
Development of Loop-Mediated Isothermal Amplification Assay for Rapid Detection and Analysis of the Root-Knot Nematode Meloidogyne hapla in Soil


Abstract
:1. Introduction
2. Materials and Methods
2.1. Nematodes and DNA Samples
2.2. The Design of LAMP Primers
2.3. DNA Extraction from Second-Stage Juveniles
2.4. Specificity Tests
2.5. Soil Sampling and Soil Preparation
2.5.1. Artificially Infested Soils
2.5.2. Naturally Infested Soils
2.6. Development of Manual Soil DNA Extraction Procedure
2.7. Validation of the HSP LAMP Assay for Analyzing Meloidogyne hapla in Soil
2.7.1. DNA Extraction from 250 g Soil (SKMM Procedure)
2.7.2. DNA Extraction from 250 g of Soil (Commercial Kit)
2.7.3. Quantification of Gene Copies and Juveniles in Soil Samples
2.8. Real-Time qPCR
2.9. Statistics
3. Results
3.1. Primer Design
3.2. Specificity of the HSP-LAMP Primer Set
3.3. Optimization of the Manual DNA Extraction Procedure
3.4. Evaluation of the Manual DNA Extraction Procedure
3.4.1. Artificially Infested Soil
3.4.2. Naturally Infested Soil
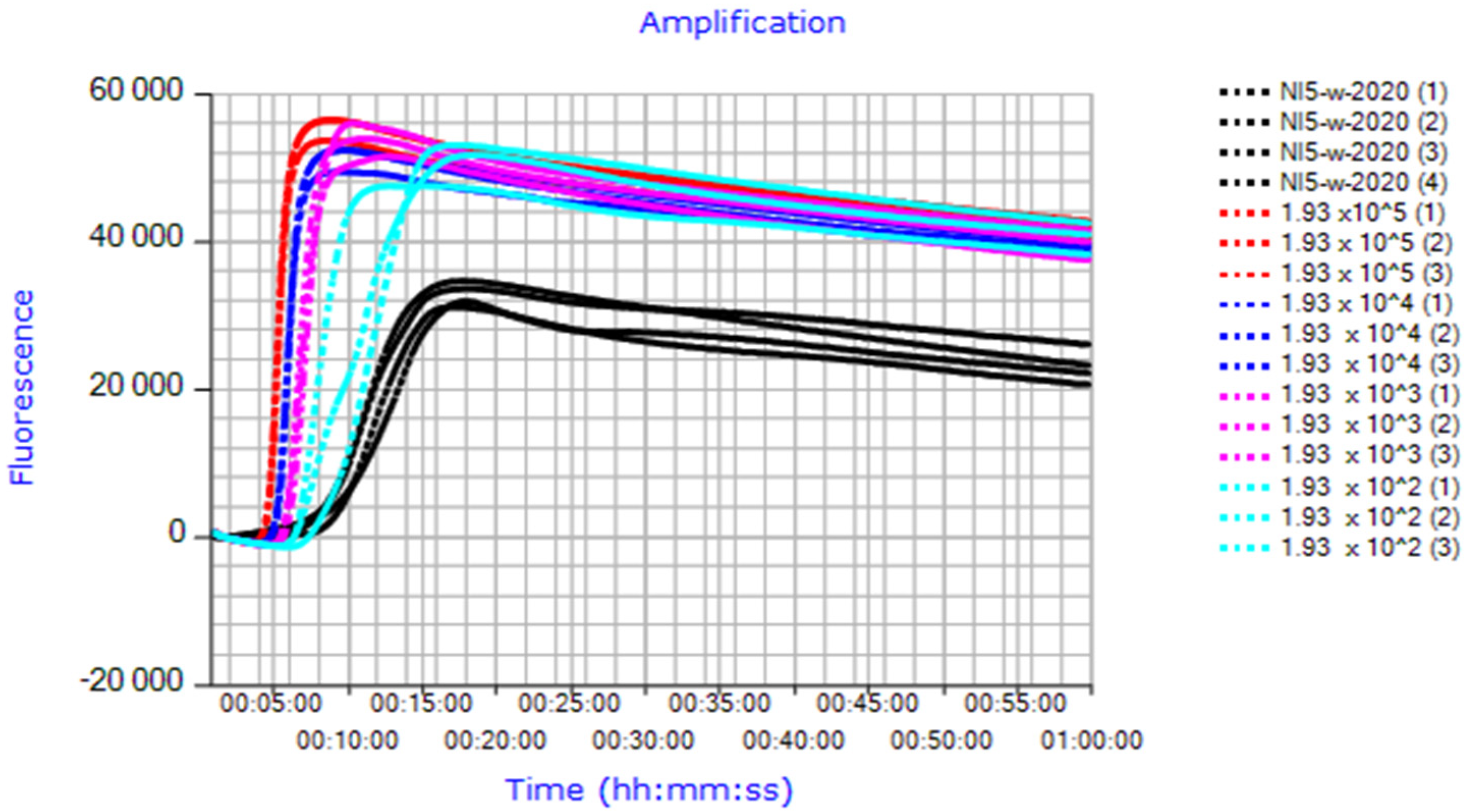
3.5. Verification of the HSP-LAMP Assay for Analyzing Meloidogyne hapla in Soil
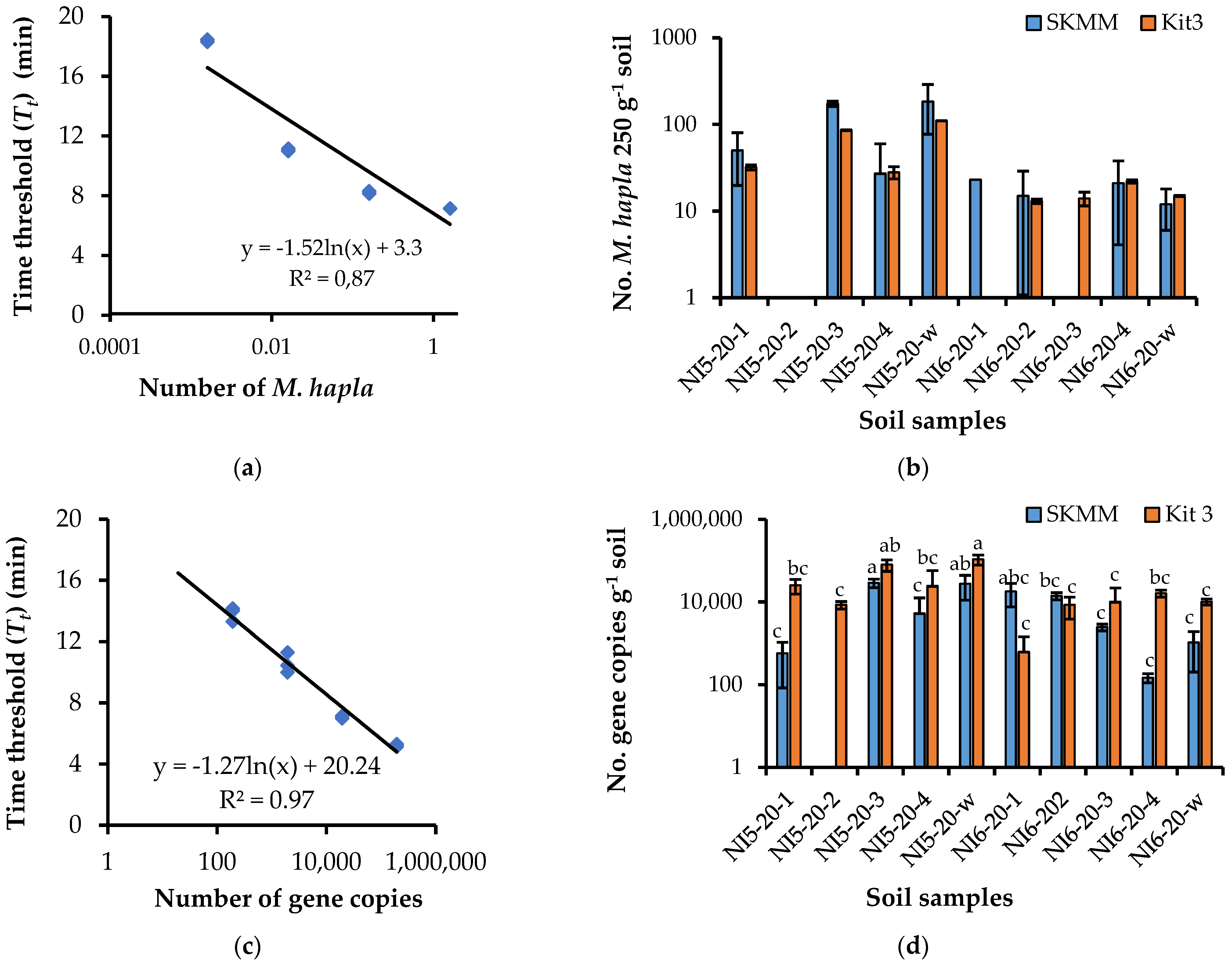
3.6. Within-Field Distribution of Meloidogyne hapla in Commercial Carrot Fields
4. Discussion
4.1. The Choice of Hsp90 Gene for Primers Design
4.2. LAMP Specificity and Sensitivity
4.3. DNA Concentrations and Quality: Comparison between SKMM and Commercial Kits
4.4. Artificial Soil Inoculation and Detection Limit in Soil
4.5. Verification of Real-Time LAMP Assay for Analyzing Meloidogyne hapla in Naturally Infested Soil
4.6. Within-Field Distribution of Meloidogyne hapla in Commercial Carrot Fields
4.7. Future Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castagnone-Sereno, P.; Danchin, E.G.J.; Perfus-Barbeoch, L.; Abad, P. Diversity and evolution of root-knot nematodes, genus Meloidogyne: New insights from the genomic era. Annu. Rev. Phytopathol. 2013, 51, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Wesemael, W.M.L.; Viaene, N.; Moens, M. Root-knot nematodes (Meloidogyne spp.) in Europe. Nematology 2011, 13, 3–16. [Google Scholar] [CrossRef]
- Andersson, S. Rotgallnematod, Ett Ökande Problem I Morotsodling; Skånska Lantbr: Svedala, Sweden, 2009; Volume 2, ISSN 1653-2368. (In Swedish) [Google Scholar]
