
Identification of QTLs Controlling Radish Root Shape Using Multiple Populations

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Phenotypic Analysis
2.3. DNA Isolation and Sequencing of Pooled Samples
2.4. QTL-Seq Analysis
2.5. Gene Annotation and Expression Analysis of QTL Regions
3. Results
3.1. Phenotypic Data Analysis of the Radish Root Shape Characteristics
3.2. Inheritance of the Root Shape Index and Mixed Pools
3.3. Correlation among the Root Shape Characteristics in Radish
3.4. Sequencing of the Parents and Mixed Pools
3.5. Detection of QTLs
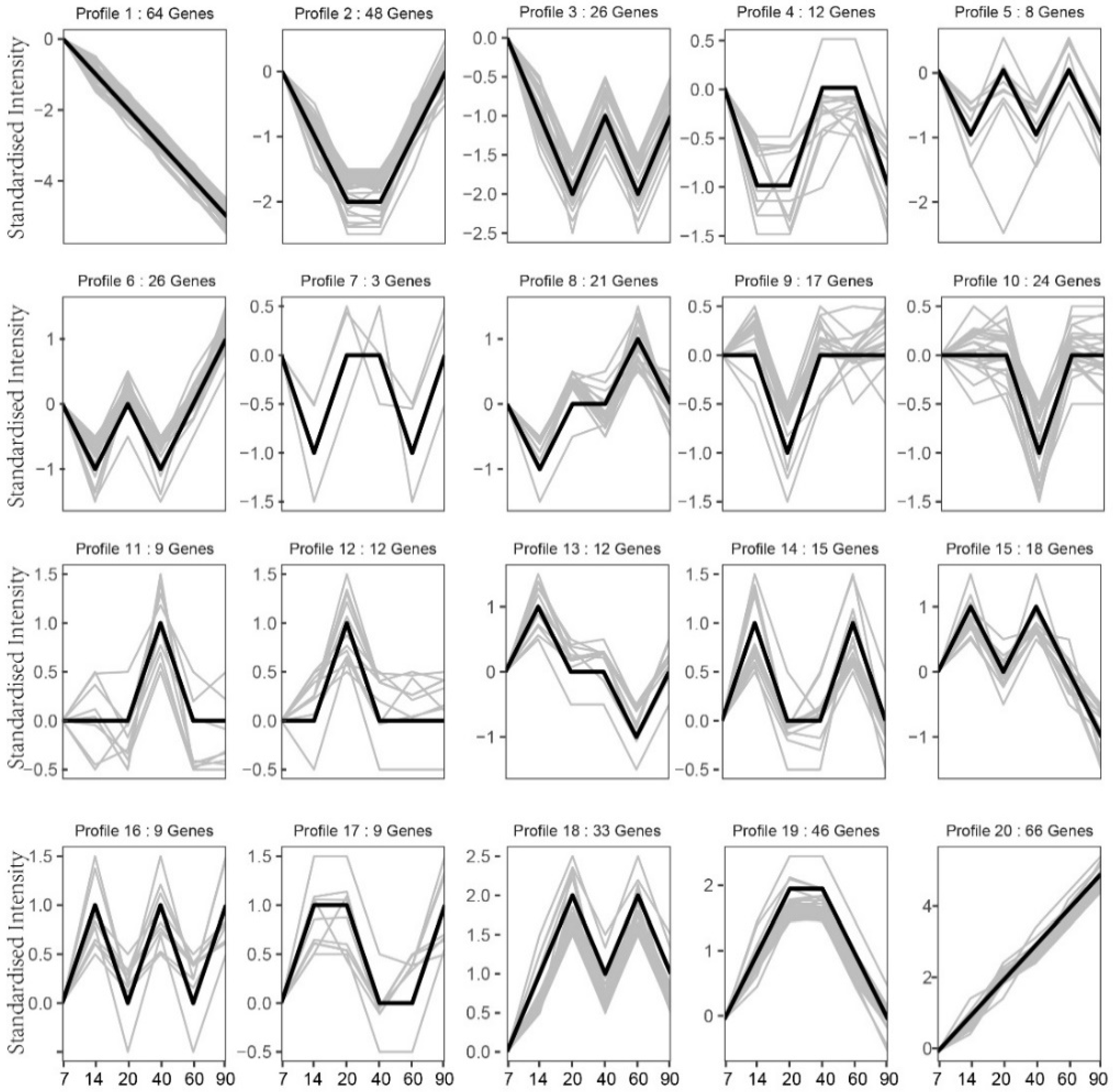
3.6. Gene Expression Trend Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tsuro, M.; Suwabe, K.; Kubo, N.; Matsumoto, S.; Hirai, M. Mapping of QTL controlling root shape and red pigmentation in radish, Raphanus sativus L. Breed. Sci. 2008, 58, 55–61. [Google Scholar] [CrossRef][Green Version]
- Iwata, H.; Niikura, S.; Matsuura, S.; Takano, Y.; Ukai, Y. Interaction between genetic effects and soil type in diallel analysis of root shape and size of Japanese radish (Raphanus sativus L.). Breed. Sci. 2004, 54, 313–318. [Google Scholar] [CrossRef]
- Mitsui, Y.; Shimomura, M.; Komatsu, K.; Namiki, N.; Shibata-Hatta, M.; Imai, M.; Katayose, Y.; Mukai, Y.; Kanamori, H.; Kurita, K.; et al. The radish genome and comprehensive gene expression profile of tuberous root formation and development. Sci. Rep. 2015, 5, 10835. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Shen, D. The Description Criterion of Radish Germplasm; China Agriculture Press: Beijing, China, 2008; pp. 25–26. [Google Scholar]
- Zaki, H.E.M.; Yokoi, S.; Takahata, Y. Identification of genes related to root shape in radish (Raphanus sativus) using suppression subtractive hybridization. Breed. Sci. 2010, 60, 130–138. [Google Scholar] [CrossRef]
- Hashida, T.; Nakatsuji, R.; Budahn, H.; Schrader, O.; Peterka, H.; Fujimura, T.; Kubo, N.; Hirai, M. Construction of a chromosome-assigned, sequence-tagged linkage map for the radish, Raphanus sativus L. and QTL analysis of morphological traits. Breed. Sci. 2013, 63, 218–226. [Google Scholar] [CrossRef][Green Version]
- Yu, X.N.; Choi, S.R.; Dhandapani, V.; Rameneni, J.J.; Li, X.N.; Pang, W.X.; Lee, J.Y.; Lim, Y.P. Quantitative Trait Loci for Morphological Traits and their Association with Functional Genes in Raphanus sativus. Front. Plant Sci. 2016, 7, 255. [Google Scholar] [CrossRef]
- Luo, X.B.; Xu, L.; Wang, Y.; Dong, J.H.; Chen, Y.L.; Tang, M.J.; Fan, L.X.; Zhu, Y.L.; Liu, L.W. An ultra-high-density genetic map provides insights into genome synteny, recombination landscape and taproot skin colour in radish (Raphanus sativus L.). Plant Biotechnol. J. 2020, 18, 274–286. [Google Scholar] [CrossRef]
