
A Preliminary Study for Identifying Quantitative Trait Loci Associated with Seed Production in Radish Using Genotyping-by-Sequencing


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Genomic DNA Extraction
2.2. Genotyping-by-Sequencing Library Preparation
2.3. Sequencing Data Analysis and Single Nucleotide Polymorphism Calling
2.4. Linkage Map Consturction and QTL Analysis
2.5. The Prediction of Orthologous Genes
3. Results
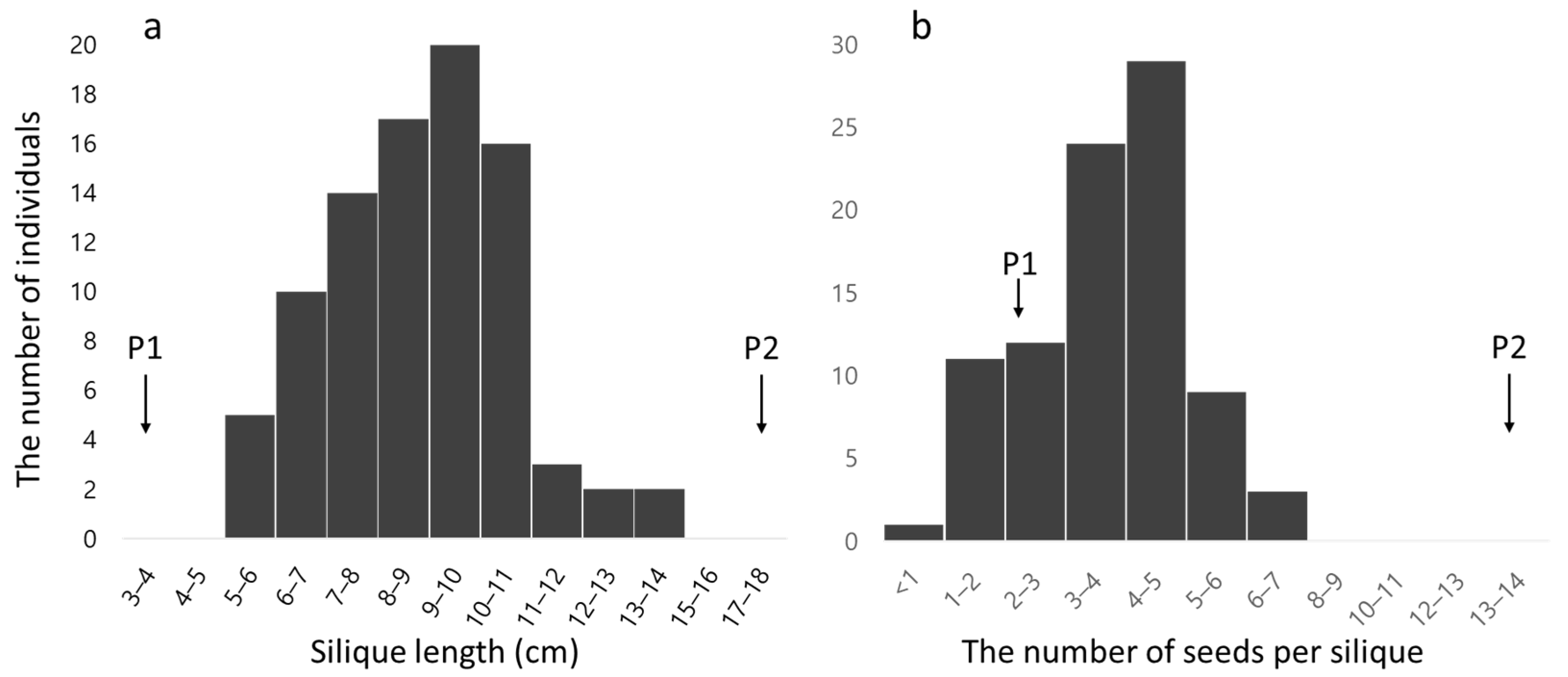
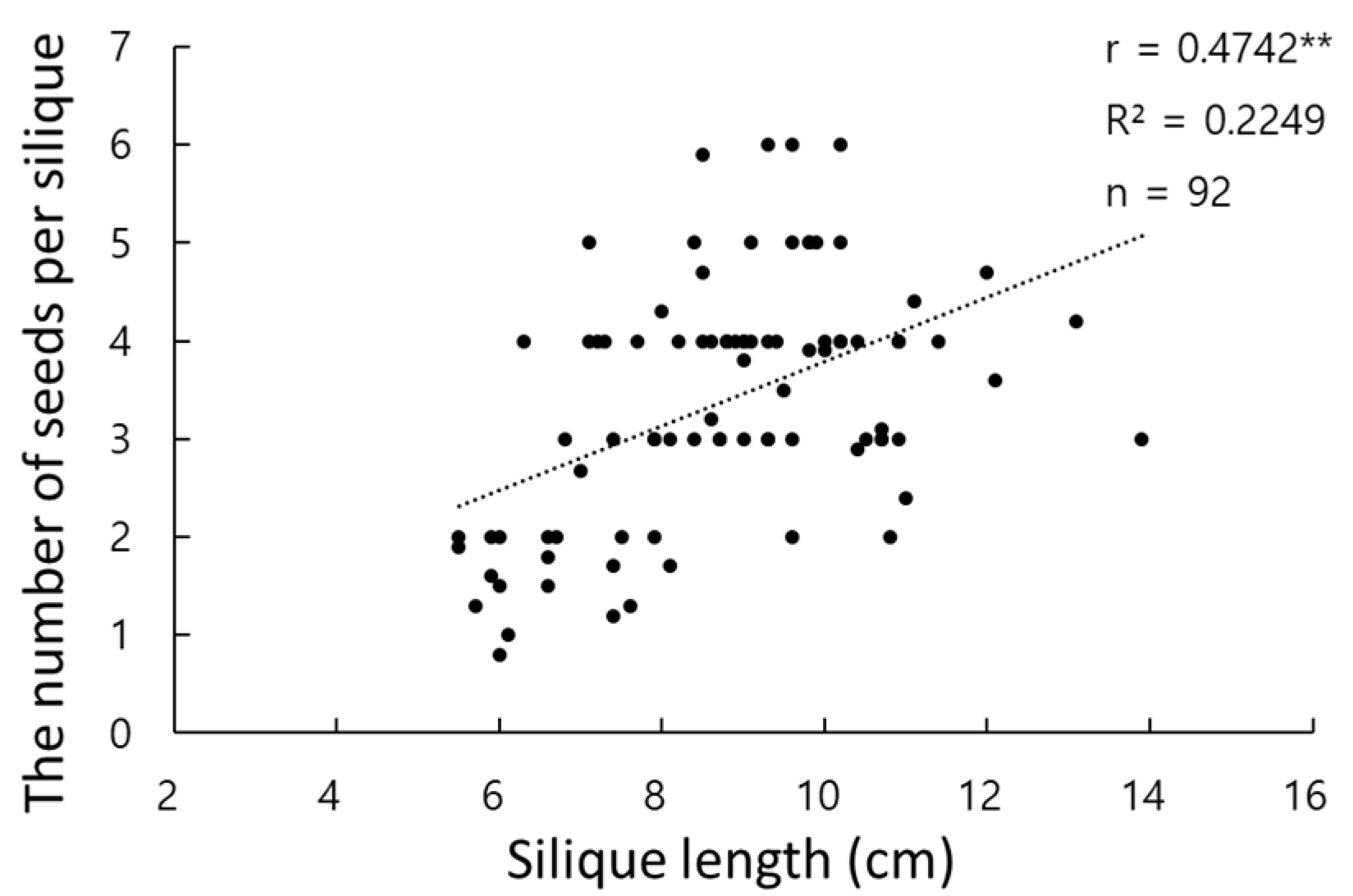
3.1. Evaluation of Phenotypic Data
3.2. Identification of Single Nucleotide Polymorphisms Using Gentyping-by-Sequencing
3.3. Linkage Map Construction
3.4. QTL Analysis
3.5. The Prediction of Orthologs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Muuoz, M.C.; Bermejo, J.E.H. La corola en la tribu Brassiceae. Anales Inst. Bot. Cavanilles 1978, 35, 297–334. [Google Scholar]
- Kaneko, Y.; Kimizuka-Takagi, C.; Bang, S.; Matsuzawa, Y. Radish. In Genome Mapping and Molecular Breeding in Plants; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 5, pp. 141–160. [Google Scholar]
- Go’mez-Campo, C. (Ed.) Taxonomy. In Biology of Brassica Coenospecies; Elsevier: New York, NY, USA, 1999; pp. 3–32. [Google Scholar]