- SJV (Swedish Board of Agriculture). Trädgårdsproduktion 2017. Statistiska Meddelanden, JO 33 SM 1801; SJV: Jönköping, Sweden, 2017. (In Swedish) [Google Scholar]
- Saint Louis University. Meloidogyne chitwoodi, M. fallax and M. hapla-Resistance of Plants Relevant in Swedish Cropping Systems; SLU: The Unit for Risk Assessment of Plant Pests: Uppsala, Sweden, 2018; 54p. [Google Scholar]
- Baermann, G. Eine einfache Methode zur Auffindung von Anklostomum (Nematoden) Larven in Erdproben. Tijdschr Diergeneeskd 1917, 57, 131–137. [Google Scholar]
- Cunha, T.G.; Visôtto, L.E.; Lopes, E.A.; Oliveira, C.M.G.; God, P.I.V.G. Diagnostic methods for identification of root-knot nematodes species from Brazil. Ciência Rural 2018, 48, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Eisenbach, J.D. Morphological comparison of head shape and stylet morphology of the second stage juveniles of Meloidogyne species. J. Nematol. 1982, 14, 339–343. [Google Scholar]
- Ingham, R.E. Nematodes. In Methods of Soil Analysis. Part 2. Microbiological and Biochemical Properties; Weaver, R.W., Angle, S., Bottomley, P., Bezdicek, D., Smith, S., Tabatabai, A., Wollum, A., Eds.; American Society of Agronomy: Madison, WI, USA, 1994; Volume 1, pp. 459–490. [Google Scholar]
- Esbenshade, P.R.; Triantaphyllou, A.C. Isozyme Phenotypes for the Identification of Meloidogyne Species. J. Nematol. 1990, 22, 10–15. [Google Scholar]
- Zijlstra, C.; Donkers-Venne, D.T.H.M.; Fargette, M. Identification of Meloidogyne incognita, M. javanica and M. arenaria using sequence characterised amplified region (SCAR) based PCR assays. Nematology. 2000, 2, 847–853. [Google Scholar]
- Sapkota, R.; Skantar, A.M.; Nicolaisen, M. A TaqMan real-time PCR assay for detection of Meloidogyne hapla in root galls and in soil. Nematology. 2015, 18, 147–154. [Google Scholar] [CrossRef]
- Tebbe, C.C.; Vajhen, W. Interference of humic acids and DNA extracted directly from soil in detection and transformation of recombinant DNA from bacteria and a yeast. Appl. Environ. Microbiol. 1993, 59, 2657–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, M.; Honda, E.; Ogura, A.; Nomoto, A.; Hanaki, K.I. Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxyl naphthol blue. Biotechniques 2009, 46, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Omer, Z.; Wallenhammar, A.-C. Development of Loop-mediated Isothermal Amplification assays for rapid detection of blackleg pathogens in Swedish winter oil seed rape. Euro. J. Plant Pathol. 2020, 157, 353–365. [Google Scholar] [CrossRef]
- Ammour, M.S.; Bilodeau, G.J.; Tremblay, D.M.; Van der Heyden, H.; Yaseen, T.; Varvaro, L.; Carisse, O. Development of Real-Time Isothermal Amplification Assays for On-Site Detection of Phytophthora infestans in Potato Leaves. Plant Dis. 2017, 101, 1269–1277. [Google Scholar] [CrossRef] [Green Version]
- Aglietti, C.; Luchi, N.; Pepori, A.L.; Bartolini, P.; Pecori, F.; Raio, A.; Capretti, P.; Santini, A. Real-time loop-mediated isothermal amplification: An early-warning tool for quarantine plant pathogen detection. AMB Express 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Deguo, W.; Guicheng, H.; Fugui, W.; Yonggang, L.; Daxi, R. Drawback of loop-mediated isothermal amplification. Afr. J. Food Sci. 2008, 2, 83–86. [Google Scholar]
- Denschlag, C.; Vogel, R.F.; Niessen, L. Hyd5 gene-based detection of the major gushing-inducing Fusarium spp. in a loop-mediated isothermal amplification (LAMP) assay. Int. J. Food Microbiol. 2012, 156, 189–196. [Google Scholar] [CrossRef]
- Moradi, A.; Almasi, M.A.; Jafary, H.; Mercado-Blanco, J. A novel and rapid loop-mediated isothermal amplification assay for the specific detection of Verticillium dahliae. J. Appl. Microbiol. 2014, 116, 942–954. [Google Scholar] [CrossRef]
- Leal, I.; Allen, E.; Foord, B.; Anema, J.; Reisle, C.; Uzunovic, A.; Varga, A.; James, D. Detection of living Bursaphelenchus xylophilus in wood, using reverse transcriptase loop-mediated isothermal amplification (RT-LAMP). For. Pathol. 2015, 45, 134–148. [Google Scholar] [CrossRef]
- Zhang, L.; Gleason, C. Loop-Mediated Isothermal Amplification for the Diagnostic Detection of Meloidogyne chitwoodi and M. fallax. Plant Dis. 2019, 103, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.-H.; Guo, Q.-X.; Jian, H.; Chen, C.-L.; Yang, D.; Liu, Q.; Guo, Y.-D. Rapid detection of Meloidogyne spp. by LAMP assay in soil and roots. Crop Prot. 2011, 30, 1063–1069. [Google Scholar] [CrossRef]