- Wang, S.F.; Wang, X.F.; He, Q.W.; Liu, X.X.; Xu, W.L.; Li, L.B.; Gao, J.W.; Wang, F.D. Transcriptome analysis of the roots at early and late seedling stages using Illumina paired-end sequencing and development of EST-SSR markers in radish. Plant Cell Rep. 2012, 31, 1437–1447. [Google Scholar] [CrossRef]
- Yu, R.G.; Wang, J.; Xu, L.; Wang, Y.; Wang, R.H.; Zhu, X.W.; Sun, X.C.; Luo, X.B.; Xie, Y.; Everlyne, M.; et al. Transcriptome profiling of taproot reveals complex regulatory networks during taproot thickening in radish (Raphanus sativus L.). Front. Plant Sci. 2016, 7, 1210. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Lu, H.F.; Lin, T.; Klein, J.; Wang, S.H.; Qi, J.J.; Zhou, Q.; Sun, J.J.; Zhang, Z.H.; Weng, Y.Q.; Huang, S.W. QTL-seq identifies an early flowering QTL located near Flowering Locus T in cucumber. Theor. Appl. Genet. 2014, 127, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Illa-Berenguer, E.; Van Houten, J.; Huang, Z.J.; van der Knaap, E. Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor. Appl. Genet. 2015, 128, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Anurag, D.; Sweta, D.; Rishi, S.; Saurabh, B.; Singh, A.K.; Pinky, A.; Parida, S.K.; Tyagi, A.K. An efficient strategy combining SSR markers and advanced QTL-seq-driven QTL mapping unravels candidate genes regulating grain weight in rice. Front. Plant Sci. 2016, 7, 1535. [Google Scholar]
- Wei, Q.Z.; Fu, W.Y.; Wang, Y.Z.; Qin, X.D.; Wang, J.; Li, J.; Lou, Q.F.; Chen, J.F. Rapid identification of fruit length loci in cucumber (Cucumis sativus L.) using next-generation sequencing (NGS)-based QTL analysis. Sci. Rep. 2016, 6, 27496. [Google Scholar] [CrossRef]
- Liu, T.J.; Wang, J.L.; Wu, C.H.; Zhang, Y.J.; Zhang, X.H.; Li, X.M.; Wang, H.P.; Song, J.P.; Li, X.X. Combined QTL-seq and traditional linkage analysis to identify candidate genes for purple skin of radish fleshy taproots. Front. Genet. 2019, 10, 808. [Google Scholar] [CrossRef]
- Wang, Q.B.; Wang, Y.P.; Sun, H.H.; Sun, L.; Zhang, L. Transposon-induced methylation of the RsMYB1 promoter disturbs anthocyanin accumulation in red-fleshed radish. J. Exp. Bot. 2020, 71, 2537–2550. [Google Scholar] [CrossRef]
- Kitashiba, H.; Li, F.; Hirakawa, H.; Kawanabe, T.; Zou, Z.W.; Hasegawa, Y.; Tonosaki, K.; Shirasawa, S.; Fukushima, A.; Yokoi, S.; et al. Draft sequences of the radish (Raphanus sativus L.) genome. DNA Res. 2014, 21, 481–490. [Google Scholar] [CrossRef]
- Moghe, G.D.; Hufnagel, D.E.; Tang, H.B.; Xiao, Y.L.; Dworkin, I.; Town, C.D.; Conner, J.K.; Shiu, S.H. Consequences of whole-genome triplication as revealed by comparative genomic analyses of the wild radish Raphanus raphanistrum and three other Brassicaceae Species. Plant Cell 2014, 26, 1925–1937. [Google Scholar] [CrossRef]
- Zhang, X.H.; Yue, Z.; Mei, S.Y.; Qiu, Y.; Yang, X.H.; Chen, X.H.; Cheng, F.; Wu, Z.Y.; Sun, Y.Y.; Jing, Y.; et al. A de novo genome of a Chinese Radish Cultivar. Hortic. Plant J. 2015, 1, 155–164. [Google Scholar]
