuan, Y.; Zhang, Y.; Yang, S.; Zhang, X. Extreme expansion of NBS-encoding genes in Rosaceae. BMC Genet. 2015, 16, 48–59. [Google Scholar] [CrossRef]
- Xu, Q.; Wen, X.; Deng, X. Phylogenetic and evolutionary analysis of NBS-encoding genes in Rosaceae fruit crops. Mol. Phylogenet. Evol. 2007, 44, 315–324. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, Z.; Zhang, Y.; Li, Q.; Jiang, X.; Jiang, Z.; Tang, J.; Chen, D.; Wang, Q.; Chen, J.; et al. An angiosperm NLR Atlas reveals that NLR gene reduction is associated with ecological specialization and signal transduction component deletion. Mol. Plant 2021, 14, 2015–2031. [Google Scholar] [CrossRef]
- Meyers, B.; Dickerman, A.; Michelmore, R.; Sivaramakrishnan, S.; Sobral, B.; Young, N. Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant 1999, 20, 317–332. [Google Scholar] [CrossRef]
- Bai, J.; Pennill, L.; Ning, J.; Lee, S.; Ramalingam, J.; Webb, C.; Zhao, B.; Sun, Q.; Nelson, J.; Leach, J.; et al. Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res. 2002, 12, 1871–1884. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, M.; Sun, L.; Wang, Y.; Yin, J.; Liu, J.; Sun, X.; Hang, Y. Genome-Wide Identification and Evolutionary Analysis of NBS-LRR Genes From Dioscorea rotundata. Front. Genet. 2020, 11, 484. [Google Scholar] [CrossRef] [PubMed]
- Bashir, S.; Rehman, N.; Zaman, F.; Naeem, M.; Jamal, A.; Tellier, A.; Ilyas, M.; Silva Arias, G.; Khan, M. Genome-wide characterization of the NLR gene family in tomato (Solanum lycopersicum) and their relatedness to disease resistance. Front. Genet. 2022, 13, 931580. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S. Evolution of Gene Duplication in Plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef]
- Lehti-Shiu, M.; Zou, C.; Hanada, K.; Shiu, S. Evolutionary history and stress regulation of plant receptor-like kinase/pelle genes. Plant Physiol. 2009, 150, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xun, Q.; Guo, Y.; Zhang, J.; Cheng, K.; Shi, T.; He, K.; Hou, S.; Gou, X.; Li, J. Genome-Wide Expression Pattern Analyses of the Arabidopsis Leucine-Rich Repeat Receptor-Like Kinases. Mol. Plant 2016, 9, 289–300. [Google Scholar] [CrossRef]
- Zuo, C.; Liu, Y.; Guo, Z.; Mao, J.; Chu, M.; Chen, B. Genome-wide annotation and expression responses to biotic stresses of the WALL-ASSOCIATED KINASE-RECEPTOR-LIKE KINASE (WAK-RLK) gene family in Apple (Malus domestica). Eur. J. Plant Pathol. 2019, 153, 771–785. [Google Scholar] [CrossRef]
- Zuo, C.; Liu, H.; Lv, Q.; Chen, Z.; Tian, Y.; Mao, J.; Chen, B. Genome-wide analysis of the apple (Malus domestica) cysteine-rich receptor-like kinase (CRK) family: Annotation, genomic organization, and expression profiles in response to fungal infection. Plant Mol. Biol. Rep. 2020, 38, 14–24. [Google Scholar] [CrossRef]
- Bellande, K.; Bono, J.; Savelli, B.; Jamet, E.; Canut, H. Plant Lectins and Lectin Receptor-Like Kinases: How Do They Sense the Outside? Int. J. Mol. Sci. 2017, 18, 1164. [Google Scholar] [CrossRef]
- Mao, X.; Ding, S.; Tian, Y.; Chen, B.; Mao, J.; Ma, Z.; Zuo, C. Transcriptomic Analysis Revealed Hormone-Related and Receptor-Like Kinase Genes Involved in Wound Healing of ‘Duli’ and its Resistance to Valsa Pyri. Plant Mol. Biol. Rep. 2022, 40, 271–283. [Google Scholar] [CrossRef]
- Zuo, C.; Zhang, W.; Ma, Z.; Chu, M.; Mao, J.; An, Z.; Chen, B. Genome-wide identification and expression analysis of the CrRLK1L gene family in apple (Malus domestica). Plant Mol. Biol. Rep. 2018, 36, 844–857. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Yu, H.; Tian, D.; Sun, E.; Zuo, L.; Jiang, D.; Zuo, C.; Fan, R. Phylogeny, Expression Profiling, and Coexpression Networks Reveals the Critical Roles of Nucleotide-BindingLeucine-Rich Repeats on Valsa Canker Resistance. Horticulturae 2023, 9, 345. https://doi.org/10.3390/horticulturae9030345
Cao Y, Yu H, Tian D, Sun E, Zuo L, Jiang D, Zuo C, Fan R. Phylogeny, Expression Profiling, and Coexpression Networks Reveals the Critical Roles of Nucleotide-BindingLeucine-Rich Repeats on Valsa Canker Resistance. Horticulturae. 2023; 9(3):345. https://doi.org/10.3390/horticulturae9030345
Chicago/Turabian StyleCao, Yanan, Hongqiang Yu, Dan Tian, E. Sun, Longgang Zuo, Daji Jiang, Cunwu Zuo, and Ruiyi Fan. 2023. "Phylogeny, Expression Profiling, and Coexpression Networks Reveals the Critical Roles of Nucleotide-BindingLeucine-Rich Repeats on Valsa Canker Resistance" Horticulturae 9, no. 3: 345. https://doi.org/10.3390/horticulturae9030345
APA StyleCao, Y., Yu, H., Tian, D., Sun, E., Zuo, L., Jiang, D., Zuo, C., & Fan, R. (2023). Phylogeny, Expression Profiling, and Coexpression Networks Reveals the Critical Roles of Nucleotide-BindingLeucine-Rich Repeats on Valsa Canker Resistance. Horticulturae, 9(3), 345. https://doi.org/10.3390/horticulturae9030345