- Peng, H.; Long, H.; Huang, W.; Liu, J.; Cui, J.; Kong, L.; Hu, X.; Gu, J.; Peng, D. Rapid, simple and direct detection of Meloidogyne hapla from infected root galls using loop-mediated isothermal amplification combined with FTA technology. Sci. Rep. 2017, 7, 44853. [Google Scholar] [CrossRef]
- Haan, E.G.; Dekker, C.C.E.M.; Tameling, W.I.L.; den Nijs, L.J.M.F.; van den Bovenkamp, G.W.; Kooman-Gersmann, M. The MeloTuber Test: A real-time TaqMan PCR-based assay to detect the root-knot nematodes Meloidogyne chitwoodi and M. fallax directly in potato tubers. Bull. OEPP/EPPO Bull. 2014, 44, 166–175. [Google Scholar] [CrossRef]
- Gorny, A.M.; Wang, X.; Hay, F.S.; Pethybridge, S.J. Development of a Species-Specific PCR for Detection and Quantification of Meloidogyne hapla in Soil Using the 16D10 Root-Knot Nematode Effector Gene. Plant Dis. 2019, 101, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Stedtfeld, R.D.; Stedtfeld, T.M.; Samhan, F.; Kanitkar, Y.H.; Hatzinger, P.B.; Cupples, A.M.; Hashshama, S.A. Direct loop mediated isothermal amplification on filters for quantification of Dehalobacter in groundwater. J. Microbiol. Methods 2016, 131, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, A.G.; Hemming, J.R. A comparison of some quantitative methods of extracting small vermiform nematodes from soil. Ann. App. Biol. 1965, 55, 25–38. [Google Scholar] [CrossRef]
- Song, Z.Q.; Cheng, J.U.; Cheng, F.X.; Zhang, D.Y.; Liu, Y. Development and Evaluation of Loop-Mediated Isothermal Amplification assay for rapid detection of Tylenchulus semipenetrans using DNA extracted from soil. Plant Pathol. J. 2017, 33, 184–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verschoor, B.; De Goede, R.G.M. The nematode extraction efficiency of the Oostenbrink elutriator-cottonwool filter method with special reference to nematode body size and life strategy. Nematology 2000, 2, 325–342. [Google Scholar] [CrossRef]
- Huynh, O.A.; Jankowicz-Cieslak, J.; Saraye, B.; Hofinger, B.; Till, B.J. Low-cost for DNA extraction and quantification. In Biotechnologies for Plant Mutation Breeding; Jankowicz-Cieslak, J., Tai, T.H., Kumlehn, J., Till, B.J., Eds.; Springer Nature: Basel, Switzerland, 2017; Volume 1, pp. 227–239. [Google Scholar]