- Jeong, Y.M.; Kim, N.; Ahn, B.O.; Oh, M.; Chung, W.H.; Chung, H.; Jeong, S.; Lim, K.B.; Hwang, Y.J.; Kim, G.B.; et al. Elucidating the triplicated ancestral genome structure of radish based on chromosome-level comparison with the Brassica genomes. Theor. Appl. Genet. 2016, 129, 1357–1372. [Google Scholar] [CrossRef]
- Shirasawa, K.; Hirakawa, H.; Fukino, N.; Kitashiba, H.; Isobe, S. Genome sequence and analysis of a Japanese radish (Raphanus sativus) cultivar named ‘Sakurajima Daikon’ possessing giant root. DNA Res. 2020, 27, dsaa010. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Mckenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.Y.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Cherry, J.M. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Choi, E.Y.; Seo, T.C.; Lee, S.G.; Cho, I.H.; Stangoulis, J. Growth and physiological responses of Chinese cabbage and radish to long-term exposure to elevated carbon dioxide and temperature. Hortic. Environ. Biotechnol. 2011, 52, 376–386. [Google Scholar] [CrossRef]
- Samuolien, G.; Sirtautas, R.; Brazaityt, A.; Sakalauskait, J.; Sakalauskien, S.; Duchovskis, P. The impact of red and blue light-emitting diode illumination on radish physiological indices. Cent. Eur. J. Biol. 2011, 6, 821–828. [Google Scholar] [CrossRef]
- Yu, R.G.; Wang, Y.; Xu, L.; Zhu, X.W.; Zhang, W.; Wang, R.H.; Gong, Y.Q.; Limera, C.; Liu, L.W. Transcriptome profiling of root microRNAs reveals novel insights into taproot thickening in radish (Raphanus sativus L.). BMC Plant Biol. 2015, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Geupil, J.; Jung-Hun, L.; Khushboo, R.; Suhyoung, P.; Sang-Hun, O.; Ji-Young, L. Cytokinin-dependent secondary growth determines root biomass in radish (Raphanus sativus L.). J. Exp. Bot. 2015, 66, 4607–4619. [Google Scholar]
- Bett, K.E.; Lydiate, D.J. Genetic analysis and genome mapping in Raphanus. Genome 2003, 46, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, K.; Kitashiba, H. Genetic maps and whole genome sequences of Radish. In The Radish Genom; Nishio, T., Kitashiba, H., Eds.; Springer: Cham, Switzerland, 2017; Volume 3, pp. 31–42. [Google Scholar]
- Muvva, C.; Tewari, L.; Aruna, K.; Ranjit, P.; Zahoorullah, S.; Matheen, K.; Veeramachaneni, H. In silico identification of miRNAs and their targets from the expressed sequence tags of Raphanus sativus. Bioinformation 2012, 8, 98–103. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F2 Population | Parent | Root Length/cm | Root Diameter/cm | Root Shape Index |
|---|---|---|---|---|
| 1902 | CZ | / | / | / |
| CLA | 18.66 ± 0.37 | 3.87 ± 0.09 | 4.82 ± 0.03 | |
| 1908 | R05 | 5.23 ± 0.39 | 5.18 ± 0.39 | 1.01 ± 0.09 |
| CLA | 18.66 ± 0.37 | 3.87 ± 0.09 | 4.82 ± 0.03 | |
| 1909 | LLYH | 2.87 ± 0.06 | 3.46 ± 0.17 | 0.82 ± 0.04 |
| BY | 20.10 ± 0.51 | 3.64 ± 0.08 | 5.52 ± 0.05 |