- Li, X.; Kong, C.; Yu, H.; Liu, X.; Fang, Z.; Liu, Y.; Yang, L.; Zhuang, M.; Wang, Y.; Lv, H.; et al. Identification of a major QTL for seed number per silique in cabbage (Brassica oleracea L. var. capitata) using genotyping by sequencing. Euphytica 2019, 215, 133. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Zhan, J.; Shi, J.; Wang, X.; Liu, X.; Wang, H. Genetic and cytological analyses of the natural variation of seed number per pod in rapeseed (Brassica napus L.). Front. Plant Sci. 2017, 8, 1890. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Ha, S.; Ko, H.; Yu, H.; Chae, W. Reproductive traits and molecular evidence related to the global distribution of cultivated radish (Raphanus sativus L.). Plant Syst. Evol. 2016, 302, 1367–1380. [Google Scholar] [CrossRef]
- Bonnema, G.; Del Carpio, D.P.; Zhao, J. Diversity analysis and molecular taxonomy of Brassica vegetable crops. In Genetics, Genomics and Breeding of Vegetable Brassicas; Sadowski, J., Kole, C., Eds.; CRC Press: Boca Raton, FA, USA, 2011; pp. 81–124. [Google Scholar]
- Jatoi, S.A.; Javaid, A.; Iqbal, M.; Sayal, O.U.; Masood, M.S.; Siddiqui, S.U. Genetic diversity in radish germplasm for morphological traits and seed storage proteins. Pak. J. Bot. 2011, 43, 2507–2512. [Google Scholar]
- Zhang, L.W.; Li, S.P.; Chen, L.; Yang, G.S. Identification and mapping of a major dominant quantitative trait locus controlling seeds per silique as a single Mendelian factor in Brassica napus L. Theor. Appl. Genet. 