- Niessen, L.; Gräfenhan, T.; Vogel, R.F. ATP citrate lyase 1 (acl1) gene-based loop-mediated amplification assay for the detection of the Fusarium tricinctum species complex in pure cultures and in cereal samples. Int. J. Food Microbiol. 2012, 158, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Skantar, A.M.; Carta, L.K. Multiple displacement amplification (MDA) of total genomic DNA from Meloidogyne spp. and comparison to crude DNA extracts in PCR of ITS1, 28S D2-D3 rDNA and Hsp90. Nematology 2005, 7, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Handoo, Z.A.; Skantar, A.M.; Carta, L.K.; Schmitt, D.P. Morphological and Molecular Evaluation of a Meloidogyne hapla Population Damaging Coffee (Coffea arabica) in Maui, Hawaii. J. Nematol. 2005, 37, 136–145. [Google Scholar]
- Tomlinson, J.A.; Dickinson, M.J.; Boonham, N. Detection of Botrytis cinerea by loop-mediated isothermal amplification. Lett. Appl. Microbiol. 2010, 51, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.Y.; Toyota, K.; Sato, E. A novel nematode diagnostic method using the direct quantification of major plant-parasitic nematodes in soil by real-time PCR. Nematology 2012, 14, 265–276. [Google Scholar] [CrossRef]
- Cheng, F.; Houa, L.; Woestec, K.; Shang, Z.; Penge, X.; Zhaof, P.; Zhang, S. Soil pretreatment and fast cell lysis for direct polymerase chain reaction from forest soils for terminal restriction fragment length polymorphism analysis of fungal communities. Braz. J. Microbiol. 2016, 47, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Wallenhammar, A.-C.; Arwidsson, O. Detection of Plasmodiophora brassicae by PCR in naturally infested soils. Eur. J. Plant Pathol. 2001, 107, 313–321. [Google Scholar] [CrossRef]
- Knauth, S.; Schmidt, H.; Tippkötter, R. Comparison of commercial kits for the extraction of DNA from paddy soils. Lett. Appl. Microbiol. 2012, 56, 222–228. [Google Scholar] [CrossRef]
- Leite, D.C.A.; Balieiro, F.C.; Pires, C.A.; Madari, B.E.; Rosado, A.S.; Coutinho, H.L.C.; Peixoto, R.S. Comparison of DNA extraction protocols for microbial communities from soil treated with biochar. Braz. J. Microbiol. 2014, 45, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallenhammar, A.-C.; Almquist, C.; Söderström, M.; Jonsson, A. In-field distribution of Plasmodiophora brassicae measured using quantitative real-time PCR. Plant Pathol. 2012, 61, 16–28. [Google Scholar] [CrossRef]