| F2 Population | Number of Plants | The Minimum Value of the Root Shape Index | The Maximum Value of the Root Shape Index | Number of Plants in the Extreme Pools | Mean Value of the Extremely Small Pool | Mean Value of the Extremely Large Pool |
|---|---|---|---|---|---|---|
| 1902 | 432 | 0.608 | 4.109 | 35 | 1.105 | 3.253 |
| 1908 | 397 | 0.847 | 3.643 | 40 | 1.142 | 2.880 |
| 1909 | 453 | 0.69 | 4.468 | 45 | 0.893 | 2.957 |
| Name | Clean Reads | Clean Base | Q30 (%) | GC (%) | Mapped (%) | Coverage Ratio 1× (%) | Average Depth |
|---|---|---|---|---|---|---|---|
| CZ | 40,770,993 | 12,203,297,052 | 92.81 | 38.00 | 92.54 | 84.62 | 16 |
| LLYH | 38,237,712 | 11,441,633,036 | 93.57 | 37.40 | 95.91 | 82.79 | 17 |
| BY | 35,629,299 | 10,664,593,300 | 92.31 | 37.54 | 96.86 | 83.06 | 16 |
| CLA | 34,695,079 | 10,383,258,026 | 92.89 | 37.45 | 96.64 | 82.82 | 15 |
| R05 | 37,984,421 | 11,363,953,126 | 92.95 | 37.40 | 96.73 | 81.95 | 18 |
| S-pool-1 | 53,006,850 | 15,872,724,858 | 92.36 | 37.43 | 96.67 | 90.20 | 22 |
| L-pool-1 | 52,964,015 | 15,850,467,634 | 93.92 | 37.34 | 96.66 | 90.25 | 23 |
| S-pool-2 | 56,828,841 | 17,010,808,844 | 93.20 | 37.26 | 96.83 | 90.23 | 24 |
| L-pool-2 | 53,796,882 | 16,102,268,428 | 93.74 | 37.32 | 96.84 | 90.26 | 23 |
| S-pool-3 | 52,592,836 | 15,736,253,794 | 93.85 | 37.30 | 96.58 | 90.10 | 23 |
| L-pool-3 | 51,721,719 | 15,485,509,054 | 92.75 | 37.36 | 96.28 | 90.71 | 22 |
| Population | QTL | Chr. | Start | End | Interval (Mb) | No. of SNPs in the Interval | No. of Genes with Nonsynonymous Mutations |
|---|---|---|---|---|---|---|---|
| 1902 | rs1.1 | R1 | 13.06 | 16.37 | 3.31 | 4753 | 222 |
| rs4.1 | R4 | 40.51 | 42.59 | 2.08 | 5597 | 215 | |
| rs5.1 | R5 | 27.54 | 28.60 | 1.06 | 2279 | 94 | |
| 1908 | rs7.1 | R7 | 9.92 | 14.81 | 4.89 | 7471 | 368 |
| 1909 | rs2.1 | R2 | 32.07 | 33.43 | 1.36 | 5514 | 132 |
| rs4.2 | R4 | 42.71 | 45.81 | 2.10 | 2106 | 122 | |
| rs7.2 | R7 | 13.79 | 15.35 | 1.56 | 4954 | 137 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Q.; Wang, J.; Wang, W.; Hu, H.; Yan, Y.; Bao, C.; Hu, T. Identification of QTLs Controlling Radish Root Shape Using Multiple Populations. Horticulturae 2022, 8, 931. https://doi.org/10.3390/horticulturae8100931
Wei Q, Wang J, Wang W, Hu H, Yan Y, Bao C, Hu T. Identification of QTLs Controlling Radish Root Shape Using Multiple Populations. Horticulturae. 2022; 8(10):931. https://doi.org/10.3390/horticulturae8100931
Chicago/Turabian StyleWei, Qingzhen, Jinglei Wang, Wuhong Wang, Haijiao Hu, Yaqin Yan, Chonglai Bao, and Tianhua Hu. 2022. "Identification of QTLs Controlling Radish Root Shape Using Multiple Populations" Horticulturae 8, no. 10: 931. https://doi.org/10.3390/horticulturae8100931
APA StyleWei, Q., Wang, J., Wang, W., Hu, H., Yan, Y., Bao, C., & Hu, T. (2022). Identification of QTLs Controlling Radish Root Shape Using Multiple Populations. Horticulturae, 8(10), 931. https://doi.org/10.3390/horticulturae8100931