2012, 125, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ye, J.; Zhan, J.; Zheng, X.; Zhang, J.; Shi, J.; Wang, X.; Liu, G.; Wang, H. Validation and characterization of a seed number per silique quantitative trait locus qSN.A7 in rapeseed (Brassica napus L.). Front. Plant Sci. 2020, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Shu, C.; Chen, L.; Xu, J.; Wu, J.; Liu, K. Identification of a major QTL for silique length and seed weight in oilseed rape (Brassica, napus, L.). Theor. Appl. Genet. 2012, 125, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Shi, J.; Wang, X.; Liu, G.; Wang, H. A combined linkage and regional association mapping validation and fine mapping of two major pleiotropic QTLs for seed weight and silique length in rapeseed (Brassica napus L.). BMC Plant Biol. 2014, 14, 1–14. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, E.S. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Oh, S.K.; Lee, J.H.; Lee, B.M.; Jo, S.H. Genome-wide SNP calling using next generation sequencing data in tomato. Mol. Cells 2014, 37, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Basten, C.J.; Zeng, Z.B. Windows QTL Cartographer 2.5. Department of Statistics; North Carolina State University: Raleigh, NC, USA, 2012; Available online: http://statgen.ncsu.edu/qtlcart/WQTLCart.htm (accessed on 22 September 2021).