- Waite, I.S.; O’Donnell, A.G.; Harrison, A.; Davies, J.T.; Colvan, S.R. Design and evaluation of nematode 18S rDNA primers for PCR and denaturing gradient gel electrophoresis (DGGE) of soil community DNA. Soil Biol. Biochem. 2003, 35, 1165–1173. [Google Scholar] [CrossRef]
- Macmillan, K.; Blok, V.; Young, I.; Crawford, J.; Wilson, M.J. Quantification of the slug parasitic nematode Phasmarhabditis hermaphrodita from soil samples using real time PCR. Int. J. Parasitol. 2006, 36, 1453–1461. [Google Scholar] [CrossRef]
- Brierley, J.L.; Stewart, J.A.; Lees, A.K. Quantifying potato pathogen DNA in soil. Appl. Soil Ecol. 2009, 41, 234–238. [Google Scholar] [CrossRef]
- Baidoo, R.; Yan, G.; Nagachandrabose, S.; Skantar, A.M. Developing a Real-Time PCR Assay for Direct Identification and Quantification of Pratylenchus penetrans in Soil. Plant Dis. 2017, 101, 1432–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhu, W.; Wang, Y.; Liu, X.; Chen, L.; Duan, Y. The cold tolerance of the northern root-knot nematode, Meloidogyne hapla. PLoS ONE 2018, 13, e0190531. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA 1 (ng µL−1) | Absorbance Ratios 2 | |||||
|---|---|---|---|---|---|---|
| (A260/A280) | (A260/A230) | |||||
| Silica | ||||||
| Celite 545 WA | 59 ± 6.8 | a | 1.5 ± 0.07 | a | 0.07 ± 0.02 | a |
| Celite 545 FA | 70 ± 7.3 | a | 1.4 ± 0.04 | a | 0.07 ± 0.10 | a |
| p | ns | ns | ns | |||
| CV | 34 | 13 | 55 | |||
| SDS volume (µL) | ||||||
| 25 | 54 ± 2.2 | b | 1.5 ± 0.03 | a | 0.05 ± 0.12 | b |
| 50 | 58 ± 1.6 | b | 1.5 ± 0.11 | a | 0.06 ± 0.01 | ab |
| 100 | 81 ± 2.4 | a | 1.4 ± 0.06 | a | 0.11 ± 0.02 | a |
| p | p < 001 | ns | 0.03 | |||
| CV | 33 | 13 | 55 | |||
| Skim milk concentration (% w/v) | ||||||
| 3.2 | 22 ± 1.7 | a | 1.7 ± 0.01 | a | 0.2 ± 0.06 | a |
| 4 | 23 ± 2.4 | a | 1.6 ± 0.02 | a | 0.2 ± 0.03 | a |
| 5 | 20 ± 0.5 | a | 1.6 ± 0.02 | ab | 0.2 ± 0.03 | a |
| 6 | 24 ± 1.3 | a | 1.5 ± 0.01 | b | 0.2 ± 0.04 | a |
| p | ns | p < 001 | ns | |||
| CV | 13 | 4 | 35 | |||
| DNA (µg g−1 Soil) | Absorbance Ratios | Corr. Coeff 8 | |||||
|---|---|---|---|---|---|---|---|