- Stuber, C.W.; Edwards, M.D.; Wendel, J.F. Molecular marker-facilitated investigations of quantitative trait loci in maize. II. Factors influencing yield and its component traits 1. Crop. Sci. 1987, 27, 639–648. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, J.; Wang, X.; Liu, G.; Wang, H. Genetic architecture and mechanism of seed number per pod in rapeseed: Elucidated through linkage and near-isogenic line analysis. Sci. Rep. 2016, 6, 24124. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, K.; Hirakawa, H.; Fukino, N.; Kitashiba, H.; Isobe, H. Genome sequence and analysis of a Japanese radish (Raphanus sativus) cultivar named ‘Sakurajima Daikon’ possessing giant root. DNA Res. 2020, 27, dsaa010. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kang, Y.; Lee, S.; Choi, S.; Jeon, D.; Park, M.; Park, S.; Lim, Y.; Kim, C. Quantitative trait loci (QTLs) associated with microspore culture in Raphanus sativus L. (radish). Genes 2020, 11, 337. [Google Scholar] [CrossRef]
- Gan, C.; Deng, X.; Cui, L.; Yu, X.; Yuan, W.; Dai, Z.; Yao, M.; Pang, W.; Ma, Y.; Yu, X.; et al. Construction of a high-density genetic linkage map and identification of quantitative trait loci associated with clubroot resistance in radish (Raphanus sativus L.). Mol. Breed. 2019, 39, 116. [Google Scholar] [CrossRef]
- Mun, J.; Chung, H.; Chung, W.; Oh, M.; Jeong, Y.; Kim, N.; Ahn, B.; Park, B.; Park, S.; Lim, K.; et al. Construction of a reference genetic map of Raphanus sativus based on genotyping by whole-genome resequencing. Theor. Appl. Genet. 2015, 128, 259–272. [Google Scholar] [CrossRef]
- Luo, X.; Xu, L.; Wang, Y.; Dong, J.; Chen, Y.; Tang, M.; Fan, L.; Zhu, Y.; Liu, L. An ultra-high-density genetic map provides insights into genome synteny, recombination landscape and taproot skin colour in radish (Raphanus sativus L.). Plant Biotechnol. J. 2020, 18, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Charmet, G. Power and accuracy of QTL detection: Simulation studies of one-QTL models. Agronomie 2000, 20, 309–323. [Google Scholar] [CrossRef][Green Version]
- Wang, X.; Chen, L.; Wang, A.; Wang, H.; Tian, J.; Zhao, X.; Chao, H.; Zhao, Y.; Zhao, W.; Xiang, J.; et al. Quantitative trait loci analysis and genome-wide comparison for silique related traits in Brassica napus. BMC Plant Biol. 2016, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- López-Bucio, J.S.; Dubrovsky, J.G.; Raya-González, J.; Ugartechea-Chirino, Y.; López-Bucio, J.; de Luna-Valdez, L.A.; Ramos-Vega, M.; León, P.; Guevara-García, A.A. Arabidopsis thaliana mitogen-activated protein kinase 6 is involved in seed formation and modulation of primary and lateral root development. J. Exp. Bot. 2014, 65, 169–183. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Linkage Groups | No. SNPs | Genetic Distance (cM) | Average Interval | |
|---|---|---|---|---|
| Genetic Map (cM) | Physical Map (kb) 1 | |||
| R1 | 23 | 51.8 | 2.25 | 1205.2 |
| R2-1 | 50 | 88.0 | 1.76 | 346.3 |
| R2-2 | 74 | 111.5 | 1.51 | |
| R3 | 127 | 119.9 | 0.94 | 247.3 |
| R4 | 126 | 116.9 | 0.93 | 448.4 |
| R5 | 88 | 75.9 | 0.86 | 481.3 |
| R6 | 159 | 149.7 | 0.94 | 339.2 |
| R8 | 100 | 66.3 | 0.66 | 283.2 |
| R9 | 101 | 90.6 | 0.90 | 381.4 |
| Total | 848 | 870.6 | 1.20 | 379.4 |
| Traits | QTL Names | LGs | SNPs | Position in LG | LOD | Additive | Dominant | R2 | |d/a|Value 1 |
|---|---|---|---|---|---|---|---|---|---|
| Silique length | qSLr4 | R4 | R4_38169053–R4_40520291 | 48.781–55.211 | 9.59 | 1.061 | −1.647 | 0.296 | OD |
| No. seeds per silique | qNSPSr3 | R3 | R3_27235075 | 95.092 | 4.47 | −0.611 | 0.132 | 0.184 | A |
| qNSPSr6 | R6 | R6_52261344 | 93.323 | 4.30 | −0.614 | −0.953 | 0.144 | A |
| QTL Names | Gene ID | A.T. Orthologs | Gene Name | Traits Related |
|---|---|---|---|---|
| qNSPSr3 | RSAskr1.0R3g41067 | AT1G07890 | APX1 | Embryo Development |
| RSAskr1.0R3g41068 | AT3G11440 | ATMYB65 | pollen development | |
| RSAskr1.0R3g41068 | AT4G18770 | ATMYB98 | Embryo Sac Development | |
| RSAskr1.0R3g41068 | AT1G14350 | ATMYB124 | Embryo Sac Development | |
| RSAskr1.0R3g41071 | AT1G09100 | RPT5B | Embryo Sac Development | |
| RSAskr1.0R3g41071 | AT3G05530 | ATS6A.2 | Embryo Sac Development | |
| qNSPSr6 | RSAskr1.0R6g71396 | AT5G57800 | CER3 | Double Fertilization |
| RSAskr1.0R6g71397 | AT1G71830 | ATSERK1 | Embryo development ending in seed dormancy | |
| RSAskr1.0R6g71397 | AT3G02130 | CLI1 | Embryo development ending in seed dormancy | |
| RSAskr1.0R6g71397 | AT5G44700 | EDA23 | Embryo Sac Development | |
| RSAskr1.0R6g71397 | AT1G08590 | ATPXL1 | Embryo Development | |
| RSAskr1.0R6g71397 | AT1G11130 | SCM | Ovule development | |
| RSAskr1.0R6g71397 | AT1G63700 | EMB71 | Embryo development ending in seed dormancy | |
| RSAskr1.0R6g71397 | AT2G43790 | ATMAPK6 | Ovule development | |
| RSAskr1.0R6g71397 | AT5G20930 | TSL | Ovule development |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Park, S.; Lee, J.; Han, K.; Jang, Y.; Chae, W. A Preliminary Study for Identifying Quantitative Trait Loci Associated with Seed Production in Radish Using Genotyping-by-Sequencing. Horticulturae 2022, 8, 268. https://doi.org/10.3390/horticulturae8030268
Kim J, Park S, Lee J, Han K, Jang Y, Chae W. A Preliminary Study for Identifying Quantitative Trait Loci Associated with Seed Production in Radish Using Genotyping-by-Sequencing. Horticulturae. 2022; 8(3):268. https://doi.org/10.3390/horticulturae8030268
Chicago/Turabian StyleKim, Jinhee, Suhyoung Park, Junho Lee, Koeun Han, Yoonah Jang, and Wonbyoung Chae. 2022. "A Preliminary Study for Identifying Quantitative Trait Loci Associated with Seed Production in Radish Using Genotyping-by-Sequencing" Horticulturae 8, no. 3: 268. https://doi.org/10.3390/horticulturae8030268
APA StyleKim, J., Park, S., Lee, J., Han, K., Jang, Y., & Chae, W. (2022). A Preliminary Study for Identifying Quantitative Trait Loci Associated with Seed Production in Radish Using Genotyping-by-Sequencing. Horticulturae, 8(3), 268. https://doi.org/10.3390/horticulturae8030268