| (A260/A280) | (A260/A230) | (r) | |||||
| NF-17 1 | |||||||
| Kit 1 4 | 15 ± 0.59 | a | 1.80 ± 0.0 | b | 0.22 ± 0.017 | c | |
| Kit 2 5 | 11 ± 0.51 | b | 1.83 ± 0.01 | a | 0.36 ± 0.017 | b | |
| Kit 3 6 | 8 ± 0.38 | c | 1.81 ± 0.0 | b | 0.90 ± 0.03 | a | 0.98 |
| SKMM 7 | 8 ± 0.53 | c | 1.80 ± 0.0 | b | 0.99 ± 0.04 | a | 0.83 |
| p | <0.0001 | 0.0005 | <0.0001 | ||||
| CV | 37.1 | 1.9 | 66.1 | ||||
| NI-19 2 | |||||||
| Kit 1 | 28 ± 5.0 | a | 1.65 ± 0.02 | c | 0.20 ± 0.02 | c | |
| Kit 2 | 10 ± 0.8 | b | 1.78 ± 0.01 | a | 0.17 ± 0.01 | c | |
| Kit 3 | 6 ± 0.4 | d | 1.74 ± 0.01 | b | 0.57 ± 0.02 | b | 0.93 |
| SKMM | 7 ± 0.3 | c | 1.76 ± 0.01 | a | 0.81 ± 0.02 | a | 0.83 |
| p | <0.0001 | <0.0001 | <0.0001 | ||||
| CV | 167.9 | 6.2 | 69.3 | ||||
| NI-20 3 | |||||||
| Kit 1 | 13 ± 1.4 | a | 1.75 ± 0.02 | b | 0.10 ± 0.01 | c | |
| Kit 2 | 5 ± 0.37 | b | 1.79 ± 0.01 | a | 0.13 ± 0.01 | c | |
| Kit 3 | 3 ± 0.25 | c | 1.80 ± 0.01 | a | 0.49 ± 0.03 | b | 0.60 |
| SKMM | 6 ± 0.26 | b | 1.72 ± 0.01 | b | 0.62 ± 0.02 | a | 0.86 |
| p | <0.0001 | <0.0017 | <0.0001 | ||||
| CV | 106 | 7 | 82 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omer, Z.S.; Wallenhammar, A.-C.; Viketoft, M. Development of Loop-Mediated Isothermal Amplification Assay for Rapid Detection and Analysis of the Root-Knot Nematode Meloidogyne hapla in Soil. Horticulturae 2022, 8, 87. https://doi.org/10.3390/horticulturae8020087
Omer ZS, Wallenhammar A-C, Viketoft M. Development of Loop-Mediated Isothermal Amplification Assay for Rapid Detection and Analysis of the Root-Knot Nematode Meloidogyne hapla in Soil. Horticulturae. 2022; 8(2):87. https://doi.org/10.3390/horticulturae8020087
Chicago/Turabian StyleOmer, Zahra Saad, Ann-Charlotte Wallenhammar, and Maria Viketoft. 2022. "Development of Loop-Mediated Isothermal Amplification Assay for Rapid Detection and Analysis of the Root-Knot Nematode Meloidogyne hapla in Soil" Horticulturae 8, no. 2: 87. https://doi.org/10.3390/horticulturae8020087
APA StyleOmer, Z. S., Wallenhammar, A.-C., & Viketoft, M. (2022). Development of Loop-Mediated Isothermal Amplification Assay for Rapid Detection and Analysis of the Root-Knot Nematode Meloidogyne hapla in Soil. Horticulturae, 8(2), 87. https://doi.org/10.3390/horticulturae8020087